Synthesis of 6,6′-(S)-cyclo-2′-deoxyuridine featuring a unique Barbier-style cyclization†
Christopher S.
Theile
and
Larry W.
McLaughlin
*
Boston College, Department of Chemistry, Merkert Chemistry Center, 2609 Beacon Street Chestnut Hill, MA 02467, USA. E-mail: larry.mclaughlin@bc.edu; Fax: +01 617 552 2705; Tel: +01 617 552 3622
First published on 9th October 2012
Abstract
Here we present the synthesis of the novel nucleoside 6,6′-(S)-cyclo-2′-deoxyuridine (1). This efficient synthesis is carried out starting from 2′-deoxyuridine and reaches the deprotected product in few steps and high yield. Crystal structures of the product indicate that it is a very good structural mimic of thymidine as found in DNA duplexes. This rigidified nucleoside locks the uracil base in the anti conformation, positioning the nucleobase for Watson–Crick duplex formation at significant entropic savings. The structure of 6,6′-(S)-cyclo-2′-deoxyuridine maps more favorably onto naturally occurring nucleosides than previous cyclo nucleosides. The synthesis features a unique Barbier-style cyclization, expanding on Luche and Sarandeses allyl zinc additions, to create a 7-membered ring in good yield.
Due to their complex structural motifs and high functionalization, non-natural nucleosides present opportunities for the development of novel chemical strategies that can benefit the synthetic community at large. Cyclonucleosides have been targets of the nucleoside community due to their rigid geometry. A second linkage between the 5′-carbon on the sugar to the nucleobase fixes the base in the anti conformation, lowering the entropy associated with rotation of the base. Previous work on cyclo-uridine and cyclo-2′-deoxyuridine (compound 2, Fig. 1) has featured a direct linkage between the 5′ position of the sugar and the C6 position of the pyrimidine base.1–8 This bond “pulls” the base back from its native Watson and Crick bonding position, so that it can no longer effectively hydrogen bond in a duplex.1 The torsion angles are also not optimized for 6,5′-cyclo-2′-deoxyuridine (2). The χ angle, which relates the position of the base to the C1′–O5′ bond, adopts a position closer to that of a ribonucleoside.
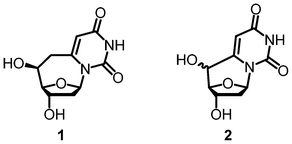 | ||
| Fig. 1 6,6′-(S)-cyclo-2′-deoxyuridine (1) and 6,5′-cyclo-2′-deoxyuridine (2). | ||
We predicted that by inserting a methylene group into the linkage, we could effectively “push” the base back toward its preferred position. The spacer would allow for a minimal amount of flexibility so the base can adopt a χ angle similar to what is observed in B-form DNA.9 The base will still be locked into the anti position. When inserted into oligonucleotide strands, we predict that preorganization of the base into its Watson and Crick conformation will lower the entropic costs associated with rotation around the glycosidic bond.
Here we present the first synthesis of 6,6′-(S)-cyclo-2′-deoxyuridine (1). Previous work from Ueda et al. produced the ribonucleoside, 6,6′-cyclouridine, lacking a 5′ OH and 8,6′-cyclo-adenosine.10–13 Otter et al. were able to produce a modified, ribose variant of 6,6′-cyclouridine with a 5′ OH, but the work relies on the use of diazomethane, which results in methylation of the N3 position, making studies related to duplex formation impossible.10–12,14–16 Eventually they were able to make the 6,6′-cyclouridine, but the synthesis was long and suffered from low yields.16 Our synthesis of 6,6′-(S)-cyclo-2′-deoxyuridine takes only seven steps, highlighted by a cyclization, yielding the newly formed 7-membered ring. Due to synthetic limitations the uracil base was utilized instead of the naturally occurring thymidine base, which prevents access to the cyclo bridge. Previous work has shown that deoxy-uridine is a sufficient mimic for thymidine.17–19
Our synthesis begins with a protection of the 3′ and 5′ hydroxyls of 3 with TBSCl (Scheme 1). We then carry out a one-pot multistep reaction to install a methyl–hydroxyl linkage on the C6 position of the base of 4.20 LDA is used to deprotonate the C6 position, which subsequently adds to dimethylformamide. After neutralization with acid, the aldehyde is reduced to the alcohol with NaBH4 to form 5. The resulting alcohol is then brominated in a two-step, one-pot reaction. The 5′ TBS group is removed under mild conditions with PPTS to make 7.21 This allows for the 5′ OH to be selectively oxidized with Dess Martin periodinane, yielding aldehyde 8. This product decomposes on silica, so the crude material is carried forward for the cyclization reaction.
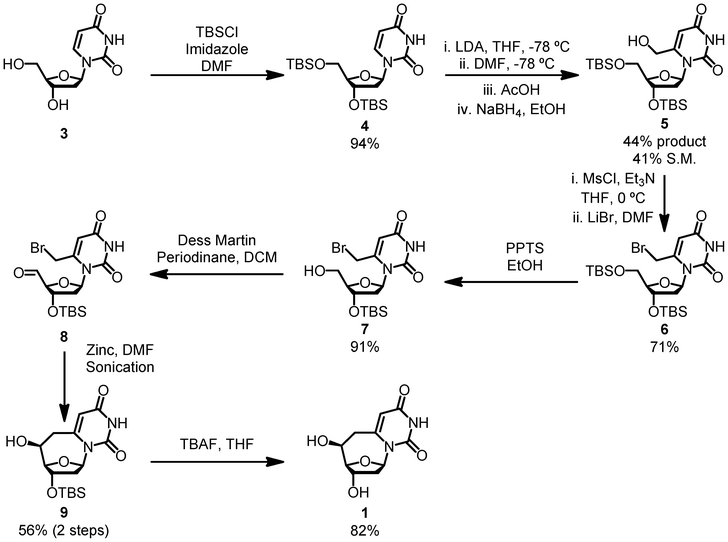 | ||
| Scheme 1 The synthesis of 6,6′-(S)-cyclo-2′-deoxyuridine. | ||
To form the 7-membered ring 9 in good yield and excellent purity we modified Luche and Sarandeses' zinc allylation Barbier-style reaction.22,23 This chemistry has exclusively been used to perform intermolecular addition of allyl halides to aldehydes. Applying this chemistry towards an intramolecular cyclization significantly increases the scope of the reaction.
Seven-membered rings have often created challenges in syntheses, and this reaction allows for an expansion of the toolbox to tackle molecules featuring this molecular framework.
A sonicator is employed to activate the zinc powder, allowing the cyclization to occur under mild conditions and with inexpensive reagents. Although the 56% 2-step yield is moderate, we hypothesize that the cyclization step is high yielding and the lower yield is due to the poor stability of compound 8. Wittig reactions on other 5′ aldehyde nucleosides in our lab have proceeded in approximately 50% yields over the 2-step procedure, reinforcing our hypothesis (unpublished results). TBAF deprotection of the 3′ position results in the desired product 1. Only a single diastereomer was observed by NMR and LC/MS analyses.
We were able to obtain a crystal structure of the final deprotected product, 1. This confirmed that the stereochemistry of the 5′ OH was in the S configuration, which was formed exclusively during the Barbier cyclization. Presumably the selectivity is controlled by zinc chelation; however, it is unclear which functional groups interact with the zinc metal to give the observed product, but further studies are being done in our lab to test the selectivity of this reaction on other substrates. Several attempts were made to invert the hydroxyl to give the R stereochemistry, but they either gave elimination products or starting material.
When the atoms on the nucleobase, as well as the 3′ and 5′ oxygen atoms of 1 were overlaid with the corresponding atoms from a crystal structure of thymidine that was cut from a Dickerson dodecamer B-form double helix, our product matches quite closely (Fig. 2).24 Conveniently the S diastereomer compares more favorably than the unobserved R, as the R configuration would cause the 5′ OH to be in front of the C4′–O bond of the sugar ring. The root mean squared deviation of the relevant atoms averaged 0.293 Å. The χ angle of our crystal is −112.59°, which is in the middle of the range usually adopted by deoxynucleotides of about −95° to −125°. Comparatively, the previously synthesized R and S diastereomers of 6,5′-cyclo-2′-deoxyuridine monomers were very poor mimics.1 Since the base is “pulled” backwards from the Watson and Crick bonding position, either the position of the backbone hydroxyls or the base is compromised to the extent that it cannot form stable duplexes. The χ angle is also −152.11°, which is closer to a ribonucleoside than a deoxynucleoside.
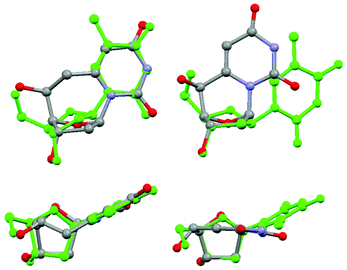 | ||
| Fig. 2 A front and top view of 6,6′-(S)-cyclo-2′-deoxyuridine, shown on the left, compared to 6,5′-(S)-cyclo-2′-deoxyuridine on the right. Both are overlayed with thymidine. | ||
Conclusions
In summary, we present a rapid and high yielding synthesis of 6,6′-(S)-cyclo-2′-deoxyuridine. This molecule locks the base in the anti position while maintaining proper positions of the hydrogen bonding face and the 3′ and 5′ hydroxyls. The synthesis featured a unique Barbier cyclization, expanding the scope of Luche and Sarandeses' zinc allylation chemistry beyond intermolecular additions to an intramolecular cyclization of a medium sized ring. The cyclization is carried out in high yield using low cost reagents and mild conditions without affecting other regions of complex functionality. Ongoing investigations in our lab will indicate how the ring-expanded cyclo linkage will affect the stability of an oligonucleotide strand. We are also currently exploring what features influence the selectivity observed during the cyclization. This molecule will allow us to probe enzymes associated with pyrimidine synthesis, such as thymidylate synthase, after monophosphorylation of the 5′ OH. Ring expanded cyclo-dU also has great potential to test the promiscuity of bacterial and eurkaryotic DNA polymerase enzymes after incorporation into the template strand, or as a triphosphate monomer.Acknowledgements
We would like to thank Dr Bo Li and Marek Domin for their assistance with the crystal structure and mass spectroscopy respectively, Christopher Pace and Azade Hosseini for LC/MS analysis, and Tyler Mann for assistance with optical rotation measurements. We would also like to acknowledge Dr Carl Christianson and Dr Mark Schlegel for providing useful insight in the preparation of the manuscript. Lastly, we would like to thank the NSF for funding this research.References
- H. Yueh, H. Yu, C. S. Theile, A. Pal, A. Horhota, N. J. Greco, C. V. Christianson and L. W. McLaughlin, Nucleosides, Nucleotides Nucleic Acids, 2012, 31, 661–679 CAS.
- C. S. Theile and L. W. McLaughlin, Chem. Commun., 2012, 48, 5587–5589 RSC.
- B. A. Otter, E. A. Falco and J. J. Fox, J. Org. Chem., 1976, 41, 3133–3137 CrossRef CAS.
- V. T. Perchyonok, Lett. Org. Chem., 2011, 8, 337–340 CrossRef CAS.
- Y. Suzuki, A. Matsuda and T. Ueda, Chem. Pharm. Bull., 1987, 35, 1808–1811 CrossRef CAS.
- T. Ueda, H. Usui, S. Shuto and H. Inoue, Chem. Pharm. Bull., 1984, 32, 3410–3416 CrossRef CAS.
- Y. Yoshimura, Y. Yamazaki, K. Wachi, S. Satoh and H. Takahata, Synlett, 2007, 111–114 CrossRef CAS.
- T. Ueda, S. Shuto, T. Sano, H. Usui and H. Inoue, Nucleic Acids Symp. Ser., 1982, 11, 5–8 CAS.
- W. Saenger, Principles of nucleic acid structure, Springer-Verlag, New York, 1984 Search PubMed.
- Y. Yamagata, K. Tomita, H. Usui, T. Sano and T. Ueda, Chem. Pharm. Bull., 1989, 37, 1971–1976 CrossRef CAS.
- T. Sano, H. Inoue and T. Ueda, Chem. Pharm. Bull., 1985, 33, 1856–1860 CrossRef CAS.
- Y. Yoshimura, A. Matsuda and T. Ueda, Chem. Pharm. Bull., 1989, 37, 660–664 CrossRef CAS.
- A. Matsuda and T. Ueda, Chem. Pharm. Bull., 1986, 34, 1573–1578 CrossRef CAS.
- B. A. Otter and E. A. Falco, Tetrahedron Lett., 1978, 19, 4383–4386 CrossRef.
- I. M. Sasson and B. A. Otter, J. Org. Chem., 1981, 46, 1114–1120 CrossRef CAS.
- I. M. Sasson and B. A. Otter, J. Heterocycl. Chem., 1987, 24, 1439–1444 CrossRef CAS.
- Z. H. Sun and L. W. McLaughlin, Biopolymers, 2007, 87, 183–195 CrossRef CAS.
- B. Golos, J. M. Dzik, Z. Kazimierczuk, J. Ciesla, Z. Zielinski, J. Jankowska, A. Kraszewski, J. Stawinski, W. Rode and D. Shugar, Biol. Chem., 2001, 382, 1439–1445 CrossRef CAS.
- T. Utagawa, H. Morisawa, S. Yamanaka, A. Yamazaki, F. Yoshinaga and Y. Hirose, Agric. Biol. Chem., 1985, 49, 3239–3246 CrossRef CAS.
- A. J. Matthews, P. K. Bhardwaj and A. Vasella, Helv. Chim. Acta, 2004, 87, 2273–2295 CrossRef CAS.
- M. P. Groziak and D. W. Thomas, J. Org. Chem., 2002, 67, 2152–2159 CrossRef CAS.
- C. Einhorn and J. L. Luche, J. Organomet. Chem., 1987, 322, 177–183 CrossRef CAS.
- J.-L. Luche and L. A. Sarandeses, Organozinc Reagents, 1999, pp. 307–323 Search PubMed.
- E. Johansson, G. Parkinson and S. Neidle, J. Mol. Biol., 2000, 300, 551–561 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and characterization data. CCDC 882419. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra21559d |
| This journal is © The Royal Society of Chemistry 2012 |