Synthesis, crystal structure and properties of a novel framework aluminium diphosphonate†
Martin P.
Attfield
*a,
Carlos
Mendieta-Tan
a,
Ryan N.
Telchadder
a and
Mark A.
Roberts
b
aCentre for Nanoporous Materials, School of Chemistry, The University of Manchester, Manchester, United Kingdom M13 9PL. E-mail: m.attfield@manchester.ac.uk; Fax: +44 161 275459; Tel: +44 161 3064467
bCLRC Daresbury Laboratory, Daresbury, Warrington, United Kingdom WA4 4AD
First published on 31st August 2012
Abstract
We report here the hydrothermal synthesis, crystal structure and properties of the hybrid inorganic–organic framework material Al2[O3PC6H4PO3](H2O)2F2·2H2O which is the seventh member of the group 13 metal diphosphonate framework series of formula M(III)2[O3PRPO3](H2O)2F2. The structure is formed from linear chains of corner-sharing AlO4F2 octahedra in which the two fluorine atoms are present in a trans configuration. The diphosphonate groups link the chains together through Al–O–P–O–Al bridges and through the phenylene groups to form a three-dimensional framework structure containing a one-dimensional channel system containing eight non-framework water molecules per unit cell. The framework structure remains crystalline with respect to the loss of the non-framework water molecules but removal of the framework water molecules results in a rapid loss of framework stability and crystallinity. The work demonstrates the potential to design the structure and void volume of a hybrid framework material through directed formation of the extended two-dimensional inorganic component and the packing of the organic component within the interlayer region.
1. Introduction
Inorganic–organic hybrid materials now form one of the largest families of functional material within the area of materials chemistry due to their diverse array of properties and the potential for the rational design of their structure and chemical functionality. The incorporation of the organic groups within these materials specifically allows for the possibility of synthesising materials with predetermined chemical functionality, properties and structures. The ability to design the structure of hybrid materials is of particular importance for framework hybrid materials where the actual framework geometry of the resultant material, for instance the pore and void size and shape, and the internal surface area, impart significant influence on the resultant performance for applications such as gas storage, separations, ion-exchange and catalysis.1–6 This specific need has fuelled work on the structural design of framework hybrid materials, in addition to the enduring desire of chemists to synthesise crystalline extended solid state materials with the type of precision already practised in synthetic organic chemistry.Structural design of framework hybrid materials has been demonstrated to differing extents by the groups of Yaghi and Férey amongst others, through the formation of a huge array of individual and isoreticular series of materials constructed from different zero-dimensional inorganic clusters, for example octahedra of zinc centred tetrahedra,7 twelve-coordinate clusters of zirconium centred square antiprisms,8 or triangular prisms of metal(III) centred octahedra,9–12 linked by an array of organic groups.
Less work has been reported on formation of designed framework hybrid materials containing inorganic components of higher dimensionality. Examples of isoreticular framework hybrid materials containing one-dimensional inorganic components include the many members of the MIL-53 metal dicarboxylate family of materials,13–15 the MOF-74 series,16 the metal piperazinylphosphonates STA-12, -1617,18 and zinc diphosphonates ZAG-4, -6.19,20 Metal diphosphonates provide a rich source of framework hybrid materials that contain two-dimensional inorganic components and include several designed isoreticular series that contain identical two-dimensional inorganic components connected into framework materials by diphosphonate groups of varying length.20,21
One area of design for which less research has been reported is the structural control of the extended inorganic components in framework hybrid systems for which the inorganic section may exist as isomers of different configuration. In our continuing investigation of group 13 metal diphosphonate framework hybrid materials we have synthesized six framework materials of formula M(III)2[O3PRPO3](H2O)2F2.22–25 Four of these materials (M = Al, R = C2H4, C3H6, C4H8 and M = Ga, R = C2H4)22–24 contain two dimensional inorganic components consisting of corrugated chains of corner-sharing MO4F2 octahedra. Three of these four materials (M = Al, R = C2H4, C4H8 and M = Ga, R = C2H4)22,24 form part of an isoreticular series. The remaining two materials (M = Ga, R = C2H4, CH2C6H4CH2)25 of the six mentioned form the first two members of another isoreticular series that has a more open framework structure because the two dimensional inorganic component consists of relatively linear chains of corner-sharing MO4F2 octahedra. Five of these materials have been synthesised from identical reagent components treated under similar reaction conditions. Our results suggest that both configurations of the chains of corner-sharing MO4F2 octahedra may easily be formed under the reported synthetic conditions and we have also shown that it is possible to direct formation of the linear chain only by design of the organic group within the diphosphonic acid (H2O3PCH2C6H4CH2PO3H2) forming the framework.25 However, this example of the design strategy yielded a framework structure without the void space necessary to contain non-framework species due to the inherent flexibility within the organic group. Here we demonstrate that by judicial design of the diphosphonic acid it is possible to synthesise the new material Al2[O3PC6H4PO3](H2O)2F2·2H2O (1) that is the third member of the series of framework formula M2[O3PRPO3](H2O)2F2 that contains linear chains of corner-sharing MO4F2 octahedra. The directed formation of the linear chains of octahedra and the greater void volume in (1) arise from the non-flexible nature of the phenylene group within the diphosphonic acid.
2. Experimental
2.1 Materials and methods
The reagents used to synthesise (1) were Al2(SO4)3·18H2O (Aldrich), 1, 4-benzenediphosphonic acid (Epsilon Chimie), pyridine (Aldrich), HF (48% wt in H2O, Aristar) and distilled water. All the reagents were used without further purification.Microprobe measurements were made using a FEI Quanta 200/EDAX Genesis system with an X-ray cone fitted, operating with an accelerating voltage of 15 kV, a beam diameter of 1 μm, under a vacuum of 0.3 Torr. Analysis of separate regions of the samples gave quantitative results for Al![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P ratios and qualitative results for fluorine.
:P ratios and qualitative results for fluorine.
The magic angle spinning solid state nuclear magnetic resonance (MAS SSNMR) 19F spectrum for (1) was recorded using a Varian Unity Inova spectrometer operating at a frequency of 282.08 MHz, a sample spinning speed of 14.5 KHz, with recycle delays of 1 s and using CFCl3 as a reference.
Room temperature X-ray diffraction patterns were collected using a Panalytical X'Pert diffractometer employing Cu-Kα1+2 radiation and a RTMS X'Celerator detector. Synchrotron X-ray data were collected on a sample contained in a 0.5 mm diameter Lindemann glass capillary tube mounted on the high resolution X-ray powder diffractometer at station 9.1, Daresbury, SRS, UK. The incident X-ray wavelength was 0.99999 Å, selected using a Si(111) monochromator and the capillary tube was spun during data collection to minimize preferred orientation and sampling effects. Data were collected with a step size of 0.01° 2θ for 4 s per step between 3 and 16° 2θ, 6 s per step between 16 and 40° 2θ, and 12 s per step between 40 and 80° 2θ. Corrections were made for synchrotron beam intensity decay through comparison with a beam flux monitor.
Thermogravimetric analysis (TGA) data were collected using a Seiko TG/DTA 220 thermogravimetric analyser with the sample heated in an open platinum crucible under flowing nitrogen from 25 to 1000 °C at a heating rate of 5 °C min−1.
Thermodiffraction patterns were collected using a Panalytical X'Pert Pro diffractometer, in reflection geometry employing Cu-Kα1+2 radiation and an RTMS X'Celerator detector, fitted with an Anton Paar XRK 900 high-temperature furnace stage. The sample was heated in static air at a heating rate of 10 °C min−1, with patterns being collect at 20, 25 or 50 °C intervals within the 30 to 700 °C temperature range of the experiment. The X-ray diffraction patterns were all collected in the 3–50° 2θ range with a scan time of 15.7 min per scan.
2.2 Synthesis and preliminary characterisation
Compound (1) was synthesized by mixing together Al2(SO4)3·18H2O (1.120 g, 1.68 mmol), H2O3PC6H4PO3H2 (0.863 g, 3.62 mmol), pyridine (7.245 g, 91.71 mmol), HF (0.602 g , 14.45 mmol 48% wt HF in H2O) and H2O (4.047 g, 225 mmol) to form a synthesis gel Al:H2O3PC6H4PO3H2:pyridine:HF:H2O of molar ratio 1:1.08:27.3:4.30:81.1. The reagent mixture was loaded into a 23 ml Teflon-lined steel autoclave and heated for 4 days at 200 °C. The autoclave was allowed to cool to room temperature in air after the heating period. The resultant crystalline product was separated by suction filtration, washed with distilled water, and dried under ambient conditions to reveal a white polycrystalline powder. EDXA analysis on the product showed that the powder contained Al:P in the ratio 1:1.1 and the presence of fluorine.
2.3 Ab initio powder X-ray structure determination of (1)
The laboratory X-ray data used to determine the unit cell parameters of (1) were collected during a 18 h scan. The first 20 low angle Bragg reflections were used to determine the monoclinic unit cell parameters using the auto-indexing program ITO (FOM(20) = 32).26 Combined use of the Chekcell program within the LMGP suite27 and visual inspection of the synchrotron X-ray diffraction pattern revealed systematic absences consistent with the space group C2/c. Observed structure factors were extracted from this diffraction data by the Le Bail method28 as implemented in the GSAS suite of programs.29 The background of the diffraction profile was fitted with a linear interpolation function running through 50 fixed points in the profile and the peak profiles were described by a pseudo–Voigt function. The extracted structure factors were used in the direct methods program SIRPOW97,30via the EXPO interface,31 to provide the Al, P and some of the O atoms of the structure. This output was used as the starting model for the Rietveld refinement, again using the GSAS suite of programs.29 The remaining atoms of the structure were located from difference Fourier maps. This includes the four C atoms of the phenylene rings that are disordered over two possible positions. The occupancy of the C atoms that are disordered over the two positions of the phenylene rings were fixed at values of a half. Initially, soft constraints were applied to the Al–O/F, P–O/C, and C–C distances within the structure with the soft constraint weighting factor fixed at a high value. The soft constraint weighting factor was reduced as the refinement progressed to yield a value for which the bond distances remained chemically sensible. The final cycle of least squares refinement included the background coefficients, zero point, lattice parameters, peak profile parameters, and positional and isotropic displacement parameters for all atoms in the structure. The displacement parameters of all the atoms were constrained to have the same value during refinement. The Rietveld refinement gave reasonable bond lengths and angles. However the final residuals and overall quality of fit were higher than expected. The crystallographic data and structure refinement parameters are given in Table 1, atomic coordinates, isotropic thermal parameters and fractional occupancies are provided in Table 2, and selected bond distances are presented in Table 3. The final observed, calculated and difference profiles are plotted in Fig. 1 and the asymmetric unit is shown in Fig. 2. CCDC-887707 contain the supplementary crystallographic data, including full geometric data, for this paper.†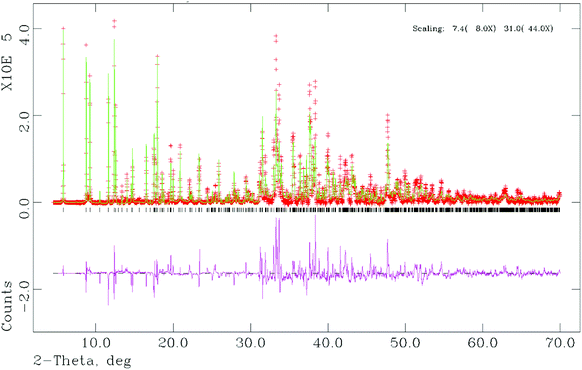 | ||
| Fig. 1 The final observed (crosses) calculated (lines) and difference profiles (lines) for (1). Tick-marks show reflection positions and scaling of the data is × 1 for 0–7.4 2θ, × 8 for 7.4–31.0 2θ and × 44 for 31.0–70.0 2θ. | ||
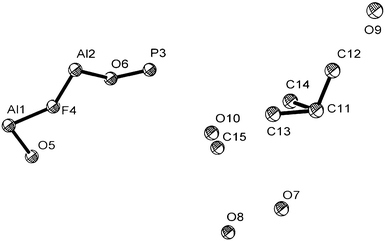 | ||
| Fig. 2 The asymmetric unit of (1). All displacement ellipsoids are shown at the 50% probability level. | ||
| Empirical formula | C3H6AlPO5F |
|---|---|
| M r | 199.03 |
| Crystal system | Monoclinic |
| a | 4.9530(1) Å |
| b | 13.0559(3) Å |
| c | 19.7444(5) Å |
| b | 93.651(2)° |
| V | 1274.19(5) Å3 |
| Temperature | 298 K |
| Space group | C2/c |
| Z | 8 |
| No. of reflections | 1004 |
| R wp | 10.19 |
| R(F) | 13.20 |
| R exp | 3.60 |
| Atom | x | y | z | Uiso (Å2) | Occupancy |
|---|---|---|---|---|---|
| Al1 | −0.250000 | −0.250000 | 0.000000 | 0.027(1) | 1 |
| Al2 | 0.000000 | 0.000000 | 0.000000 | 0.027(1) | 1 |
| P3 | 0.5137(12) | 0.0668(4) | 0.08556(21) | 0.027(1) | 1 |
| F4 | −0.0437(18) | −0.1365(4) | 0.0253(4) | 0.027(1) | 1 |
| O5 | −0.0626(24 | −0.3183(6) | 0.07242(47) | 0.027(1) | 1 |
| O6 | 0.3256(14) | −0.0139(7) | 0.05159(48) | 0.027(1) | 1 |
| O7 | 0.0037(22) | 0.2903(10) | 0.43265(51) | 0.027(1) | 1 |
| O8 | 0.1766(17) | 0.0417(9) | 0.42179(34) | 0.027(1) | 1 |
| O9 | 0.000000 | 0.8648(14) | 0.250000 | 0.027(1) | 1 |
| O10 | 0.000000 | 0.2547(13) | 0.250000 | 0.027(1) | 1 |
| C11 | −0.0014(24) | 0.5645(9) | 0.32530(27) | 0.027(1) | 1 |
| C12 | 0.1514(80) | 0.6395(24) | 0.29039(41) | 0.027(1) | 0.5 |
| C13 | −0.1323(82) | 0.4850(20) | 0.28499(40) | 0.027(1) | 0.5 |
| C14 | 0.1351(76) | 0.4858(20) | 0.28988(40) | 0.027(1) | 0.5 |
| C15 | 0.3564(80) | 0.1406(22) | 0.28393(39) | 0.027(1) | 0.5 |
| a −0.5 − x, −0.5 − y, −z; b −x − 0.5, y − 0.5, 0.5−z; c −x + 1, y, −z + 0.5; d 0.5 − x, −0.5 + y, −z + 0.5; e x − 0.5, y + 0.5, z. | |||
|---|---|---|---|
| Al1_F4 × 2a | 1.851(5) | Al2_F4 × 2 | 1.867(5) |
| Al1_O5 × 2a | 1.880(5) | Al2_O6 × 2 | 1.861(5) |
| All_O7 × 2b | 1.959(5) | Al2_O8 × 2 | 1.902(5) |
| P3_O5 | 1.564(7) | P3_O8c | 1.584(6) |
| P3_O6 | 1.533(6) | P3_C11d | 1.765(5) |
| C11_C12 | 1.440(7) | C11_C13 | 1.437(7) |
| C11_C14 | 1.436(7) | C11_C15e | 1.441(7) |
| C12_C15 | 1.466(7) | C13_C14 | 1.477(7) |
3. Results and discussion
The structure of (1) is shown in Fig. 3. The structure consists of chains of corner sharing AlO4F2 octahedra with bridging fluorine atoms (F4) linking the octahedra as shown in Fig. 3. These chains consist of two types of AlO4F2 octahedra that both contain aluminium atoms coordinated to two fluorine atoms in a trans configuration. The remaining corners of one of the aluminium centred octahedra (Al2) are occupied by four oxygen atoms (O6 and O8) from four different diphosphonate groups, while the remaining apices of the other aluminium centred octahedra (Al1) are occupied by two oxygen atoms (O5) from two different diphosphonate groups and by two water molecules (O7). The benzenediphosphonate groups link the chains of octahedra in the [100] and the [001] directions, as shown in Fig. 3, to form a framework structure with a one-dimensional channel system. The channels of this framework structure run along the [100] direction and are large enough (7.21 Å [O7−O7] × 4.64 Å [C14−C15]) to locate four pairs of non-framework water molecules, O9 and O10, per unit cell with each pair being situated in the same horizontal plane relative to one another. The structure and form of the chains of AlO4F2 octahedra linked by the tetrahedral PCO3 moieties of the diphosphonate groups in (1) are analogous to the chains of FeO4(OH)2 octahedra connected by tetrahedral PO4 moieties found in the mineral Laueite [MnFe2(PO4)2(OH)2(H2O)2(H2O)6].32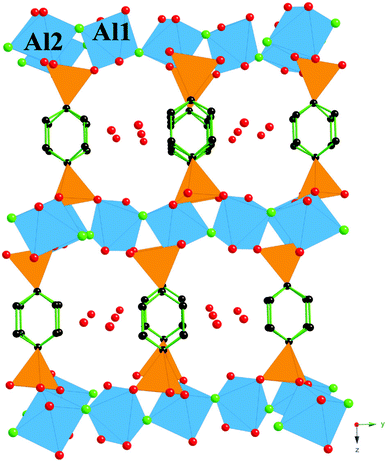 | ||
| Fig. 3 A ball and stick and polyhedral perspective representation of the framework structure of (1) viewed along the a axis. Atom representation: blue octahedra (Al), orange tetrahedra (P), green balls (F), red balls (O) and black balls (C). Hydrogen atoms are omitted for clarity. | ||
The framework structure of (1) is isomeric with all the other members of the family of materials with framework formula M2[O3PRPO3](H2O)2F2, however it is different to the members of the series that contain corrugated chains of corner-sharing octahedra as it contains linear chains of corner-sharing octahedra. Compound (1) is also not a complete member of the isoreticular series that contain linear chains of corner-sharing octahedra.25 This is because the chains of octahedra are directed along the [110] and [−110] directions in adjacent layers along the c axis in (1) while the linear chains of corner-sharing octahedra in the other materials are aligned in one direction only.
Compound (1) is also unlike all the other members of this family in that the organic group has two possible orientations, as shown in Fig. 4, and is statically disordered between the orientations. Only one of the two possible orientations of the phenylene ring is present per diphosphonate group. Such a phenomenon has also been observed and inferred in other crystal structures of metal phenylene-, biphenylenediphosphonates and phenylphosphonates.33–35 It is likely that the structure reported here is based upon a crystallographic subcell of the true structure of this material. This is reflected in the poorer than usual fit of the calculated diffraction pattern derived from the structural model to the observed X-ray data and the presence of a major broad resonance in the 19F MAS SSNMR spectrum of (1) centred at −144.6 ppm (see ESI Fig. 1†). The latter is not in agreement with the one sharp resonance for the 19F nucleus suggested from the crystal structure. The complete structure of (1) is likely to contain an ordered arrangement of the two orientations of the phenylene rings as has been observed in some lanthanum phenylphosphonates. Little trace of X-ray diffraction peaks related to a supercell for this structure are present in the synchrotron X-ray data making a full structure determination of the material difficult. However, the basic chemical connectivity of the material is fully determined within the reported structural model. The chemical shift of the major resonance arising from the 19F nuclei is consistent with those previously observed for bridging fluorine atoms.22,36 The other smaller resonances at −121.9 and −154.6 ppm in the 19F MAS SSNMR spectrum of (1) indicate the presence of additional small amounts of impurity phases in the sample. However, there is no evidence for the impurity in the synchrotron diffraction pattern indicating the impurity is of low concentration or amorphous in nature.
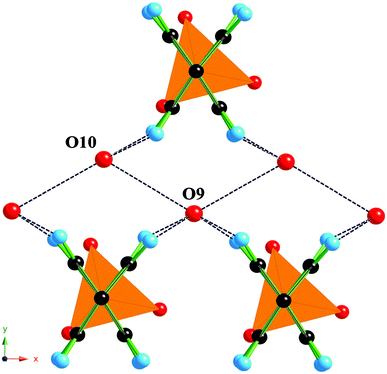 | ||
| Fig. 4 A ball and stick and polyhedral representation of the pore section of (1) displaying the two possible orientations of the phenylene rings for each disphosphonate linker and the intermolecular interactions between the oxygen atoms of the non-framework water molecules and the hydrogen atoms of the phenylene rings. Atom representation: orange tetrahedra (P), red balls (O), black balls (C) and blue balls (H). Intermolecular bonding interactions are represented by black dotted lines. | ||
The non-framework water molecules, O9 and O10, are bound tightly within the void volume of the framework by a variety of intermolecular hydrogen-bonding interactions as shown in Fig. 4. The non-framework water molecules are ideally arranged to form moderately strong hydrogen bonds between themselves with a O9⋯O10 hydrogen-bonding distance of 2.864 Å and O9–O10–O9 angles of 119.72°. Intermolecular bonding interactions with framework atoms also contribute significantly to the overall interactions of the non-framework water molecules evidenced by the hydrogen-bonding distances of 3.635 Å for O10⋯O7 and the C–H⋯O9/O10 interactions for which the H⋯O9/O10 distances lie in the range 1.813–2.093 Å. The H atoms were geometrically placed on the carbon atoms of the phenylene rings for these calculations.
The successful synthesis of (1) demonstrates two elements of structural design that may be appreciated when the structure is compared to other members of the family of materials with framework formula M2[O3PRPO3](H2O)2F2. Several of these isomeric materials (M = Al, R = C2H4, C3H6, C4H8 and M = Ga, R = C2H4)22–24 contain two dimensional inorganic components consisting of corrugated chains of corner-sharing MO4F2 octahedra as is seen in Fig. 5a for the framework of Al2[O3PC2H4PO3](H2O)2F2.22 Use of a large, rigid organic linker such as 1,4-benzenediphosphonic acid prevents the corrugated chains of corner-sharing MO4F2 octahedra from forming as the diphosphonate linker is too rigid and long to allow connection of the corrugated chains at the appropriate points in adjacent layers without causing huge strain within the chain of octahedra. Use of the rigid benzenediphosphonate linker in the synthesis directs formation of the inorganic layers containing the linear chains of corner-sharing MO4F2 octahedra as the geometric configuration of the latter allows more readily for connection of the inorganic layers to form the framework of the material. Formation of the layers containing the linear chains forms more open frameworks as the horizontal separation between the CPO3 groups of adjacent diphosphonate groups is greater in the inorganic layers containing the linear chains of MO4F2 octahedra. This element of design was demonstrated in the formation of Ga2[O3PCH2C6H4CH2PO3](H2O)2F225 in which the [1, 4-phenylenebis(methylene)]diphosphonate linker contains a relatively long rigid component, the phenylene ring. However, this linker also contains the methylene groups which allow the diphosphonate linker to adopt a suitable conformation to pack within the interlayer space between inorganic components such that no void volume is present within the resultant framework as shown in Fig. 5b. Use of a straight linker such as the 1,4-benzenediphosphonate prevents such a close packing of the diphosphonate groups to occur by design and so directs formation of a framework with void volume that is occupied by non-framework water molecules.
2F2·H2O and (b) Ga2[O3PCH2C6H4CH2PO3](H2O)2F2 viewed along the a axis. Atom representation: blue octahedra (Al/Ga), orange tetrahedra (P), green balls (F), red balls (O) and black balls (C). Hydrogen atoms are omitted for clarity.](/image/article/2012/RA/c2ra21930a/c2ra21930a-f5.gif) | ||
| Fig. 5 A ball and stick and polyhedral representation of the framework structure of (a) Al2[O3PC2H4PO3](H2O)2F2·H2O and (b) Ga2[O3PCH2C6H4CH2PO3](H2O)2F2 viewed along the a axis. Atom representation: blue octahedra (Al/Ga), orange tetrahedra (P), green balls (F), red balls (O) and black balls (C). Hydrogen atoms are omitted for clarity. | ||
The thermogravimetric data for (1) is shown in ESI Fig. 2.† The first mass loss of 9.2%, measured between 202 and 337 °C, corresponds to the loss of the two non-framework water molecules (calculated 9.0%). A slightly slower mass loss of 9.7% occurs subsequently between 337 and 512 °C that corresponds to the loss of the two framework water molecules (calculated 9.0%). Finally a more gradual mass loss of 12.3% occurs between 512 and 977 °C. The latter loss arises from loss of fluoride from the system and incomplete degradation of the organic components.
The thermodiffraction data of (1) are shown in Fig. 6. From room temperature up to 220 °C there are only minimal changes in the X-ray powder diffraction patterns of the material. From 220 °C to 320 °C a structural transition is seen to occur with some peaks decreasing in intensity and re-emerging at higher 2θ values and other peaks disappearing. Overall the new material is less crystalline than the original material. The new phase remains crystalline up to 550 °C before becoming X-ray amorphous by 600 °C.
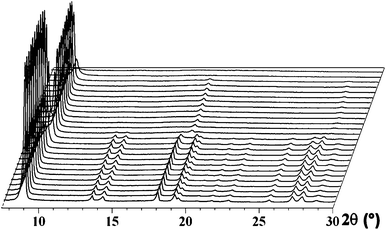 | ||
| Fig. 6 The thermodiffraction patterns of (1). The diffraction profiles are recorded at temperatures 30–150 °C in 20 °C intervals, 150–200 °C in 25 °C intervals, 200–400 °C in 20 °C intervals, 400–500 °C in 25 °C intervals and 500–650 °C in 50 °C intervals from the front to the back of the figure. | ||
The crystal structure, thermogravimetric and thermodiffraction data suggest that the framework structure of (1) contains tightly bound non-framework water molecules that do not leave the structure until the relatively high temperature of 200 °C. At this temperature rapid loss of the non-framework water molecules begin to occur accompanied by a structural transformation of the framework. The shifting of the diffraction peaks to higher 2θ values indicates the unit cell is decreasing as would be expected for removal of the non-framework water molecules from the structure. Between 337 and 512 °C the loss of the two framework water molecules occurs. The framework remains crystalline during this process but becomes amorphous at 550 °C when all the water is removed. Overall the framework structure of (1) remains stable with respect to the loss of the non-framework water molecules but removal of the framework water molecules results in a rapid loss of framework stability and crystallinity of the material. The thermal behaviour of (1) is similar to that observed for other members of this family of isomeric framework materials.22–25
The gas adsorption and solvent exchange properties of (1) were tested with little success for either property. This reflects the tight binding of the two non-framework water molecules within the pores of the material that hinders solvent exchange. Gas adsorption within the void volume of (1) was unsuccessful as there is not a suitable temperature range between loss of the non-framework water and the framework water to be able to activate the material for successful gas adsorption. Also, the thermodiffraction data indicates a structural change during loss of the non-framework water molecules that results in formation of a less crystalline material with a smaller unit cell volume. The structural changes arising upon removal of the tightly bound non-framework water molecules presumably coincide with a loss of, or access to, the void volume that prevent gas adsorption into the internal void volume of the material.
4 Conclusions
In this work we demonstrate that careful design of the constituent diphosphonic acid enables a new member of the family of materials with framework formula M2[O3PRPO3](H2O)2F2 to be formed in which formation of the two-dimensional extended inorganic component is directed and close packing of the organic linker is prevented. The work further exemplifies that a design approach can be applied to dual aspects, the extended inorganic component and the packing within the interlayer region, of the structural control of framework hybrid systems and demonstrates further the potential for full rational design of the structure and functionality of hybrid materials.Acknowledgements
The authors thank the EPSRC and CCLRC for financial support and beamtime allocation for the synchrotron component of the work using Station 9.1, Daresbury Laboratory and Dr D. Apperley of the EPSRC Solid State NMR Service, University of Durham, UK for collection of the SS MAS NMR data. MPA thanks the Royal Society for provision of a University Research Fellowship.References
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213 RSC.
- L. R. MacGillivray, Metal–organic frameworks: Design and application, Wiley, 2010 Search PubMed.
- S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276 RSC.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3 CrossRef CAS.
- M. J. Rosseinsky, Microporous Mesoporous Mater., 2004, 73, 15 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS.
- J. Hafizovic Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850 CrossRef CAS.
- G. Ferey, C. Serre, C. Mellot-Draznieks, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296 CrossRef CAS.
- G. Ferey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040 CrossRef CAS.
- A. Sonnauer, F. Hoffmann, M. Froba, L. Kienle, V. Duppel, M. Thommes, C. Serre, G. Ferey and N. Stock, Angew. Chem., Int. Ed., 2009, 48, 3791 CrossRef CAS.
- C. Serre, F. Millange, S. Surble and G. Ferey, Angew. Chem., Int. Ed., 2004, 43, 6285 CrossRef.
- C. Volkringer, T. Loiseau, N. Guillou, G. Ferey, M. Haouas, F. Taulelle, E. Elkaim and N. Stock, Inorg. Chem., 2010, 49, 9852 CrossRef CAS.
- T. Ahnfeldt, D. Gunzelmann, T. Loiseau, D. Hirsemann, J. Senker, G. Ferey and N. Stock, Inorg. Chem., 2009, 48, 3057 CrossRef CAS.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Ferey, Chem.–Eur. J., 2004, 10, 1373 CrossRef CAS.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gandara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018 CrossRef CAS.
- J. A. Groves, S. R.Miller, S. J.Warrender, C. Mellot-Draznieks, P. Lightfoot and P. A. Wright, Chem. Commun., 2006, 3305 RSC.
- M. T. Wharmby, J. P. S. Mowat, S. P. Thompson and P. A. Wright, J. Am. Chem. Soc., 2011, 133, 1266 CrossRef CAS.
- R. B.Fu, X. T.Wu, S. M. Hu, J. J. Zhang, Z. Y.Fu and W. X. Du, Polyhedron, 2003, 22, 2739 CrossRef.
- K. J. Gagnon, H. P. Perry and A. Clearfield, Chem. Rev., 2012, 112, 1034 CrossRef CAS.
- W. Ouellette, M. H. Yu, C. J. O'Connor and J. Zubieta, Inorg. Chem., 2006, 45, 3224 CrossRef CAS.
- H. G. Harvey, S. J. Teat and M. P. Attfield, J. Mater. Chem., 2000, 10, 2632 RSC.
- H. G. Harvey, B. Slater and M. P. Attfield, Chem.–Eur. J., 2004, 10, 3270 CrossRef CAS.
- Z. Yuan, W. Clegg and M. P. Attfield, J. Porous Mater., 2006, 13, 207 CrossRef CAS.
- H. G. Harvey, A. C. Herve, H. C. Hailes and M. P. Attfield, Chem. Mater., 2004, 16, 3756 CrossRef CAS.
- J. Visser, J. Appl. Crystallogr., 1969, 2, 89 CrossRef CAS.
- J. Laugier and B. Bochu, LMGP-Suite of Programs for the interpretation of X-ray Experiments, ENSP/Laboratoire des Matériaux et du Génie Physique, Saint Martin d'Hères, France, 1995 Search PubMed.
- A. Le Bail, H. Duroy and J. Fourquet, Mater. Res. Bull., 1988, 23, 447 CrossRef CAS.
- R. B. Von Dreele and A. C. Larson, GSAS, General Structure Analysis System, Regents of the University of California, LANSCE, Los Alamos National Laboratory, 1995 Search PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, SIRPOW97, Istituto di Ricerca per lo Sviluppo di Metodologie Cristallografiche (IRMEC), Bari, Italy, 1997 Search PubMed.
- A. Altomare, M. C.Burla, M. Camalli, B. Carrozzini, G. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Rizzi, J. Appl. Crystallogr., 1999, 32, 339 CrossRef CAS.
- P. B. Moore, Am. Min., 1965, 50, 1884 CAS.
- D. M. Poojary, B. Zhang, P. Bellinghausen and A. Clearfield, Inorg. Chem., 1996, 35, 5254 CrossRef CAS.
- K. J. Martin, P. J. Squattrito and A. Clearfield, Inorg. Chim. Acta, 1989, 155, 7 CrossRef CAS.
- R. C.Wang, Y. Zhang, H. Hu, R. R. Frausto and A. Clearfield, Chem. Mater., 1992, 4, 864 CrossRef.
- M. P. Attfield, C. Mendieta-Tan, Z. Yuan and W. Clegg, Solid State Sci., 2008, 10, 1124 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: 19F MAS SSNMR spectrum, thermogravimetric data and crystallographic information file CCDC 887707 for (1). See DOI: 10.1039/c2ra21930a |
| This journal is © The Royal Society of Chemistry 2012 |