Generally applicable procedure for in situ formation of fluorescent protein-gold nanoconstructs
Sondre
Volden
a,
Sina M.
Lystvet
a,
Øyvind
Halskau
b and
Wilhelm R.
Glomm
*a
aUgelstad Laboratory, Department of Chemical Engineering, Norwegian University of Science and Technology (NTNU), N-7491 Trondheim, Norway. E-mail: glomm@nt.ntnu.no; Tel: (+47) 735594158
bDepartment of Molecular Biology, University of Bergen, Thormøhlensgt 55 90, 5008 Bergen, Norway
First published on 22nd October 2012
Abstract
Small noble metal nanoclusters can be formed in situ by direct reduction and stabilization of a metal precursor by biomolecules such as proteins. Considering the diversity in amino acid composition of proteins, and hence their reductive ability, a general method for synthesis of gold nanoclusters using proteins is presented here. A range of proteins (bovine serum albumin, fibrinogen, α-lactalbumin, lysozyme, cytochrome c, myoglobin, β-lactoglobulin and α-chymotrypsin) have been studied, based on size, isoelectric point, flexibility and 3-dimensional structure. Results show protein-gold nanoconstructs with complex protein-specific photophysical properties. The effect on the 3-dimensional conformation of the proteins upon formation of gold nanoclusters and/or nanoparticles within the protein structure is also shown to be highly protein-dependent. A general mechanism for the formation of protein-gold nanoconstructs is proposed, based on charge density matching, yielding a high local concentration of the metal precursor on the protein structure which in turn can nucleate, grow and be stabilized by amino acid residues in the protein.
Introduction
Protein-stabilized fluorescent gold and silver nanoclusters (NCs) of subnanometer size have attracted a great deal of interest, owing to their potential use in a wide range of applications such as sensing,1 biolabeling and imaging,2–4 and even as enzyme mimics.5 Unlike gold and silver nanoparticles, NCs are fluorescent, and do not exhibit localized surface plasmon resonance (LSPR) or the surface chemistry of the bulk metal. The NCs are typically of sizes comparable to the Fermi wavelength (∼0.5 nm for Ag and Au) and display molecular-like, size-dependent fluorescence likely due to the transition between discrete molecule-like electronic states. Absorption and emission properties of the NCs can be described as a function of the number of atoms N as EFermi/N1/3 according to the Jellium energy scaling law.6Recently, Xie et al.7 reported a one-pot, “green” synthetic route for the preparation of Au NCs using the intrinsic reduction potential of bovine serum albumin (BSA) at physiological temperature and basic conditions (pH > 10). The resulting Au NCs were found to consist mostly of 25 gold atoms (Au25), with red emission (λmax = 640 nm) and a quantum yield of ∼6%. Since this seminal paper, other authors have reported AuNC formation using a similar biomineralization-inspired procedure using BSA3,5,8 as well as other proteins such as transferrin, apoferritin,4 and horseradish peroxidase.1 Modifications to the original synthesis procedure typically involve the addition of a reducing agent such as ascorbic acid2,3,8 in order to trigger nucleation of NCs at lower protein concentrations than initially reported. Procedures relying on the addition of an extrinsic reducing agent (hereafter referred to as “extrinsic” protocols) also offer the advantage of short reaction times and negligible competition from amino acid residue-induced reduction of the metal precursor, leading to a well-defined reduction and growth mechanism. Conversely, biomineralization-inspired synthetic procedures (hereafter referred to as “intrinsic” protocols) rely solely on the reduction potential of amino acid residues in the polypeptide chain under the reaction conditions (pH, temperature). Here, tyrosine7 and histidine4 have been proposed as the amino acids responsible for reduction of gold precursor in BSA and apoferritin, respectively.
As the amino acid composition varies greatly between proteins, the capacity to produce nanoclusters as well as the nature of the produced nanoclusters is expected to be protein-dependent. Here, we present a generally applicable synthetic procedure for in situ modification of proteins with gold nanoclusters, yielding protein-AuNCs and AuNPs with tuneable complex fluorescence. A small library of eight proteins of different molecular weights, charge properties and conformational flexibilities has been utilized, showing that nanocluster/nanoparticle formation, as well as the accompanying conformational changes and photophysical properties, are highly protein-dependent. As both the stability and function of the final protein-gold construct depend critically on the degree of change in protein conformation as well as the characteristics of the gold nanoclusters, the results obtained here can be used for the rational design of materials for a wide range of applications, including catalysis, bio-sensing and -labeling, as well as for imaging purposes. We also present a general mechanism for the in situ nanocluster formation, which could be beneficial for research in the aforementioned areas.
Materials and methods
Materials and solutions
Bovine serum albumin (BSA), bovine milk α-lactalbumin type I (BLA, >85%), (horse heart) myoglobin (Mb, >90%), (chicken egg white) lysozyme (Lyz), bovine pancreas α-chymotrypsin (CTR), bovine plasma fibrinogen (Fib) and bovine milk β-lactoglobulin (BLG) were purchased from Sigma. Bovine heart cytochrome c (Cyt, >95%) was purchased from BioChemika, and hydrogen tetrachloroaurate (TCAA) was purchased from Acros.PBS-buffer was made by mixing two solutions; i) K2HPO4 (50 mM) and KCl (150 mM) and ii) KH2PO4 (50 mM) and KCl (150 mM), until pH was 7.4. Protein-nanoclusters were made by mixing protein (0.746 mM, 1 ml, 37 °C) with TCAA (10 mM, 1 ml, 37 °C). After stirring for 2 min NaOH (1 M, 100 μl, 37 °C) was added.7 After one week at 37 °C, the nanocluster solutions were dialyzed against PBS (400 ml) for about 60 h, using a membrane with a molecular weight cut-off of 12.4 kDa. After measuring UV-vis, the solutions were diluted with PBS to 3.6 μM for UV-vis and fluorescence measurements. Stock solutions of native protein were made by dissolving protein in PBS to 25 μM. For UV-vis and steady-state measurements the protein stock solutions were diluted to 3.6 μM using PBS-buffer.
UV-Visible spectroscopy
UV-vis spectra were collected using a Shimadzu UV-2401PC spectrophotometer, over the wavelength range of 190–1100 nm. The path length of the quartz cuvettes used was 2 mm.Fluorescence
The fluorescence measurements were performed on a Fluorolog-3 HORIBA Jobin Yvon apparatus. For the steady-state measurements, excitation was done at λ = 295 nm (slit width = 3 nm), λ = 370 nm (slit width = 5 nm) and λ = 495 nm (slit width = 10 nm) in order to probe the emission contributions from tryptophan (Trp), AuNCs (Au8 and Au25) and large AuNCs (Au25), respectively. Time-correlated single photon counting (TCSPC) emission measurements to probe the local environment of Trp residues was done using excitation at λ = 280 nm as previously described.9Circular dichroism (CD)
Measurements were conducted at room temperature on an Olis DSM 1000CD apparatus. The lamp power supply was a LPS-220B from Photon Technology International, the user interface was an Olis online instrument system, and the software was OLIS GlobalWorks. The 300 μL cuvette had a path-length of 1 mm, the grid was 2400 lines/mm, slit width was 6.32 mm, the scan range was from 290 nm to 200 nm, and the increment number was 720. Each sample was scanned three times. The concentration of BSA, BSA NC, CTR, and CTR NC was 5 μM, for Fib and Fib NC the concentration was 2.5 μM, and for BLA, BLA NC, Lyz, Lyz NC, Cyt c, Cyt c NC, Mb, Mb NC, BLG and BLG NC the concentration was 25.4 μM. The buffer was used as blank.Results and discussion
In this study, a single-step procedure for the synthesis of protein-stabilized gold nanoclusters was investigated for a small library of eight proteins, chosen on the basis of the following. Bovine serum albumin (BSA) is much studied as an important constituent of blood. Albumin is a carrier protein responsible for transport of thyroid and fat-soluble hormones via the bloodstream,10 and is also involved in other physiological functions such as control of serum osmotic pressure and pH buffering.11 Fibrinogen (Fib) is part of the final step of the blood clotting cascade, wherein Fib is converted into fibrin by the protolytic enzyme thrombin.12 Fib is made up of three globular units connected by two rod regions, each of which consists of triple-stranded α-helical coiled coils. Bovine α-lactalbumin (BLA) and lysozyme (Lyz) are homologous proteins with similar tertiary structures and primary sequence identities of approximately 35%.13–15 Apart from biological function, BLA and Lyz differ with respect to their folding and calcium binding properties. Notably, BLA has the ability to form a stable molten globule and can strongly bind Ca2+,16 whereas no such stable state or metal binding has been reported for (chicken egg white) Lyz. Cytochrome c (Cyt c) shuttles electrons between the two membrane-associated protein complexes cytochrome c reductase and cytochrome c oxidase inside the inner mitochondrial membrane and participates in mitochondrial production of ATP. Molten globule states of Cyt c have been reported under various conditions both at interfaces and in bulk.17–22 Myoglobin (Mb) is a monomeric heme-binding protein involved in oxygen-carrying and can be found in muscle and blood cells. Mb is known to form a stable molten globule state in the absence of its heme group under moderately low pH conditions.23 β-Lactoglobulin (BLG) is a globular milk protein which binds small ligands including palmitic and oleic acids.24,25 Unlike the other major whey protein BLA, the exact biological function of BLG has yet to be elucidated. BLG forms an eight-stranded, antiparallel β-barrel with a three-turn α-helix on the outer surface and a ninth β-strand flanking the first strand.24 The main ligand-binding site is located within the central cavity of the protein. The proteolytic enzyme α-chymotrypsin (CTR) participates in the breakdown of proteins in the digestive system of mammals and other organisms.12 Specifically, CTR cleaves peptide bonds selectively on the carboxyl-terminal side of large hydrophobic amino acids such as tryptophan, tyrosine, phenylalanine and methionine.A summary of the molecular properties of the proteins used in this study is shown in Table 1. By using the same synthesis procedure for the eight proteins studied here, we can probe the effects of protein size, overall protein charge (as determined by their isoelectric point; pI), conformational flexibility as well as the occurrence of specific amino acid residues on the ability to reduce and stabilize gold nanostructures – both subnanometer clusters and nanoparticles.
| Protein | Abbreviation | Molecular weight (kDa) | pI |
|---|---|---|---|
| Bovine serum albumin | BSA | 67.0 | 4.7–4.9 |
| Bovine plasma fibrinogen | Fib | 340.0 | N/A |
| Bovine milk holo α-lactalbumin (type I) | BLA | 14.2 | 4.5 |
| Chicken egg white lysozyme | Lyz | 14.3 | 10.5 |
| Bovine heart cytochrome c | Cyt c | 12.4 | 10.1 |
| Horse heart myoglobin | Mb | 17.2 | 6.8–7.0 |
| Bovine milk β-lactoglobulin | BLG | 18.0 | 3.5–5.2 |
| Bovine pancreas α-chymotrypsin | CTR | 25.0 | 8.8 |
The results will be presented in the following order 1) photophysical properties of the gold nanoclusters, 2) effect of nanocluster/nanoparticle formation on protein conformation, and 3) a suggested mechanism for protein-directed intrinsic synthesis of gold nanoclusters.
Photophysical properties of gold nanoclusters are highly protein-dependent
Following incubation at 37 °C for one week with gold precursor (tetrachloroauric acid; TCAA), the solutions changed color from light yellow (TCAA) to yellow, brown or dark red, according to the nature of the gold structures present and depending on the protein in question. Upon excitation with UV light (λex = 365 nm), most of the incubated solutions display an intense red photoluminescence, with the intensity and hue being protein-dependent, and varying from orange to purple (see Fig. 1), indicating formation of gold nanoclusters.7 Two of the proteins – β-lactoglobulin (BLG) and cytochrome c (Cyt c) – emitted a much weaker blue or purple fluorescence, which could be due to aromatic amino acid side groups (tryptophan, tyrosine and phenylalanine), or low concentrations of gold nanoclusters in the final product (not shown).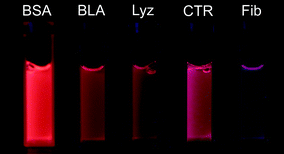 | ||
| Fig. 1 Photograph of aqueous solutions for five of the proteins used here (concentration 35.5 mM) upon excitation at λex = 365 nm. | ||
UV-vis spectra of the protein-AuNCs as well as the respective native proteins are shown in Fig. 2. For all the systems studied here, the spectra of the protein-AuNCs differ significantly from the native proteins both with respect to lineshape and intensity. Incorporation of gold nanoclusters results in a broad absorption below ∼400 nm which significantly distort or hide the 280 nm absorption band of the proteins. This can be attributed to scattering contributions upon formation of supramolecular gold nanoconstructs, with the resulting lineshape being highly protein-dependent. For Cyt c and Mb (Fig. 2, lower left panel), the absorption band at ∼410 nm is also blue-shifted and weakened, indicating that in situ formation of gold nanostructures distorts the environment surrounding the heme groups. Three of the proteins studied here – BLA, Cyt c and BLG – display strong absorbance in the 520–550 nm region upon incorporation of gold. This feature is consistent with localized surface plasmon resonance (LSPR), revealing that AuNPs of diameters larger than 3 nm are present upon incorporation of gold in BLA, Cyt c and BLG (see e.g. Glomm26 and references therein). As the three proteins in question have similar or smaller dimensions to the AuNPs formed (for example, the dimensions of BLA are approximately 4 nm × 3 nm × 3 nm), and the samples remained stable in solution, the systems formed are likely to be AuNPs stabilized by multiple proteins. Thus, under otherwise identical experimental conditions, the protein used determines the resulting gold nanostructure, from nanoclusters embedded in single proteins to gold nanoparticles stabilized by multiple proteins.
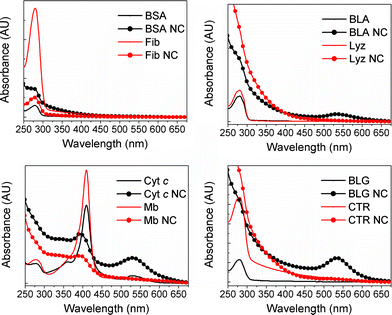 | ||
| Fig. 2 UV-vis spectra of the proteins and protein-AuNCs used in this study. For all spectra, protein concentration was 3.6 mM. | ||
From the observation that the protein-AuNCs formed display fluorescence upon excitation with UV light (see Fig. 1 for an illustration for BLA), there is no clear separation between proteins supporting AuNCs or AuNPs, respectively. Rather, the three protein-gold systems displaying LSPR likely contain AuNCs as well. In order to investigate this, we measured steady-state fluorescence upon excitation of the gold nanostructures at 370 nm and 495 nm (Fig. 3 and 4, respectively). Excitation wavelengths were chosen in order to distinguish between blue-emitting small (Au8) and red-emitting larger (Au25) AuNCs, respectively.2,8
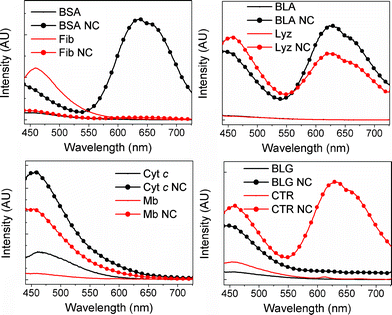 | ||
| Fig. 3 Fluorescence spectra of proteins and protein-nanoclusters upon excitation at 370 nm. Protein concentration was 3.6 mM for all samples (in the top right panel, BLA and Lyz are completely overlapping). | ||
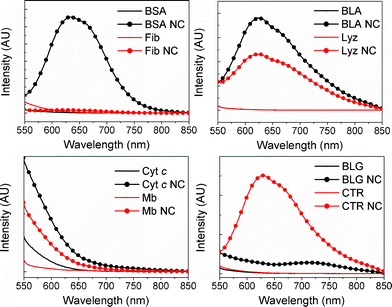 | ||
| Fig. 4 Fluorescence spectra of proteins and protein-nanoclusters upon excitation at 495 nm. Protein concentration was kept constant at 3.6 μM (in the top right panel, BLA and Lyz are completely overlapping). | ||
Steady-state fluorescence spectra of the proteins and protein-AuNCs upon excitation at 370 nm are shown in Fig. 3. The excitation wavelength used for the spectra in Fig. 3 corresponds to what was used to obtain the photograph in Fig. 1. All emission profiles of the protein-AuNCs are markedly different from those of the native proteins, confirming that the reaction has occurred between the metal precursor and the proteins. As was found for the UV-vis results above, emission lineshape and intensity is highly protein-dependent despite otherwise identical reaction conditions. With the exception of Fib NC, all the protein-AuNCs display clearly defined emission maxima around 450 nm, where the emission intensity far exceeds that of the native protein. The emission at 450 nm has been reported to emanate from small nanoclusters reported to consist of ∼8 atoms2,8 – hereafter referred to as “small AuNCs”. Four of the proteins studied – BSA, BLA, Lyz and CTR – also produce protein-AuNCs with a clearly defined second (bimodal) emission maximum between 625 and 675 nm, which has been reported to correspond to larger AuNCs reported to consist of ∼25 atoms,2,7,8 hereafter referred to as “large AuNCs”. Additionally, the emission of BLG is significantly higher than baseline levels above 600 nm, possibly indicating the presence of large AuNCs. In order to rank the proteins according to their capacity for forming small or large AuNCs, the ratio between emission intensities at 650 nm and 450 nm (I650/I450) upon excitation at 370 nm for each protein-AuNC system, as well as the type of gold nanostructure formed, have been listed in Table 2. Here, the relative population of large AuNCs increases with increasing values of I650/I450. From Table 2 as well as from Fig. 3, it is evident that the proteins' capacity for forming large AuNCs can be listed in increasing order as (BLG) ≪ Lyz < BLA ≤ CTR ≪ BSA. From the UV-vis results (Fig. 2) and the steady-state emission spectra upon excitation at 370 nm (Fig. 3), the three protein-AuNC systems found to contain AuNPs – BLA, Cyt c and BLG – also contain small, blue-emitting AuNCs. Among the eight proteins studied here, BLA NC and possibly BLG NC are the only protein-AuNC systems where small and large AuNCs are represented in addition to AuNPs. We have recently shown that the conformation of BLA can be tuned/unfolded more gradually than its homologue Lyz via incorporation of gold nanostructures using an extrinsic reducing agent. This observation is likely to be related to the greater conformational flexibility, which in turn is linked to its marginal folding barrier and stable molten globule. The behaviour may bear a semblance to the phenomenon of HAMLET, where multiple oleic acid molecules hold the anticancer HAMLET complex in a molten globule state, which can thereby interact effectively with cellular and artificial membranes and affect their morphology and integrity significantly.27,28 The relevance with respect to NC modifications may be that these provide an alternative way of loosening the fold and providing an increased affinity and effect on the lipid membrane. Initial studies suggest that BLA NC is indeed cytotoxic.
| Protein-AuNC | I 650/I450 | Gold nanostructure(s) formeda |
|---|---|---|
| a Small AuNCs and large AuNCs denote Au8 and Au25, respectively. AuNPs denotes gold nanoparticles >3 nm displaying localized surface plasmon resonance (Fig. 2), stabilized by multiple proteins. | ||
| BSA NC | 3.00 | Small AuNCs, Large AuNCs |
| Fib NC | 0.24 | N/A |
| BLA NC | 1.24 | Small AuNCs, Large AuNCs, AuNPs |
| Lyz NC | 0.72 | Small AuNCs, Large AuNCs |
| Cyt c NC | 0.04 | Small AuNCs, AuNPs |
| Mb NC | 0.04 | Small AuNCs |
| BLG NC | 0.13 | Small AuNCs, Large AuNCs, AuNPs |
| CTR NC | 1.25 | Small AuNCs, Large AuNCs |
In order to further characterize the large AuNCs, we measured the steady-state emission upon excitation at higher wavelengths, where the emission properties of the large AuNCs are expected to dominate the emission properties of the small AuNCs. Steady-state fluorescence spectra of the proteins and protein-AuNCs upon excitation at 495 nm are shown in Fig. 4. Comparing the emission profiles in Fig. 3 and 4, it is evident that a clearly defined bimodal emission band around 650 nm exists for BSA NC, BLA NC, Lyz NC and CTR NC, with the relative intensities between the systems being identical to what was found for excitation at 370 nm (Fig. 3), confirming the presence of large AuNCs in these proteins. Additionally, excitation at 495 nm reveals a similar emission band for BLG NC, albeit at significantly lower intensities, indicating the presence of large AuNCs for this system as well. Thus, the proteins' capacity for forming large AuNCs can be listed in increasing order as BLG ≪ Lyz < BLA ≤ CTR ≪ BSA.
Formation of gold nanoclusters induces highly protein-dependent conformational changes
Tryptophan (Trp) is an intrinsic fluorophore frequently used to monitor changes in the local protein environment such as unfolding.29 Partial unfolding of a protein and a concomitant increase in solvent exposure is typically accompanied by a shift of the emission maximum towards longer wavelengths and/or a decrease in emission intensity. Whereas red-shifts of the emission profile can often be directly attributed to partial protein unfolding, interpreting changes in emission intensity can be more ambiguous. However, the UV-vis results for the protein-AuNCs studied here (Fig. 2) reveal that the gold NCs absorb around 350 nm, enabling quenching of Trp emission via distance-dependent energy transfer into the AuNCs. Consequently, the changes in Trp emission profile make it possible to follow conformational changes in a protein as well as the presence of gold nanoconstructs using steady-state Trp fluorescence (Fig. 5). The excitation wavelength λex used was 295 nm.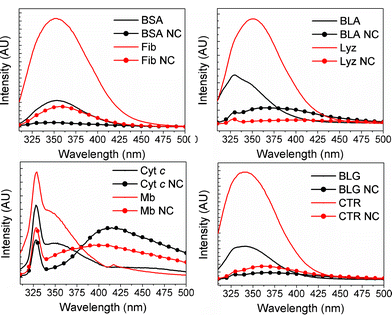 | ||
| Fig. 5 Trp emission spectra of proteins and protein-nanoclusters upon excitation at 295 nm. Protein concentration was 3.6 μM for all samples. | ||
The Trp emission profiles depicted in Fig. 5 reveal a decrease in Trp emission intensity compared to the native protein for all the protein-AuNC systems studied here, with the most severe relative intensity decrease being observed for BSA NC. Another common feature for all the protein-AuNC systems studied here is a red-shift, i.e. a shift towards longer wavelengths, of the Trp emission relative to the native protein, with the smallest red-shifts observed for the two largest proteins studied here, BSA and Fib (Fig. 5, upper left panel). For the two heme-containing proteins used here, Cyt c and Mb (Fig. 5, lower left panel) the red-shift is manifested by a large (>50 nm) red-shift of the secondary emission peak at 350 nm upon incorporation of AuNCs. While energy transfer into the AuNC band (as seen from the UV-vis spectra in Fig. 2) could contribute to the quenching of the Trp emission intensity, this mechanism cannot account for the observed red-shift of the emission maxima. Moreover, there is no clear correlation between the absorption intensity at ∼350 nm and the distortions in the corresponding Trp emission spectra for the different systems. Thus, we conclude that the quenched and red-shifted Trp emission indicates partial unfolding of the protein upon functionalization with AuNCs.
In order to further probe the local Trp environment, fluorescence lifetime measurements (time-correlated single photon counting (TCSPC), Table 3) were performed for all proteins and protein-AuNC systems. While the excitation wavelength λex = 280 nm excites all three intrinsic fluorophores in the proteins, Trp will still be the main contributor to the emission lifetime profile due to a significantly higher quantum yield as compared to tyrosine and phenylalanine.29 No distinct lifetimes attributable to the AuNCs could be detected upon excitation at 280 nm for any of the systems studied here.
| Protein | System | t 1 (ns) | B 1 (%) | t 2 (ns) | B 2 (%) | MRE200/MRE222 |
|---|---|---|---|---|---|---|
| a No discernible secondary structure could be detected using CD upon modification with AuNCs. | ||||||
| BSA | Native | 5.0 ± 0.2 | 46.5 ± 0.1 | 7.5 ± 0.1 | 53.5 ± 0.1 | 1.6 |
| NC | 1.35 ± 0.02 | 48.7 ± 0.1 | 5.19 ± 0.03 | 51.3 ± 0.1 | 3.2 | |
| Fib | Native | 2.30 ± 0.04 | 41.8 ± 0.1 | 7.99 ± 0.03 | 58.2 ± 0.1 | 1.8 |
| NC | 1.63 ± 0.04 | 45.6 ± 0.1 | 5.01 ± 0.03 | 54.4 ± 0.1 | 5.3 | |
| BLA | Native | 0.9 ± 0.1 | 93.8 ± 0.1 | 4.5 ± 0.1 | 6.2 ± 0.1 | 3.7 |
| NC | 0.90 ± 0.02 | 49.7 ± 0.1 | 4.27 ± 0.03 | 50.3 ± 0.1 | 10.8 | |
| Lyz | Native | 1.52 ± 0.03 | 69.5 ± 0.1 | 3.67 ± 0.04 | 30.1 ± 0.1 | 1.4 |
| NC | 0.6 ± 0.1 | 70.0 ± 0.1 | 4.1 ± 0.8 | 30.0 ± 0.1 | 13.5 | |
| Cyt c | Native | 2.15 ± 0.04 | 60.8 ± 0.1 | 7.8 ± 0.1 | 39.2 ± 0.1 | 3.4 |
| NC | 0.6 ± 0.1 | 68.8 ± 0.1 | 5.8 ± 0.1 | 31.2 ± 0.1 | 13.3 | |
| Mb | Native | 0.3 ± 0.2 | 55.2 ± 0.1 | 4.6 ± 0.1 | 44.8 ± 0.1 | 2.0 |
| NC | 0.7 ± 0.1 | 62.1 ± 0.1 | 4.2 ± 0.1 | 37.9 ± 0.1 | 6.1 | |
| BLG | Native | 1.25 ± 0.01 | 85.4 ± 0.1 | 5.1 ± 0.1 | 14.6 ± 0.1 | 0.2 |
| NC | 1.30 ± 0.03 | 55.4 ± 0.1 | 4.24 ± 0.03 | 44.6 ± 0.1 | 8.3 | |
| CTR | Native | 1.09 ± 0.02 | 53.2 ± 0.1 | 3.94 ± 0.03 | 46.8 ± 0.1 | 8.8 |
| NC | 0.85 ± 0.02 | 47.6 ± 0.1 | 3.87 ± 0.03 | 52.4 ± 0.1 | −31.0a | |
As the proteins used here contain different amounts of Trp residues, interpretation of the TCPSC results with respect to comparison of proteins is intricate. A bimodal fitting was found to provide the best overall fit for the fluorescence decay profiles of the samples studied here, yielding one “short” (t1) and one “long” (t2) lifetime, with corresponding relative populations B1 and B2, respectively. The lifetimes and populations listed in Table 3 show that the Trp fluorescence profiles, i.e. both lifetimes and populations, were altered upon incorporation of AuNCs for all the systems studied here, indicating changes in the Trp environment following formation of gold nanoconstructs. No single trend with respect to shifts in either lifetimes or populations could be detected for the entire dataset, revealing that the impact of AuNC formation on the chemical environment surrounding Trp residues is very protein-dependent. Additionally, differences in the number of Trp residues as well as their position and orientation relative to the NC modifications for the proteins studied here likely play a significant role. Formation of gold nanostructures within the proteins results in reduction of the short lifetime (t1) as compared to the native protein for five of the systems studied here (BSA NC, Fib NC, Lyz NC, Cyt c NC, CTR NC). Three of these systems (BSA NC, Fib NC and Cyt c NC) also reveal population shifts towards shorter lifetimes, indicating either quenching from increased Trp exposure to water, energy transfer to gold (either NCs or NPs), or a combination of these effects. The largest observable changes in the Trp fluorescence lifetime profile upon formation of gold nanoconstructs was observed for the two milk proteins included in the dataset; BLA and BLG. Interestingly, BLA and BLG also display the same changes in Trp fluorescence lifetimes (Table 3); little or no changes in t1, a significant reduction of t2, and a large shift in population towards the longest lifetime for both proteins. Specifically, whereas the population is heavily shifted towards the short lifetime for both native proteins (94% and 85% for BLA and BLG, respectively), the AuNC-modified protein systems reveal an approximately even distribution of the two populations (B1 for BLA NC and BLG NC are 50% and 55%, respectively). As such, the fluorescence results illustrate that the effect of AuNC formation on the microenvironment surrounding the Trp residues – such as the degree of unfolding and NC modifications in close proximity– is highly protein-dependent.
For a more thorough investigation of the effect of AuNC formation on protein secondary structure, circular dichroism measurements were performed (Fig. 6). Here, changes in intensity and lineshape compared to the native protein indicate conformational changes. To rule out the possibility of changes due to attenuation of the signal by gold, the relationship between signal intensity at 200 nm and 222 nm was studied for all samples (Table 3). The 200 nm/222 nm ratio varies in a manner not directly correlated to the concentration of gold precursor or the corresponding absorbance at these wavelengths (Fig. 2). Thus, the spectral changes are likely due to changes in the protein, not attenuation of the signal caused by the presence of gold.
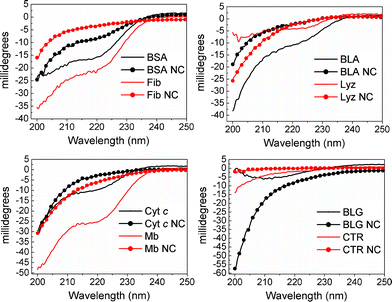 | ||
| Fig. 6 CD traces of the protein and protein-AuNCs studied here. | ||
From the CD results presented in Fig. 6, all the AuNC-containing systems display large differences as compared to the native proteins with respect to intensity and lineshape, indicating conformational changes upon incorporation of the gold nanoconstructs. With the exception of BLG, incorporation of AuNCs resulted in a relative decrease of alpha-helical motifs (i.e. mean residual ellipticity at 222 nm) compared to the native protein for the systems studied here. For two of the proteins – Lyz and BLG – incorporation of AuNCs resulted in a significant transition from alpha-helical motifs to other secondary structures (mostly random coils as seen from the increased signal at ∼200 nm), compared to the native protein. The values of the ratios between signal intensity at 200 nm and 222 nm (MRE200/MRE222, Table 3) were found to increase by more than a factor of two upon incorporation of AuNCs for all the systems studied here, further indicating that the changes in lineshapes and signal intensities, documented by the fluorescence intensities, are due to changes in protein conformation rather than attenuation of the signal from the presence of gold. No apparent trend with respect to the nature of the gold nanoconstructs formed (i.e. small or large AuNCs or AuNPs) could be identified from either Trp emission or CD data, revealing that the effect of incorporation of gold on the degree of conformational change is mainly protein-dependent. As the stability and indeed function of the protein-gold nanoconstruct for any intended application is highly dependent on the structural changes of the protein shell, the results presented here show that care must be taken to understand the nanocluster formation on a protein-specific basis. In order to be able to predict the final properties – both with respect to nanocluster population and protein conformation – for a given protein, there is a need for a general mechanism.
Protein-mediated gold nanocluster formation can be attributed to a general mechanism
Earlier biomineralization-inspired studies have pointed to single amino acid residues being responsible for reduction and nucleation of Au3+, tyrosine (Tyr) in the case of BSA,7 and histidine (His) in the case of apoferritin.4 While Tyr is known to have a high, albeit strongly pH-dependent redox potential7,30 and mixtures of His and HAuCl4 have been shown to reduce Au3+ to Au0via X-ray absorption near-edge structure (XANES),4 these are either single-protein (in the case of BSA) or single amino acid (in the case of His) studies. Additionally, the importance of cysteine (Cys) residues in immobilizing/stabilizing the resulting gold nanoconstructs within the protein via covalent Au–S bonds has been reported (see e.g. Xie7 and references therein). Here, we have compared eight different proteins varying in molecular weights, isoelectric points and conformational flexibilities with respect to their capability of forming AuNCs utilizing a single intrinsic, i.e. biomineralization-inspired, synthesis procedure. From the findings presented above, the type of gold nanoconstructs formed, the photophysical properties of the AuNCs and the resulting effect on polypeptide conformation are highly protein-dependent. No correlation could be found between the number/percentage of Tyr and His residues and the propensity for AuNC formation, or for the type of gold nanoconstruct (i.e. small and large AuNCs, AuNPs) formed. The small library employed here includes a protein without any Cys residues (Mb), which is capable of formation and stabilization of formation of small AuNCs, showing that Cys residues are not necessary for stabilization of the gold nanoconstructs.Based on the strong protein-dependence, as opposed to single amino acid residue-dependence, of AuNC formation observed here, we propose that the general protein-templated gold nanoconstruct nucleation and growth can be explained via a general mechanism. Our proposed mechanism is adapted from Berti and Burley's postulated mechanism for nucleic acid and nucleotide-mediated synthesis of inorganic nanoparticles,31 and Feldheim and Eaton's SELEX model for biomolecule-mediated crystal formation,32 and consists of the following four steps:
The general mechanism suggested here accounts for the highly protein-dependent AuNC formation in that local negative charge, availability of electron-donors and the local stability of the secondary structure and overall flexibility of the fold determines where clusters form and when formation is terminated, as well as their effect on the protein. The mechanism is also in agreement with existing literature for comparable systems.31,32
Conclusion
In this work a small library of eight proteins of different molecular weights, charge properties and conformational flexibilities has been studied with respect to their ability to form gold nanoconstructs using a single intrinsic, i.e. biomineralization-inspired, synthetic procedure wherein no extrinsic reducing agent was added. The type of gold nanostructure formed, the photophysical properties of the protein-gold constructs as well as the accompanying conformational changes were found to be highly protein-dependent. Using a combination of UV-vis spectroscopy and steady-state fluorescence measurements, we identified the three primary sets of gold nanoconstructs formed as fluorescent clusters of ∼8 and ∼25 atoms (small and large AuNCs, respectively) and nanoparticles (AuNPs) of diameters >3 nm displaying localized surface plasmon resonance (LSPR), with all three structures occurring within the same system for the two milk proteins studied here, BLA and BLG (see Table 2). We also present a general mechanism for the in situ nanocluster formation, based on nucleation, growth, termination and stabilization/solubilization events. As such, this work represents a contribution to the general understanding of how the protein shell as well as the size and properties of nanoclusters can be tuned to manipulate conformation and stability of the resulting nanomaterial, and how labelling with plasmonic and fluorescent nano-species can enable easier biolabeling and tracking in biological systems.Acknowledgements
The authors acknowledge the Department of Chemical Engineering, NTNU, for financial support. Wilhelm R. Glomm and Sondre Volden acknowledge financial support from the Research Council of Norway (NFR) within the FRINAT program, project number 177556/V30.References
- F. Wen, Y. H. Dong, L. Feng, S. Wang, S. C. Zhang and X. R. Zhang, Anal. Chem., 2011, 83, 1193 CrossRef CAS.
- X. Le Guevel, N. Daum and M. Schneider, Nanotechnology, 2011, 22, 7 Search PubMed.
- A. Retnakumari, J. Jayasimhan, P. Chandran, D. Menon, S. Nair, U. Mony and M. Koyakutty, Nanotechnology, 2011, 22, 11 CrossRef.
- C. J. Sun, H. Yang, Y. Yuan, X. Tian, L. M. Wang, Y. Guo, L. Xu, J. L. Lei, N. Gao, G. J. Anderson, X. J. Liang, C. Y. Chen, Y. L. Zhao and G. J. Nie, J. Am. Chem. Soc., 2011, 133, 8617 CrossRef CAS.
- X. X. Wang, Q. Wu, Z. Shan and Q. M. Huang, Biosens. Bioelectron., 2011, 26, 3614 CrossRef CAS.
- J. Zheng, C. W. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 4 Search PubMed.
- J. P. Xie, Y. G. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888 CrossRef CAS.
- X. Le Guevel, B. Hotzer, G. Jung, K. Hollemeyer, V. Trouillet and M. Schneider, J. Phys. Chem. C, 2011, 115, 10955 CAS.
- S. M. Lystvet, S. Volden, M. Yasuda, O. Halskau and W. R. Glomm, Nanoscale, 2011, 3, 1788 RSC.
- H. Lodish, A. Berk, P. Matsudaira, C. A. Kaiser, M. Krieger, M. P. Scott, S. L. Zipursky and J. Darnell, Molecular Cell Biology, 5th ed., W. H. Freeman and Company, New York, 2003 Search PubMed.
- V. N. Uversky, N. V. Narizhneva, T. V. Ivanova and A. Y. Tomashevski, Biochemistry, 1997, 36, 13638 CrossRef CAS.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, 5 ed., W. H. Freeman and Company, New York, 2002 Search PubMed.
- A. V. Agasoster, O. Halskau, E. Fuglebakk, N. A. Froystein, A. Muga, H. Holmsen and A. Martinez, J. Biol. Chem., 2003, 278, 21790 CrossRef CAS.
- A. C. W. Pike, K. Brew and K. R. Acharya, Structure, 1996, 4, 691 CrossRef CAS.
- S. Sugai, M. Ikeguchi, in Advances in Biophysics, Vol 30, 1994, Japan Scientific Soc Press, Tokyo, 1994, vol. 30, p 37 Search PubMed.
- E. A. Permyakov and L. J. Berliner, FEBS Lett., 2000, 473, 269 CrossRef CAS.
- P. Hildebrandt and M. Stockburger, Biochemistry, 1989, 28, 6710 CrossRef CAS.
- V. E. Bychkova, A. E. Dujsekina, S. I. Klenin, E. I. Tiktopulo, V. N. Uversky and O. B. Ptitsyn, Biochemistry, 1996, 35, 6058 CrossRef CAS.
- A. A. Moosavi-Movahedi, J. Chamani, Y. Goto and G. H. Hakimelahi, J. Biochem., 2003, 133, 93 CrossRef CAS.
- A. Muga, H. H. Mantsch and W. K. Surewicz, Biochemistry, 1991, 30, 7219 CrossRef CAS.
- P. J. R. Spooner and A. Watts, Biochemistry, 1990, 30, 3880 CrossRef.
- P. J. R. Spooner and A. Watts, Biochemistry, 1990, 30, 3871 CrossRef.
- Y. V. Griko and P. L. Privalov, J. Mol. Biol., 1994, 235, 1318 CrossRef CAS.
- G. Kontopidis, G. Holt and L. Sawyer, J. Dairy Sci., 2004, 87, 785 CrossRef CAS.
- S. E. Permyakov, E. L. Knyazeva, L. M. Khasanova, R. S. Fadeev, A. P. Zhadan, H. Roche-Hakansson, A. P. Hakansson, V. S. Akatov and E. A. Permyakov, Biol. Chem., 2012, 393, 85 CrossRef CAS.
- W. R. Glomm, J. Dispersion Sci. Technol., 2005, 26, 389 CrossRef CAS.
- A. K. Mossberg, M. Puchades, O. Halskau, A. Baumann, I. Lanekoff, Y. X. Chao, A. Martinez, C. Svanborg and R. Karlsson, PLoS One, 2010, 5, 10 Search PubMed.
- A. Baumann, A. U. Gjerde, M. Ying, C. Svanborg, H. Holmsen, W. R. Glomm, A. Martinez and O. Halskau, J. Mol. Biol., 2012, 418, 90 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2006 Search PubMed.
- A. Harriman, J. Phys. Chem., 1987, 91, 6102 CrossRef CAS.
- L. Berti and G. A. Burley, Nat. Nanotechnol., 2008, 3, 81 CrossRef CAS.
- D. L. Feldheim and B. E. Eaton, ACS Nano, 2007, 1, 154 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |