DOI:
10.1039/C2RA22048B
(Paper)
RSC Adv., 2012,
2, 12973-12982
Templating amphiphilic poly(ethylene oxide-b-ε-caprolactone) diblock copolymers provides ordered mesoporous silicas with large tunable pores
Received
4th September 2012
, Accepted 19th October 2012
First published on 30th October 2012
Abstract
In this study we used evaporation-induced self-assembly (EISA) as a facile approach for the synthesis, with amphiphilic poly(ethylene oxide-b-ε-caprolactone) (PEO-b-PCL) diblock copolymers as templates, of a family of mesoporous silicas with large, tunable pore sizes. Transmission electron microscopy (TEM), small-angle X-ray scattering (SAXS), and N2 adsorption–desorption isotherms revealed that the morphology transformed and the pore sizes of the mesoporous silica expanded upon increasing the PCL-to-PEO weight ratio in the templating PEO-b-PCL diblock copolymers. We also employed four kinds of homopolymers—two linear poly(ε-caprolactone)s [PCL20 (Mn = 2100) and PCL408 (Mn = 42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500)], a star poly(ethylene oxide)-functionalized silsesquioxane [PEO13–POSS (Mn = 5776)], and a linear poly(ethylene oxide) [PEO22 (Mn = 1000)]—as additives during the fabrication of these mesoporous silicas. Pore expansion and a unique mesophase transformation occurred when using PCL20 and PEO–POSS as the additive, respectively. Notably, we obtained highly ordered body-centered cubic (space group Im
500)], a star poly(ethylene oxide)-functionalized silsesquioxane [PEO13–POSS (Mn = 5776)], and a linear poly(ethylene oxide) [PEO22 (Mn = 1000)]—as additives during the fabrication of these mesoporous silicas. Pore expansion and a unique mesophase transformation occurred when using PCL20 and PEO–POSS as the additive, respectively. Notably, we obtained highly ordered body-centered cubic (space group Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) mesoporous silicas with large cage-like pores of 10.1 and 22.6 nm when using EO114CL20 and EO114CL20/PEO–POSS, respectively, as templates for the EISA process.
m) mesoporous silicas with large cage-like pores of 10.1 and 22.6 nm when using EO114CL20 and EO114CL20/PEO–POSS, respectively, as templates for the EISA process.
Introduction
Mesoporous silicas receive much attention because of their unique properties and various applications.1–4 Initially, their syntheses were mediated in aqueous solutions; for example, MCM-41s5,6 and the SBA-n7,8 series were fabricated using precipitation-based methods. Using this approach, however, restricted the development of mesoporous silicas to water-soluble systems, limiting the templating surfactants to low-molecular-weight species that could dissolve in aqueous solutions. In recent years, evaporation-induced self-assembly (EISA) has been used broadly for the preparation of mesoporous organic materials; in addition to the fabrication of mesoporous polymer resins2,9–15 and mesoporous carbons,12,16–23 the EISA strategy can also be used to prepare mesoporous silicas that are templated by high-molecular-weight amphiphilic block copolymers.10,20,24–32 In other words, EISA provides an approach toward the formation of ordered mesoporous nanostructures33–35 when combined with unusual AB diblock copolymers10,12,20,21,28 or ABC triblock terpolymers18,26 as templates.
The formation of self-organized morphologies through microphase separation of block copolymers is a very interesting topic in polymer science, with many research groups having discussed the phase transformations of block copolymers in terms of changes in the volume fractions or interactions within homopolymer blends.36–43 This morphology of a block copolymer–homopolymer blend system depends on critically (i) the ratio of the molar weight of the additive homopolymer to that of the associated block of the copolymer and (ii) the volume fraction and miscibility of the additive homopolymer in the blend.36,37,40,41,43 Few studies, however, have focused on the effects of homopolymer blending during the preparation of mesoporous silicas.19,29 Zhao et al. initially discussed the phase evolution of mesoporous silicas after blending with the homopolymer poly(propylene oxide) (PPO-400);29 they also investigated the formation of large-pore mesoporous carbons after adding a polystyrene homopolymer (h-PS) as a pore expander.19 Accordingly, the blending of a homopolymer during the forming process appears to be a means of controlling the morphology and/or pore size of the mesoporous silicas.
Large-pore fcc silicas and SBA-16 with a bcc structure are well known, but mesoporous silicas having cubic Imm structures with large pores (>10 nm) are rarely observed because the distribution of the mesophase diagram is very narrow; the soft templates require a high volume fraction of the hydrophilic segment of the block copolymer [e.g., poly(ethylene oxide) (PEO) segment]. To date, the most studied mesoporous silica having a body-centered cubic (bcc) structure has been SBA-16,8,44–46 which displays excellent performance in various applications, including drug delivery,47 catalysis,48 gas adsorption,49 and others,50 because of its large cage-like (or spherical) mesopores. In addition to their morphology, the pore sizes of mesopores also strongly affect the performance of mesoporous silicas in drug delivery or separation. Accordingly, we are interested in preparing new structure-directing agents for the fabrication of mesoporous silicas.
In previous studies,10,12 we used a novel double-crystalline amphiphilic block copolymer, poly(ethylene oxide-b-ε-caprolactone) (PEO-b-PCL), of unimodal distribution molecular weight as the template to prepare large-mesopore silica nanostructures, observing transformations of the mesophase upon varying the TEOS-template weight fraction. We have also examined the effect of the molecular weight of templating PEO-b-PCL on the fabrication of mesoporous phenolic resins.12 In this present study, we used ring-opening polymerization to synthesize a series of PEO-b-PCL diblock copolymers that we then used to regulate the morphologies and pore sizes of mesoporous silicas through variations in the molecular weight and hydrophilic/hydrophobic segment ratio of the template. Moreover, we were interested in using homopolymers as templates as a means of readily tuning the morphologies and pore sizes of the mesoporous silicas; accordingly, we tested the effects of four kinds of additives for the synthetic process: PCL20 (Mn = 2100), PCL408 (Mn = 42500), star PEO13–POSS (Mn = 5577), and linear PEO22 (Mn = 1000). We observed that the morphology of the mesoporous silicas transformed from bcc to cylindrical to disordered cylindrical upon increasing the PCL/PEO ratio, which should have more TEOS content to confine the PEO-b-PCL for forming an ordered mesostructure. On the other hand, we also found that the pore size expanded upon increasing the PCL/PEO ratio in the PEO-b-PCL diblock copolymer. In addition, blending with homopolymers also influenced the pore sizes of the mesoporous silicas; for example, varying the weight fraction of blended PCL20 in the EISA process resulted in the mesopore size being tunable within the ranges 10–20 nm (for templating EO114CL20) and 20–39 nm (for templating EO114CL84), consistent with the pore size variations of the mesoporous silicas templated by PEO-b-PCL at different molecular weights. In contrast, blending with PCL408 barely affected the pore sizes of the mesoporous silicas, due to the poor solubility of PCL408 in the blending system; as a result, macrophase separation occurred when the molecular weight of the homopolymer was greater than that of the corresponding segment in the diblock copolymer. In blending experiments performed with the hydrophilic homopolymer PEO–POSS, however, we observed interesting mesophase transformations and enhanced ordering upon increasing the amount of the blended homopolymer. We also obtained special bcc-type mesoporous silica with large (10.1 nm) cage-like pores after templating with EO114CL20 (Mn = 7280); to the best of our knowledge, this bcc-type mesoporous silica is the first to have been synthesized with ultra-large pores (>10 nm) without the addition of any additives during the synthetic process, compared to the large pore bcc type mesoporous silicas reported in the previous studies.51,52 In this study, we used transmission electron microscopy (TEM), small-angle X-ray scattering (SAXS), and N2 adsorption–desorption isotherms to characterize the changes in the mesophases and mesoporous sizes that occurred during the EISA process.
Experimental
Materials
Monomethoxy-poly(ethylene glycol) with a molecular weight of 5000 (PEO114) was obtained from Fluka and dried through azeotropic distillation with dry toluene. ε-Caprolactone (ε-CL, Acros) was purified through vacuum distillation over CaH2; the distillation fraction collected at 96–98 °C (5 mm-Hg) was used in all polymerization reactions. Stannous(II) octoate [Sn(Oct)2, Sigma] was used as received. CH2Cl2 was dried over CaH2 prior to use. Diblock copolymers were readily prepared through the ring-opening polymerization of ε-CL in the presence of PEO114 and Sn(Oct)2 as the catalyst. The reaction mixtures were prepared by introducing a desired volume of ε-CL monomer into a silanized flask containing a pre-weighed amount of PEO114 under a N2 atmosphere. Several drops of Sn(Oct)2 were added and then the flask was connected to a vacuum line, evacuated, sealed off, and heated at 130 °C. After 24 h, the resulting block copolymers were dissolved in CH2Cl2 and precipitated in an excess of cold n-hexane. The polymers were dried at 40 °C under vacuum. The characteristics of the diblock copolymers used in this study are summarized in Table 1. Star PEO–POSS [(C2m+3H4m+7Om+1)n(SiO1.5)n; m = 13.3; n = 8; Mn = 5577] was purchased from Hybrid Plastics (USA). PCL20, PCL408, PEO22, and benzyl alcohol (the macroinitiator for preparing the homopolymer PCL20) were obtained from Aldrich.
| Copolymer PEO-b-PCL |
M
n(NMR)
a
|
M
n(GPC)
b
|
M
w/Mn(GPC)b |
|
Obtained by 1H NMR spectra.
Obtained by GPC trace with DMF eluent of 0.6 ml min−1 and PS-standard calibration.
|
| EO114CL20 |
7280 |
8200 |
1.21 |
| EO114CL42 |
9788 |
14000 |
1.14 |
| EO114CL84 |
14576 |
22000 |
1.50 |
| EO114CL130 |
19820 |
21000 |
1.52 |
Synthesis of mesoporous silicas templated by PEO-b-PCL copolymers of various molecular weights10
Mesoporous silicas were prepared through an EISA strategy in THF, using the various copolymers PEO-b-PCL (EO114CL20, EO114CL42, EO114CL84, and EO114CL130; Table 1) as templates and TEOS as the silica precursor (Scheme 1). In a typical synthesis, a TEOS-to-EO114CL20 ratio of 2, a TEOS-to-EO114CL42 ratio of 3, a TEOS-to-EO114CL84 ratio of 5, or a TEOS-to-EO114CL130 ratio of 11 (Table 2) was used at a constant concentration of HCl (0.012 g of 0.1 M HCl/PEO-b-PCL); the mixture of TEOS and HCl at a specific composition was added into a stirred solution of PEO-b-PCL (2.0 wt%, containing 0.10 g of copolymer) in THF (5 g); stirring was continued for 30 min to form a homogeneous solution. The sample was poured into a Petri dish and then the THF was evaporated at room temperature for 48 h. The transparent film was collected, ground into a powder, transferred to a PFA bottle containing 1.0 M HCl (30 mL), and hydrothermally treated at 100 °C for 3 days. The product was washed with water and EtOH, dried at room temperature, and calcined in air at 600 °C for 6 h to produce a white mesoporous silica. Calcination processes were conducted in a furnace operated at a heating rate of 1 °C min−1.
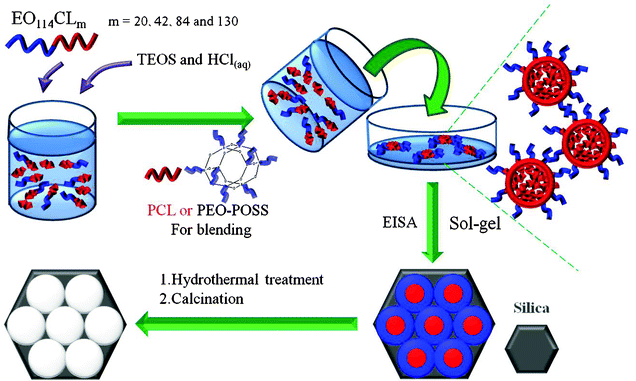 |
| | Scheme 1 Preparation of mesoporous silicas. | |
| Sample |
d (nm)a |
Pore size (nm) |
SBET (m2 g−1)b |
Pore volume (cm3 g−1) |
Micropore volume (cm3 g−1) |
TEOS/template (weight fraction) |
|
The d-spacing values were calculated from the first SAXS peak by the formula d = 2π/q*.
SBET is the total BET surface area and calculated from the adsorption branch of isotherm curve.
|
| 1 |
12.8 |
10.1 |
667 |
0.85 |
0.07 |
TEOS/EO114CL20 = 2/1 |
| 2 |
16.5 |
12.4 |
416 |
0.52 |
0.066 |
TEOS/EO114CL42 = 3/1 |
| 3 |
24.1 |
20.0 |
170 |
0.33 |
0.02 |
TEOS/EO114CL84 = 5/1 |
| 4 |
44.9 |
28.6 |
350 |
0.40 |
0.018 |
TEOS/EO114CL130 = 11/1 |
| 5 |
15.7 |
11.7 |
699 |
0.86 |
0.07 |
TEOS/EO114CL20/PCL20 = 2/1/0.1 |
| 6 |
19.6 |
17.5 |
689 |
1.05 |
0.07 |
TEOS/EO114CL20/PCL20 = 2/1/0.3 |
| 7 |
22.4 |
20.2 |
669 |
1.28 |
0.07 |
TEOS/EO114CL20/PCL20 = 2/1/0.5 |
| 8 |
14.6 |
9.0 |
748 |
0.82 |
0.07 |
TEOS/EO114CL20/PCL408 =2/1/0.1 |
| 9 |
15.3 |
9.0 |
633 |
0.82 |
0.06 |
TEOS/EO114CL20/PCL408 =2/1/0.3 |
| 10 |
15.0 |
10.3 |
654 |
0.79 |
0.07 |
TEOS/EO114CL20/PCL408 =2/1/0.5 |
| 11 |
26.2 |
20.1 |
403 |
0.46 |
0.10 |
TEOS/EO114CL84 = 3/1 |
| 12 |
31.4 |
28.4 |
401 |
0.70 |
0.09 |
TEOS/EO114CL84/PCL20 = 3/1/0.5 |
| 13 |
33.1 |
33.4 |
538 |
0.95 |
0.10 |
TEOS/EO114CL84/PCL20 =3/1/0.7 |
| 14 |
36.9 |
38.5 |
581 |
1.07 |
0.11 |
TEOS/EO114CL84/PCL20 =3/1/0.9 |
| 15 |
26.2 |
20.0 |
469 |
0.71 |
0.07 |
TEOS/EO114CL84/PEO13–POSS = 3/1/0.3 |
| 16 |
26.2 |
19.8 |
757 |
1.05 |
0.1 |
TEOS/EO114CL84/PEO13–POSS = 3/1/0.5 |
| 17 |
28.6 |
22.6 |
316 |
0.56 |
0.02 |
TEOS/EO114CL84/PEO13–POSS = 3/1/0.7 |
Synthesis of mesoporous silicas templated by PEO-b-PCL copolymers blended with homopolymer additives
The synthesis of mesoporous silicas was performed using an EISA process similar to that described above, except that homopolymer additives were added. Using PCL20 (EO114CL20 or EO114CL84 as a template), PCL408 (EO114CL20 as a template), PEO–POSS (EO114CL84 as a template), or PEO22 (EO114CL84 as a template) as the additive, a series of samples were prepared according to the following procedure. A mixture of TEOS and an additive at a specific composition (see TEOS/template/additive weight ratios in Table 2) was added into a stirred solution of PEO-b-PCL (2.0 wt%, containing 0.10 g of copolymer) in THF (5 g); stirring was continued for 30 min to form a homogeneous solution. The sample was poured into a Petri dish and then the THF was evaporated at room temperature for 48 h. The transparent film was collected, ground into a powder, transferred to a PFA bottle containing 1.0 M HCl (30 mL), and hydrothermally treated at 100 °C for 3 days. The product was washed with water and EtOH, dried at room temperature, and calcined in air at 600 °C for 6 h to produce white mesoporous silica. Calcination processes were conducted in a furnace operated at a heating rate of 1 °C min−1.
Characterization
SAXS was performed using a NANOSTAR U small-angle X-ray scattering system (Bruker AXS, Karlsruhe, Germany) and Cu-Kα radiation (30 W, 50 kV, 600 μA). The d-spacings were calculated using the formula d = 2π/q, where q is the scattering vector. TEM images were recorded using a JEOL 3010 microscope operated at 200 kV; samples for TEM measurement were suspended in EtOH and supported onto a holey carbon film on a Cu grid. Nitrogen adsorption–desorption isotherms were measured at −196 °C using an ASAP 2020 analyzer; prior to measurement, the samples were degassed under vacuum at 200 °C for at least 6 h. The Brunauer–Emmett–Teller (BET) method was used to calculate specific surface areas and pore volumes; pore size distributions were derived from the adsorption branches of the isotherms through use of the Broekhoff–de Boer (BdB) model.
Results and discussion
Body-centered cubic mesoporous silica templated by amphiphilic block copolymer EO114CL20
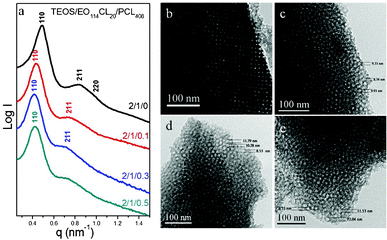
We used a simple EISA method to synthesize a new mesoporous silica having a bcc structure when applying a PEO-b-PCL (EO114CL20) copolymer, having a large volume fraction of its EO block segment, as the template at a TEOS/PEO-b-PCL ratio of 2:1. The SAXS pattern (Fig. 1a) of this bcc-type mesoporous silica exhibited a strong reflection with a large d-spacing of 12.8 nm, one strong reflection at a value of q of 0.85 nm−1, and one weaker reflection at a value of q of 0.98 nm−1; this SAXS pattern could be indexed as having (110), (211), and (220) reflections, corresponding to a cubic structure (Imm space group). In addition, we confirmed the structural ordering and cubic symmetry of this material through TEM analysis (Fig. 1b–d). The TEM images of the bcc-type mesoporous silica with different orientations ([110], [110], and [111] planes, respectively) were consistent with a three-dimensional (3D) cubic cage structure having an ultralarge d-spacing—a material having potential advantages in catalysis and separation applications. Notably, these synthesis conditions enabled the facile and highly reproducible preparation of a large-cage 3D cubic mesoporous silica having Imm symmetry. We obtained further information regarding the textural properties of the materials from N2 adsorption–desorption isotherms measured at 77 K. Fig. 1e presents the N2 physisorption isotherms of the cubic mesoporous silica sample. The sample displays individual type-IV isotherms, exhibiting an apparent H2 hysteresis loop characteristic of a cage-like mesoporous material. A sharp capillary condensation step appeared for this sample, suggesting uniform pore dimensions and high-quality ordering of the materials, in agreement with the TEM and SAXS data. Pore size distribution analysis revealed a well-ordered cubic structure having pores with an average diameter of approximately 10.1 nm. The significant width of the hysteresis loops observed for the SBA-16 samples studied herein suggested cage-like pore shapes. We used the Broekhoff–de Boer (BdB) model to determine the pore sizes while considering spherically shaped pores. The BET surface area of the samples was 667 m2 g−1; the pore volume was approximately 0.85 cm3 g−1 (Table 2, sample 1).
![(a) SAXS pattern, (b–d) TEM images viewed from [100], [110], and [111], respectively (insets: the corresponding FFT), (e) N2 adsorption–desorption isotherm, and (f) pore size distribution curve of bcc mesoporous silica templated by EO114CL20 with a TEOS/EO114CL20 ratio of 2 : 1.](/image/article/2012/RA/c2ra22048b/c2ra22048b-f1.gif) |
| | Fig. 1 (a) SAXS pattern, (b–d) TEM images viewed from [100], [110], and [111], respectively (insets: the corresponding FFT), (e) N2 adsorption–desorption isotherm, and (f) pore size distribution curve of bcc mesoporous silica templated by EO114CL20 with a TEOS/EO114CL20 ratio of 2:1. | |
Mesoporous silicas templated by PEO-b-PCL copolymers with PCL blocks of different molecular weights
We prepared the mesoporous silica samples 1–4 when using various PEO-b-PCL diblock copolymers as templates at TEOS-to-template ratios of 2:1, 3:1, 5:1, and 11:1, respectively (Table 2), and analyzed them through SAXS, TEM, and BET measurements. We found that a greater weight of the PCL block in the PEO-b-PCL copolymer required more TEOS to confine the PCL segments of PEO-b-PCL and form ordered structures in the self-assembling system. In other words, the PCL segments were surrounded by PEO segments and TEOS during the EISA process; therefore, to construct ordered mesoporous silicas, higher-weight PCL blocks required larger amounts of TEOS. Fig. 2a displays the SAXS patterns of samples 1–4; the d-spacings of the large-pore mesoporous silicas increased dramatically from 12.8 nm (sample 1) to 44.9 nm (sample 4) upon increasing the weight of the PCL block in the PEO-b-PCL copolymer. Furthermore, the SAXS reflection sets also changed from cubic characteristics [(110), (211) and (220)] for sample 1 to hexagonal cylindrical features [(100), (110) and (200)] for sample 3 upon varying the composition of the template. TEM images (Fig. 2b–e) confirmed that the morphologies of the mesoporous large-pore silicas varied from bcc for sample 1 (along [100]) to a worm-like structure for sample 2, to a hexagonal packing structure for sample 3, and finally to a disordered micelle structure for sample 4. All of the mesoporous silica samples prepared with different PEO-b-PCL weight ratios provided typical type-IV isotherms in their N2 adsorption–desorption curves (Fig. 3a) and exhibited the H2 hysteresis loops expected for cage-like mesopores, which explained the spherical mesopores of sample 1 (bcc spheres), sample 2 (wormhole-like structure), sample 3 (hexagonal packing pores) and sample 4 (disordered micelles). We discussed the hexagonal packing mesoporous nanostructure (sample 3) in a previous report.10 We calculated the pore size distributions of the mesoporous silica samples from the adsorption branches of their BET isotherms, based on the BdB sphere model for spherical mesopores (Fig. 3b). The pore size of mesoporous silica samples increased greatly from 10.1 nm in sample 1 to 28.6 nm in sample 4 as a result of the increase in the weight fraction of the PCL block; accordingly, we believe that the pore size was mainly influenced by the size of the hydrophobic PCL segment. Herein, we could readily control the morphology and pore size of the mesoporous silicas merely by adjusting the PCL-to-PEO weight ratio of the amphiphilic copolymer PEO-b-PCL through simple ring-opening polymerization. Table 2 summarizes the related textural details of these mesoporous structures, including their d-spacings, pore sizes, BET surface areas, pore volumes, and micropore volumes.
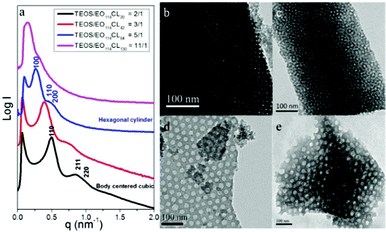 |
| | Fig. 2 (a) SAXS patterns and (b–e) TEM images of mesoporous silicas templated by EO114CLn at weight fractions of (b) TEOS/EO114CL20 = 2:1, (c) TEOS/EO114CL42 = 3:1, (d) TEOS/EO114CL84 = 5:1, and (e) TEOS/EO114CL130 = 11:1. | |
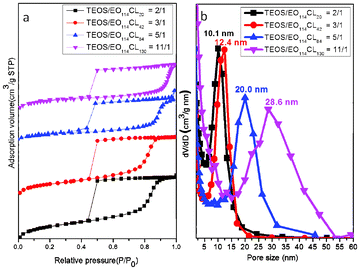 |
| | Fig. 3 (a) N2 adsorption–desorption isotherms and (b) pore size distribution curves of mesoporous silicas templated by EO114CLn at TEOS/EO114CLn weight fractions of 2:1 (n = 20), 3:1 (n = 42), 5:1 (n = 84), and 11:1 (n = 130). | |
Mesoporous silicas prepared using PCL homopolymers as a pore expander from EISA
To study the effects of blending a hydrophobic homopolymer during the EISA process for the synthesis of mesoporous silicas, we tested two PCL homopolymers with different molecular weights as additives: PCL20 (lab-made; Mn = 2100) and PCL408 (commercially available; Mn = 42500). Initially, we employed EO114CL20 as the template and PCL20 as the pore expander; the primary SAXS reflection peak of mesoporous silica shifted to the low-q position and the SAXS patterns tended toward short-range order upon increasing the amount of blended PCL20. The primary d-spacing of sample 5 (TEOS/EO114CL20/PCL20 = 2:1:0.1; 15.7 nm) was larger than that (12.8 nm) for sample 1 without the addition of PCL20 (Fig. 4a, Table 2), clearly suggesting a swollen mesostructure. The SAXS pattern of sample 5, however, displays two weaker scattering peaks (Fig. 4a, Table 2), implying that the bcc mesostructure (sample 1) became disordered, presumably because of the formation of distorted domains or defects in the mesostructure. Further increasing the amount of PCL20 (sample 6; TEOS/EO114CL20/PCL20 = 2:1:0.3) caused the primary d-spacing to increase further to 19.6 nm, reflecting a continuous structural expansion. The SAXS scattering eventually became one weak and broad peak corresponding to a large d-spacing of 22.4 nm, suggesting that the ordered structure continued to become disordered. TEM images of the mesoporous silicas confirmed the trends of expanded pore sizes and decreased order, with the ordered bcc mesostructure in Fig. 4b transforming into the disordered spheres in Fig. 4c–e upon increasing the PCL20 content, implying that large amounts of PCL20 could lead to distorted domains and even cause the mesostructure to disorder; the TEM images in Fig. 4c–e reveal that mesopores with polydisperse sizes and disordered spherical structures were formed in the presence of excessive amounts of PCL20. The N2 isotherms of samples 1, 5, 6, and 7, prepared by adding PCL20 as a pore expander, all feature typical type-IV curves with sharp capillary condensation steps in the relative pressure range 0.80–0.95 (Fig. 5a), suggesting uniform mesopores. Notably, the relative pressure for the capillary condensation increased upon increasing the amount of PCL20, indicating an increase in the mesopore size. In addition, we observe very large H2-type hysteresis loops, suggesting large caged mesopores with a small window size (<5.0 nm). Pore cavitations may have formed for the caged mesoporous materials during the desorption process. The pore size distributions derived from the adsorption branches, using the BdB sphere model, for samples 1 and 5–7 reveal that all were centered at mean values of 10–20 nm and increased upon increasing the amount of added PCL20 (Fig. 5b).
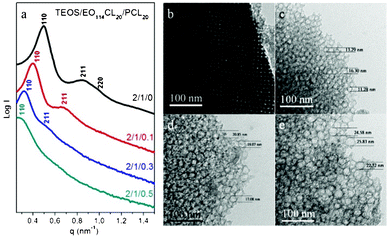 |
| | Fig. 4 (a) SAXS patterns and (b–e) TEM images of mesoporous silicas templated by EO114CL20 at TEOS/EO114CL20/PCL20 weight fractions of (b) 2:1:0, (c) 2:1:0.1, (d) 2:1:0.3, and (e) 2:1:0.5. | |
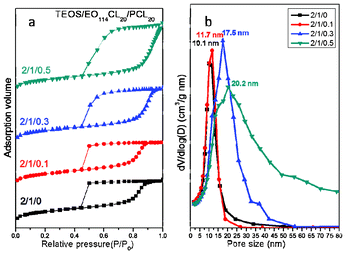 |
| | Fig. 5 (a) N2 adsorption–desorption isotherms and (b) pore size distribution curves of mesoporous silicas templated by EO114CL20 at TEOS/EO114CL20/PCL20 weight fractions of 2:1:0, 2:1:0.1, 2:1:0.3, and 2:1:0.5. | |
When we blended the higher-molecular-weight additive PCL408 with the same template EO114CL20, the SAXS patterns (Fig. 6a) did not reveal such large variations, with the primary d-spacing increasing only slightly from 12.8 nm for sample 1 (without the additive) to approximately 15 nm for samples 8–10, implying that the homopolymer PCL408 was not intimately involved into the self-assembling system during the solvent evaporation process. TEM analysis revealed that the morphology of the mesoporous silica transformed from an ordered bcc mesostructure (Fig. 6b) to a disordered sphere structure at a low addition ratio (TEOS/EO114CL20/PCL408 = 2:1:0.1; Fig. 6c), but the pore size changed only slightly upon further addition of PCL408 (Fig. 6d and e), suggesting that the molecular weight of PCL408 was too high to blend into the PCL domains of the TEOS/PEO-b-PCL self-assembled system during the EISA process. This behavior is similar to that of a typical A-b-B/A (block copolymer–homopolymer) blending system, where a homopolymer (A) having too high a molecular weight finds it difficult to blend into the A domains of the A-b-B block copolymer because of macrophase separation;43 in contrast, the blending of a homopolymer A having a molecular weight similar to that in the A-b-B block copolymer will result in dry-brush behavior, with the morphology not changing upon increasing the content of the homopolymer A.53,54 As a result, increasing the amount of PCL20 led to an increase in the mesopore size, but the long-range order of the mesopores decreased. Similar to the systems discussed above, samples 1 and 8–10 all displayed typical type-IV isotherms in their N2 adsorption–desorption curves, with large H2-type hysteresis loops and sharp capillary condensation steps in the relative pressure range from 0.80 to 0.90 (Fig. 7a). Furthermore, the pore sizes ranged between 9 and 10 nm (Fig. 7b, Table 2) with no obvious trend, suggesting that the homopolymer PCL408 did not blend well with PEO-b-PCL—as would be required for further expansion of the pore size. Table 2 lists the pore size distributions, calculated from the adsorption branches based on the spherical BdB model (Fig. 7b), and other textural details of samples 8–10.
 |
| | Fig. 6 (a) SAXS patterns and (b–e) TEM images of mesoporous silicas templated by EO114CL20 at TEOS/EO114CL20/PCL408 weight fractions of (b) 2:1:0, (c) 2:1:0.1, (d) 2:1:0.3, and (e) 2:1:0.5. | |
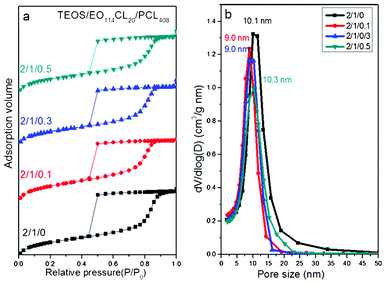 |
| | Fig. 7 (a) N2 adsorption–desorption isotherms and (b) pore size distribution curves of mesoporous silicas templated by EO114CL20 at TEOS/EO114CL20/PCL408 weight fractions of 2:1:0, 2:1:0.1, 2:1:0.3, and 2:1:0.5. | |
We also employed SAXS, TEM, and BET isotherms to characterize the mesoporous silicas prepared using EO114CL84 as the template and PCL20 as the pore expander. The primary peak in the SAXS pattern (Fig. 8a) of sample 11 (without added PCL20) featured a large d-spacing (26.2 nm); the primary d-spacings of these mesoporous silica samples increased upon increasing the amount of PCL20 in the blend, reaching a significantly expansive d-spacing of 36.9 nm for sample 14 (Table 2). TEM images suggested pore sizes of 20–33 nm for sample 11 (Fig. 8b), 25–40 nm for sample 12 (Fig. 8c), 25–45 nm for sample 13 (Fig. 8d), and 35–60 nm for sample 14 (Fig. 8e). Similar to the results discussed above, samples 11–14 all revealed typical type-IV adsorption–desorption curves with H2 hysteresis loops (Fig. 9a); we calculated the pore size distributions from the adsorption branches, based on the spherical BdB model. Fig. 9b reveals that the pore size of sample 11 (20.1 nm) expanded dramatically to 38.5 nm for sample 14 upon adding PCL20, confirming that this low-molecular-weight homopolymer could indeed be used effectively as a pore expander. When a homopolymer A having a low molecular weight is blended with an A-b-B block copolymer, it typically results in wet-brush behavior, with the morphology changing upon increasing the content of the homopolymer.51 Because the precursors of silica continue to form silica through hydrolysis and condensation during the EISA process, different to the polymer blend system, with an increase of the homopolymer PCL it is hard to change the morphology of the self-assembly system and only the increase in pore size could be observed.
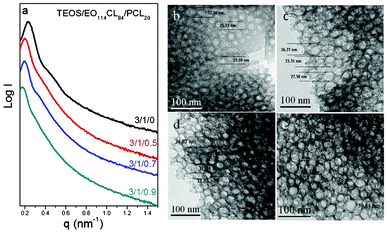 |
| | Fig. 8 (a) SAXS patterns and (b–e) TEM images of mesoporous silicas templated by EO114CL84 at TEOS/EO114CL84/PCL20 weight fractions of (b) 3:1:0, (c) 3:1:0.5, (d) 3:1:0.7, and (e) 3:1:0.9. | |
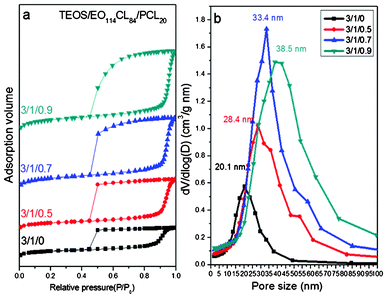 |
| | Fig. 9 (a) N2 adsorption–desorption isotherms and (b) pore size distribution curves of mesoporous silicas templated by EO114CL84 at TEOS/EO114CL84/PCL20 weight fractions of 3:1:0, 3:1:0.5, 3:1:0.7, and 3:1:0.9. | |
Mesoporous silicas prepared using PEO–POSS and PEO as modifiers for EISA
We also tested two other homopolymers—star PEO octa-functionalized polyhedral oligomeric silsesquioxane (PEO13–POSS) and linear PEO22 of low molecular weight (Mn = 1000)—as morphology modifiers for the fabrication of mesoporous silicas (templated by EO114CL84) through the EISA process. In a previous study of the mesoporous structures of phenolic resin templated by PEO-b-PCL/PEO–POSS, we found that the morphology changed and pore size increased upon increasing the PEO–POSS content.55 We were curious to discern whether these phenomena would also occur in a different matrix. First, we focused on the mesoporous silicas templated by the block copolymer EO114CL84 at various PEO–POSS contents. Fig. 10 presents the SAXS patterns and TEM images of the mesoporous silicas templated at TEOS/EO114CL84/PEO–POSS ratios of 3:1:0 (sample 11), 3:1:0.3 (sample 15), 3:1:0.5 (sample 16), and 3:1:0.7 (sample 17). The arrangement of the spherical morphology gradually tends to become ordered with the addition of PEO–POSS, however, it is hard to distinguish the appearance of the bcc structure by SAXS analysis, therefore, we need to discuss the morphology change by the combination of SAXS, TEM and BET characterizations. The primary spacing of the mesoporous silicas remained constant at 26.2 nm for samples 11, 15, and 16, but increased suddenly to 28.6 nm for sample 17. The three major scattering peaks of sample 17 at values of q of 0.22, 0.38, and 0.44 nm−1 could be indexed as (110), (211), and (220) reflections, respectively, indicating an ordered bcc mesostructure with a large primary d-spacing of 28.6 nm, which was further confirmed by TEM analysis (Fig. 10e). The mesoporous silicas in samples 11, 15, and 16 featured disordered sphere morphologies, whereas sample 17 possessed an ordered bcc structure (Fig. 10b–e). Thus, the use of PEO–POSS as an additive could lead to modification of the morphology of the mesoporous silica. The N2 sorption isotherms of the mesoporous silica samples obtained from a series of TEOS/EO114CL84/PEO–POSS blends exhibited (Fig. 11) representative type-IV curves with sharp capillary condensation steps in the relative pressure range from 0.85 to 0.95, suggesting regular mesostructures with uniform ultralarge pores. All of these mesoporous silica samples templated by EO114CL84 exhibited typical H2-like hysteresis loops, characteristic of spherical mesopores. Based on the BdB model, we determined the mean pore sizes for the mesoporous silica samples 11, 15, 16, and 17, measured from their adsorption branches, to be 20.1, 20.0, 19.8, and 22.6 nm, respectively; Table 2 summarizes the primary d-spacings, surface areas, pore volumes, and pore sizes of these samples. The primary d-spacings and pore sizes of samples 11, 15, and 16 were all similar, but both increased suddenly for sample 17, implying a transformation in morphology. In addition, the BET surface area, the micropore surface area, the pore volume, and the micropore volume all increased upon increasing the PEO–POSS content for samples 11, 15, and 16, but exhibited different trends upon further increasing the PEO–POSS content for sample 17; these experimental data also suggest a particular morphological transformation of the mesoporous silicas (from disordered spheres to an ordered bcc structure).
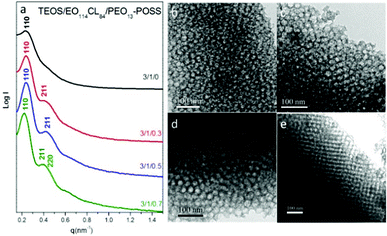 |
| | Fig. 10 (a) SAXS patterns and (b–e) TEM images of mesoporous silicas templated by EO114CL84 at TEOS/EO114CL84/PEO13–POSS weight fractions of 3:1:0, 3:1:0.3, 3:1:0.5, and 3:1:0.7. | |
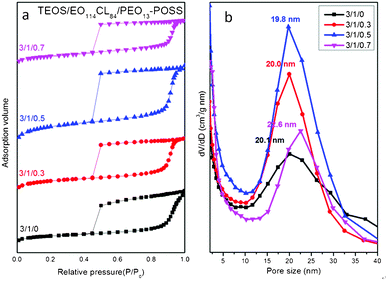 |
| | Fig. 11 (a) N2 adsorption–desorption isotherms and (b) pore size distribution curves of mesoporous silicas templated by EO114CL84 at TEOS/EO114CL84/PEO13–POSS weight fractions of 3:1:0, 3:1:0.3, 3:1:0.5, and 3:1:0.7. | |
For comparison with the effects of the star-like PEO–POSS, we also examined the linear homopolymer PEO22 as an additive blended with the EO114CL84 template for the fabrication of a series of mesoporous silicas. The features in the SAXS patterns (Fig. 12a) became more obvious and sharper upon increasing the PEO22 content, suggesting that blending with PEO22 could also enhance the ordering of the mesoporous silicas during the EISA process. The degree of modification was, however, limited and different from that of PEO–POSS: the TEM image in Fig. 12b reveals that the morphology did not ultimately change into an ordered bcc structure, suggesting that the star-like homopolymer was better at adjusting the morphology of the mesoporous silica during the self-assembly process.56,57 For brevity, we will not discuss the N2 adsorption–desorption isotherms. To our surprise, the long-range order and pore sizes both increased upon increasing the PEO–POSS content. The TEOS/template ratios for samples 1 (TEOS/EO114CL20 = 2:1) and 17 (TEOS/EO114CL84/PEG–POSS = 3:1:0.7) were similar and both revealed bcc structures in their SAXS and TEM analyses; the pore size in sample 17 (22.6 nm) was, however, much larger than that in sample 1 (10.1 nm). Therefore, our developed method allows the preparation of mesoporous silica with long-range order and large pore sizes through simple blending with homopolymers.
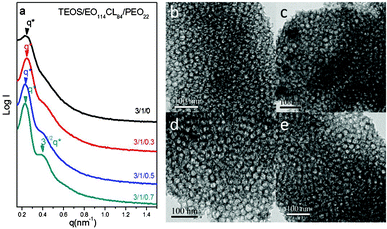 |
| | Fig. 12 (a) SAXS patterns and (b–e) TEM images of mesoporous silicas templated by EO114CL84 at TEOS/EO114CL84/PEO22 weight fractions of 3:1:0, 3:1:0.3, 3:1:0.5, and 3:1:0.7. | |
Conclusions
We have synthesized a series of PEO-b-PCL diblock copolymers, through ring-opening polymerization, that we have used as templates for the formation of mesoporous silicas. The morphology of the mesoporous silicas transformed from bcc to cylindrical structures and their pore sizes expanded upon increasing the PCL-to-PEO weight ratio in the templating PEO-b-PCL diblock copolymers. In addition, larger amounts of TEOS were necessary to construct ordered mesoporous silicas when using PEO-b-PCL diblock copolymers featuring PCL blocks of higher molecular weight. We employed four different additives (PCL20, PCL408, PEO13–POSS and PEO22) in the blends during the synthetic process. The pore size expanded upon increasing the PCL-to-PEO ratio through blending with PCL20, but it did not occur when blending with PCL408, because of wet- or dry-brush behavior in the presence of PCL20 and macrophase separation in the presence of PCL408. We observed interesting mesophase transitions and enhanced ordering upon increasing the amount of blended PEO–POSS, but not when blending with linear PEO, because this star-like homopolymer had greater miscibility with silica. Therefore, we have developed a simple method—blending with PEO–POSS—for the preparation the mesoporous silicas having bcc structures and exhibiting long-range order and large pore sizes (22.6 nm).
Acknowledgements
This study was supported financially by the National Science Council, Taiwan, Republic of China, under contracts NSC 97-2221-E-110-013-MY3 and NSC-99-2628-E-110-003.
References
- D. Arcos, A. Lopez-Noriega, E. Ruiz-Hernandez, O. Terasaki and M. Vallet-Regi, Chem. Mater., 2009, 21, 1000–1009 CrossRef CAS.
- A. P. Katsoulidis and M. G. Kanatzidis, Chem. Mater., 2012, 24, 471–479 CrossRef CAS.
- A. Popa, V. Sasca and I. Holclajtner-Antunovic, Microporous Mesoporous Mater., 2012, 156, 127–137 CrossRef CAS.
- H. J. Wang, L. Wang, T. Sato, Y. Sakamoto, S. Tominaka, K. Miyasaka, N. Miyamoto, Y. Nemoto, O. Terasaki and Y. Yamauchi, Chem. Mater., 2012, 24, 1591–1598 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- D. Y. Zhao, Q. S. Huo, J. L. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- S. Valkama, A. Nykanen, H. Kosonen, R. Ramani, F. Tuomisto, P. Engelhardt, G. ten Brinke, O. Ikkala and J. Ruokolainen, Adv. Funct. Mater., 2007, 17, 183–190 CrossRef CAS.
- J. G. Li and S. W. Kuo, RSC Adv., 2011, 1, 1822–1833 RSC.
- Y. Wan, Y. F. Shi and D. Y. Zhao, Chem. Commun., 2007, 897–926 RSC.
- J. G. Li, Y. D. Lin and S. W. Kuo, Macromolecules, 2011, 44, 9295–9309 CrossRef CAS.
- Y. Meng, D. Gu, F. Q. Zhang, Y. F. Shi, L. Cheng, D. Feng, Z. X. Wu, Z. X. Chen, Y. Wan, A. Stein and D. Y. Zhao, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS.
- Y. Wan, Y. F. Shi and D. Y. Zhao, Chem. Mater., 2008, 20, 932–945 CrossRef CAS.
- Y. Meng, D. Gu, F. Q. Zhang, Y. F. Shi, H. F. Yang, Z. Li, C. Z. Yu, B. Tu and D. Y. Zhao, Angew. Chem., Int. Ed., 2005, 44, 7053–7059 CrossRef CAS.
- L. Y. Song, D. Feng, N. J. Fredin, K. G. Yager, R. L. Jones, Q. Y. Wu, D. Y. Zhao and B. D. Vogt, ACS Nano, 2010, 4, 189–198 CrossRef CAS.
- H. Wei, Y. Y. Lv, L. Han, B. Tu and D. Y. Zhao, Chem. Mater., 2011, 23, 2353–2360 CrossRef CAS.
- J. Y. Zhang, Y. H. Deng, J. Wei, Z. K. Sun, D. Gu, H. Bongard, C. Liu, H. H. Wu, B. Tu, F. Schuth and D. Y. Zhao, Chem. Mater., 2009, 21, 3996–4005 CrossRef CAS.
- Y. H. Deng, J. Liu, C. Liu, D. Gu, Z. K. Sun, J. Wei, J. Y. Zhang, L. J. Zhang, B. Tu and D. Y. Zhao, Chem. Mater., 2008, 20, 7281–7286 CrossRef CAS.
- Y. H. Deng, T. Yu, Y. Wan, Y. F. Shi, Y. Meng, D. Gu, L. J. Zhang, Y. Huang, C. Liu, X. J. Wu and D. Y. Zhao, J. Am. Chem. Soc., 2007, 129, 1690–1697 CrossRef CAS.
- Y. Deng, C. Liu, D. Gu, T. Yu, B. Tu and D. Zhao, J. Mater. Chem., 2008, 18, 91–97 RSC.
- L. Sterk, J. Gorka, A. Vinu and M. Jaroniec, Microporous Mesoporous Mater., 2012, 156, 121–126 CrossRef CAS.
- Y. Huang, J. P. Yang, H. Q. Cai, Y. P. Zhai, D. Feng, Y. H. Deng, B. Tu and D. Y. Zhao, J. Mater. Chem., 2009, 19, 6536–6541 RSC.
- M. Mandal and M. Kruk, Chem. Mater., 2012, 24, 149–154 CrossRef CAS.
- Y. F. Lee, K. H. Chang, C. Y. Chu, H. L. Chen and C. C. Hu, RSC Adv., 2011, 1, 401–407 RSC.
- J. G. Li, R. B. Lin and S. W. Kuo, Macromol. Rapid Commun., 2012, 33, 678–682 CrossRef CAS.
- S. P. Naik, W. Fan, T. Yokoi and T. Okubo, Langmuir, 2006, 22, 6391–6397 CrossRef CAS.
- K. Yu, A. J. Hurd, A. Eisenberg and C. J. Brinker, Langmuir, 2001, 17, 7961–7965 CrossRef CAS.
- C. Liu, Y. H. Deng, J. Liu, H. H. Wu and D. Y. Zhao, Microporous Mesoporous Mater., 2008, 116, 633–640 CrossRef CAS.
- Z. D. Zhang, X. X. Yan, B. Z. Tian, C. Z. Yu, B. Tu, G. S. Zhu, S. L. Qiu and D. Y. Zhao, Microporous Mesoporous Mater., 2006, 90, 23–31 CrossRef.
- M. Mandal and M. Kruk, Chem. Mater., 2012, 24, 123–132 CrossRef CAS.
- A. F. Zhang, K. K. Hou, L. Gu, C. Y. Dai, M. Liu, C. S. Song and X. W. Guo, Chem. Mater., 2012, 24, 1005–1010 CrossRef CAS.
- E. Nisson, Y. Sakamoto and A. E. C. Palmqvist, Chem. Mater., 2011, 23, 2781–2785 CrossRef.
- D. Grosso, C. Boissiere, B. Smarsly, T. Brezesinski, N. Pinna, P. A. Albouy, H. Amenitsch, M. Antonietti and C. Sanchez, Nat. Mater., 2004, 3, 787–792 CrossRef CAS.
- E. Ortel, A. Fischer, L. Chuenchom, J. Polte, F. Emmerling, B. Smarsly and R. Kraehnert, Small, 2012, 8, 298–309 CrossRef CAS.
- S. C. Chen, S. W. Kuo, U. S. Jeng, C. J. Su and F. C. Chang, Macromolecules, 2010, 43, 1083–1092 CrossRef CAS.
- K. Dobrosielska, S. Wakao, J. Suzuki, K. Noda, A. Takano and Y. Matsushita, Macromolecules, 2009, 42, 7098–7102 CrossRef CAS.
- M. W. Matsen, Macromolecules, 1995, 28, 5765–5773 CrossRef CAS.
- M. P. Stoykovich, M. Muller, S. O. Kim, H. H. Solak, E. W. Edwards, J. J. de Pablo and P. F. Nealey, Science, 2005, 308, 1442–1446 CrossRef CAS.
- T. Hashimoto, K. Yamasaki, S. Koizumi and H. Hasegawa, Macromolecules, 1993, 26, 2895–2904 CrossRef CAS.
- V. Abetz and T. Goldacker, Macromol. Rapid Commun., 2000, 21, 16–34 CrossRef CAS.
- L. Zhu, B. R. Mimnaugh, Q. Ge, R. P. Quirk, S. Z. D. Cheng, E. L. Thomas, B. Lotz, B. S. Hsiao, F. J. Yeh and L. Z. Liu, Polymer, 2001, 42, 9121–9131 CrossRef CAS.
- H. Tanaka, H. Hasegawa and T. Hashimoto, Macromolecules, 1991, 24, 240–251 CrossRef CAS.
- T. W. Kim, R. Ryoo, M. Kruk, K. P. Gierszal, M. Jaroniec, S. Kamiya and O. Terasaki, J. Phys. Chem. B, 2004, 108, 11480–11489 CrossRef CAS.
- Y. Sakamoto, M. Kaneda, O. Terasaki, D. Y. Zhao, J. M. Kim, G. Stucky, H. J. Shim and R. Ryoo, Nature, 2000, 408, 449–453 CrossRef CAS.
- F. Kleitz, T. W. Kim and R. Ryoo, Langmuir, 2006, 22, 440–445 CrossRef CAS.
- Y. C. Hu, Z. Z. Zhi, Q. F. Zhao, C. Wu, P. Zhao, H. T. Jiang, T. Y. Jiang and S. L. Wang, Microporous Mesoporous Mater., 2012, 147, 94–101 CrossRef.
- H. Q. Yang, J. Li, J. Yang, Z. M. Liu, Q. H. Yang and C. Li, Chem. Commun., 2007, 1086–1088 RSC.
- F. Kleitz, T. Czuryszkiewicz, L. A. Solovyov and M. Linden, Chem. Mater., 2006, 18, 5070–5079 CrossRef CAS.
- S. B. Hartono, W. Y. Gu, F. Kleitz, J. Liu, L. Z. He, A. P. J. Middelberg, C. Z. Yu, G. Q. Lu and S. Z. Qiao, ACS Nano, 2012, 6, 2104–2117 CrossRef CAS.
- C. E. Tattershall, N. P. Jerome and P. M. Budd, J. Mater. Chem., 2001, 11, 2979–2984 RSC.
- C. E. Tattershall, S. J. Aslam and P. M. Budd, J. Mater. Chem., 2002, 12, 2286–2291 RSC.
- K. H.Dai and E. J. Kramer, Macromolecules, 1992, 25, 220–225 CrossRef.
- S. Koizumi, H. Hasegawa and T. Hashimoto, Macromolecules, 1994, 27, 6532–6540 CrossRef CAS.
- J. G. Li, C. Y. Chung and S. W. Kuo, J. Mater. Chem., 2012, 22, 18583–18595 RSC.
- C. F. Huang, S. W. Kuo, H. C. Lin, J. K. Chen, Y. K. Chen, H. Y. Xu and F. C. Chang, Polymer, 2004, 45, 5913–5921 CrossRef CAS.
- J. Shen and S. X. Zheng, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 942–952 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.