Capping of oligonucleotides with “clickable” m3G-CAPs†
Malgorzata
Honcharenko
*a,
Joanna
Romanowska
a,
Margarita
Alvira
a,
Martina
Jezowska
a,
Mikael
Kjellgren
a,
C. I.
Edvard Smith
ab and
Roger
Strömberg
*a
aDepartment of Biosciences and Nutrition, Karolinska Institutet, Novum, SE-14183 Huddinge, Sweden. E-mail: Roger.Stromberg@ki.se; Malgorzata.Honcharenko@ki.se
bDepartment of Laboratory Medicine, Karolinska Institutet, Karolinska University Hospital Huddinge, SE-141 86 Huddinge, Sweden
First published on 8th November 2012
Abstract
The RNA components of small nuclear ribonucleoproteins (U snRNPs) possess a characteristic 5′-terminal 2,2,7-trimethyl-guanosine CAP structure (m3G-CAP). This cap is an important component of the nuclear localization signal of U snRNPs. Here we report synthesis of four m3G-CAP constructs and the effective attachment of these onto oligonucleotides (ONs) using Cu(I) [3 + 2] cycloaddition (“click chemistry”). The four constructs (1–4, Fig. 1) are equipped with a handle that in principle allows for universal conjugation to any cargo and differ in their complexity starting from m3GpppA(linker) to m3GpppA(OMe)U(OMe)A(linker) that resembles the native m3G-CAP followed by the 2′-O-methylated sequence AUA. The four m3G-CAP containing constructs are equipped with an azide linker and by taking advantage of initial attachment of a novel activated triple bond donor p-aminomethyltoluic acid (PATA) to ONs on solid support we were able to synthesize novel bioconjugates equipped with different constructs carrying the m3G-CAP Nuclear Localisation Signal (NLS) for the investigation of nuclear delivery.
Introduction
Accessing the nucleus through the surrounding membrane is one of the major obstacles for therapeutic molecules large enough to be excluded due to nuclear pore size limits. In some therapeutic applications the large size of certain nucleic acids, like plasmid DNA, hampers their access to the nuclear compartment. Moreover, also for small ONs, achieving higher nuclear concentrations by active, in addition to passive, transport could be of great benefit.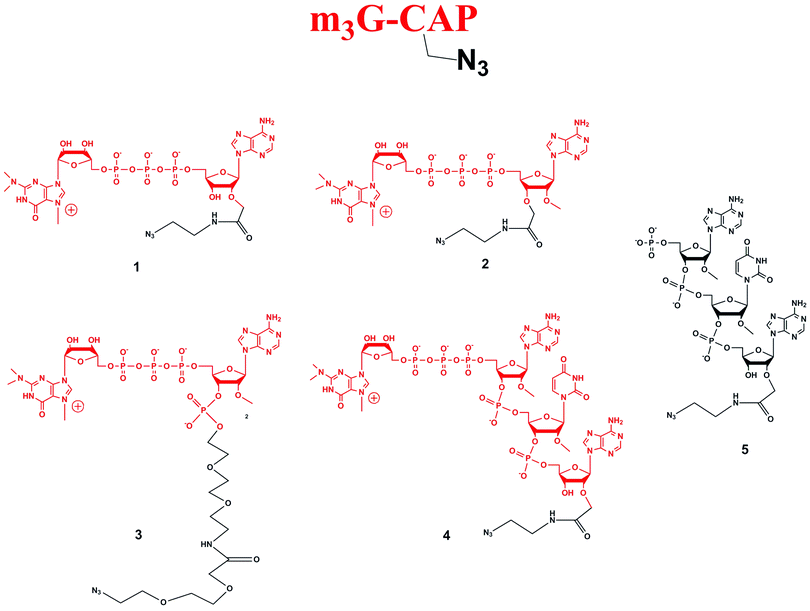 | ||
| Fig. 1 Structures of m3G-CAP analogues (1–4) and the 5′-phosphorylated AUA trimer (5). | ||
The 2,2,7-trimethylguanosine cap (m3G-CAP) is found naturally in the small nuclear RNAs that are a constitutive part of the small nuclear ribonucleoprotein complexes involved in nuclear splicing.1
As we have reported,2 increased efficiency in a splice-correction (splice-switching) assay (measured by increase in activity of a luciferase reporter construct) is achieved by using a synthetic RNA 5′-end nuclear localization signal composed of an m3G-CAP and this is presumably due to increased nuclear localization. We also investigated the usage of the m3G-CAP as an adaptor signal for the nuclear transport of the complex between a biotinylated ON and the protein streptavidin clearly showing that the m3G-CAP can promote nuclear transport.2
Use of the m3G-CAP as an NLS signal for increased nuclear delivery of a therapeutic compound is likely to require additional stability towards enzymatic degradation. For that reason introduction of stabilizing modifications into the triphosphate bridge may be beneficial. Existing capping methods that rely on reaction of 5′-phosphate ONs with activated form of m3G-pyrophosphate2,3 do, however, not permit inclusion of most feasible modifications and also do not allow attachment to a cargo (e.g., protein, PNA etc) that does not contain a nucleoside 5′-phosphate. In addition use of the m3G-CAP as an adaptor for delivery in vivo requires not only convenient synthesis but also a method that is readily scaled up for animal experiments and clinical trials.
When making a synthetic artificially attached m3G -CAP we also need to investigate the minimum structural requirement for retained nuclear transport ability of the construct. The import adaptor Snurportin1 recognizes the m3G-CAP and facilitates the nuclear transport of the UsnRNPs by binding to Importin-β. The crystal structure of the m3G-CAP-binding domain of Snurportin1 with bound m3GpppG at 2.4 Å resolution revealed that Snurportin1 binds both the hyper-methylated cap and the first nucleotide of the RNA in a stacked conformation.4 The Kd-value for the m3GpppG dinucleotide (which was present in the crystal structure) was determined to be 1.0 μM, while it is 12.1 μM for the m3GpppA dinucleotide. The crystal structure of the complex containing the m3GpppG dinucleotide reveals that the adenosine of m3GpppA could be harbored in the binding pocket as well, since there is no strong discrimination between the purines commonly caused by polar interactions. In contrast the Kd-value for m7GpppA is greater than 170 μM. Obviously, the difference in Kd-values between m3GpppA and m7GpppA must be related to the two methyl groups on N2 of the m3G-base. These two methyl groups have van der Waals (VDW) contact with Trp107, but the difference in affinities between m3GpppA/G and m7GpppA is suggested to not being caused exclusively by the contribution of these two VDW contacts to the free binding energy, but that the difference in affinity should be related to the binding process itself. Consistent with a previous report,5 the binding of the synthetic tetranucleotide m3GpppA(OMe)U(OMe)A, which more closely resembles UsnRNAs in vivo, to full-length Snurportin1 is even stronger (Kd = 0.23 mM) than that of m3GpppG, which should be related to the presence of additional nucleotides and/or due to the 2′-O-methylated riboses. Inspection of the electrostatic surface potential of the m3G-CAP-binding domain gives no hint on binding to the additional nucleotides. However, the crystallized domain lacked the C-terminal 61 residues, which could extend the RNA-binding surface. The addition of an artificial linker could also affect the ability to bind Snurportin1 and it then seemed necessary to develop a method that also allowed us to make a number of constructs that could be used to map out the requirements and that would allow attachment to a number of cargos. At the same time the method should allow for construction of m3G-CAP analogs with modified triphosphate bridges that would stabilize these towards degradation.
Cu(I) catalysed [3 + 2] cycloaddition, seems to be ideal for capping, since it can be performed under aqueous conditions and the triphosphate structure necessitates this. In addition the copper(I)-catalyzed reaction is mild, usually efficient, highly selective and requires no protecting of functional groups in biomolecules. The azide group is largely inert towards biological molecules and aqueous environments, which allows the use of the Huisgen 1,3-dipolar cycloaddition in target guided synthesis6 and activity-based protein profiling.7 In our experience click reaction with a standard alkyne linker at low concentration is often unsuccessful. Microwave heating could then be used to promote the reaction but this could be detrimental to sensitive structures, e.g., the m3G-Cap.
Here we report methodology and synthesis of different m3G-CAP constructs (1–4, Fig. 1) equipped with an azide handle allowing for conjugation through “click reaction” with a triple bond partner. We also demonstrate attachment of the different m3G-CAP constructs to ONs carrying the novel activated PATA8 triple bond donor that allows efficient conjugation at low concentration without heating giving the corresponding ON-m3G-CAP conjugates.
Experimental procedures
Acetonitrile (MeCN, HPLC grade, Merck), methanol (MeOH), dichloromethane (DCM, Fisher Scientific, Analytical Grade) and 2-(2-(2-aminoethoxy)ethoxy)ethanol (Aldrich) were of commercial grade and used as received. Dimethylformamide (DMF) and pyridine (Py) (both from Merck and analytical grade) were additionally dried over 4A molecular sieves. Silica gel column chromatography was performed on Merck G60, TLC-analysis was carried out on pre-coated Silica Gel 60 F254 (Merck), with detection by UV light and/or charring by treatment with 8% Sulfuric acid in ethanol-water followed by heating. NMR spectra were recorded on a Bruker AVANCE DRX-400 instrument (400.13 MHz for 1H, 162.00 MHz for 31P, 100.62 MHz for 13C). Reversed phase HPLC was carried out on a Jasco HPLC system using the following columns: Hypersil 5 μm (250 × 4.6 mm) with 1 mL min−1 flow rate, Kromasil 100-5-C18, 5 μm (250 × 4.6 mm) with 1 mL min−1 flow rate and Kromasil 100-5-C18, 5 μm (250 × 10 mm) at 4 mL min−1 flow rate. For ion exchange chromatography a Dionex NucleoPac PA-100 (4 × 250) column was used. Buffers for reversed phase chromatography were as follows: A: 50 mM triethylammonium acetate (TEAA), pH 6.5; B: 50 mM TEAA pH 6.5 in 50% MeCN. Buffers for ion exchange chromatography: C: 20 mM NaOAc in 30% MeCN; D: 20 mM NaOAc, 0.3 M LiClO4 in 30% MeCN. Mass spectra were obtained by use of a Micromass LCT electrospray time-of-flight (ES-TOF) instrument. Molecular weights of the conjugates were reconstructed from the m/z values of the multiply charged ions using the mass deconvolution program (MAXENT) of the instrument, which gave values with no decimal place. The oligonucleotide, 5'-ODMT-2'-OMeCCUCUUACCUCAGUUACA [18merRNA(2′-OMe)], was purchased from Rasayan Inc., Encinitas, CA, USA, still attached on solid support. Nucleobase protecting groups were benzoyl for Cytosine and Adenine and isobutyryl for Guanine. Calculation of the amount of the 5′-alkynelinker-18merRNA(2′-OMe) was done by the nearest neighbor approach9 and based on OD measurement and relative peak areas after analysis of a small fraction of crude 5′-alkynelinkerON by HPLC.2́-O-(N-(2-Azidoethyl)carbamoylmethyl-5́-O-(4-monomethoxytrityl)adenosine (7)
MMTrA (6) (1.05 mmol, 565.5 mg) was dried by evaporation of added dry tetrahydrofurane (THF). The nucleoside was dissolved in dry THF (45 mL) at r.t. under argon atmosphere. Potassium t-butoxide (t-BuOK, 1.3 eq, 1.36 mmol, 152.9 mg) was added and after 20 min also allyl bromoacetate (1.3 eq, 1.32 mmol, 236.7 mg).10–12 Stirring was continued for 6 h and then the mixture was concentrated under reduced pressure. The residue was dissolved in dry MeOH (20 mL) and 2-azidoethylamine13 (10.2 eq, 10.7 mmol, 919 mg) was added and the reaction mixture was stirred for 24 h. The reaction was quenched with NaHCO3 (aq.) and extracted with DCM (3 × 20 mL). The combined organic layers were dried with MgSO4, filtered, and the solvent evaporated giving the crude product as a yellow oil. Purification was done using flash chromatography using a gradient of methanol in DCM (from 0–10%). Concentration of collected fractions afforded compound 7 as a white solid. Yield: 348 mg, 50%. 1H NMR (400 MHz, CDCl3): δ 8.30 (1H, s), 8.03 (1H, s), 7.30 (12H, m), 6.84 (2H, m), 6.81 (1H, d, J = 3.8 Hz), 5.81 (1H, s), 4.55 (1H, t, J = 4.8 Hz), 4.47 (1H, t, J = 5.2 Hz), 4.29 (3H, m), 4.03 (2H, m), 3.79 (3H, s), 3.46 (6H, m), 2.88 (1H, bs). 13C NMR (100 MHz, CDCl3) δ 38.8, 41.5, 50.8, 54.8, 55.4, 63.2, 71.0, 70.2, 70.6, 83.9, 84.2, 87.3, 87.8, 100.2, 113.5, 120.4, 127.4, 128.0, 128.2, 128.5, 130.5, 131.3, 135.2, 138.9, 144.0, 144.1, 149.6, 153.4, 155.7, 158.9, 169.6; MS (ESI) TOF-mass m/z 666.27 calculated for C34H36N9O6; found 666.19 [M+H].2́-O-(N-(2-Azidoethyl)carbamoyl)methyl-3́-O,N,N-tribenzoyladenosine (8)
7 (0.283 mmol, 188 mg) was dried by evaporation of added dry pyridine and dissolved in the same solvent (3.0 mL). Benzoyl chloride was added (3.2 eq, 0.905 mmol, 0.105 mL) and reaction was left stirring overnight under argon atmosphere. After completion of the reaction, the solvent was removed under reduced pressure and the residue dissolved in 30 mL DCM. Sat. aqueous NaHCO3 (50 mL) was slowly added. The mixture was extracted with DCM (3 × 20 mL), the organic phase was dried over MgSO4 and filtered. The solvent was evaporated and residue dried by evaporation of added toluene (2 × 20 mL) and DCM (2 × 20 mL). The crude product was dissolved in 80% acetic acid (100 mL), and reaction was left stirring for 2 h. After completion of the reaction the acid was removed by evaporation under reduced pressure and remaining acetic acid was removed by co-evaporation with toluene (2 × 20 mL) and DCM (2 × 20 mL). Purification with flash chromatography (using a 0–1.5% gradient of methanol in dichloromethane) gave compound 8. Yield: 137 mg, 69% (after two steps). 1H NMR (400 MHz, CDCl3): δ 8.61 (1H, s), 8.15 (1H, s), 7.80–7.29 (15H, m), 6.04 (1H, d, J = 7.5 Hz), 5.85 (1H, d, J = 4.9 Hz), 5.01 (1H, m), 4.51 (1H, s), 3.98–3.80 (2H, m), 3.26–3.09 (4H, m), 1.21–1.18 (2H, m). 13C NMR (100 MHz, CDCl3): δ 37.4, 45.8, 49.4, 69.7, 72.2, 79.8, 85.0, 88.6, 126.6, 127.5 (4C), 127.9 (2C), 128.6 (4C), 128.9 (2C), 129.2, 132.3, 132.9 (2C), 133.1, 133.5 (2C), 140.2, 143.5, 146.3, 151.0, 165.9, 167.9, 171.2 (2C); MS (ESI) TOF-mass m/z 706.24 calculated for C35H32N9O8, found 706.14, [M+H]2́-O-(N-(2-Azidoethyl)carbamoyl)methyladenosine 5́-phosphate (10)
Ammonium (9H-fluoren-9-yl)methyl-H-phosphonate (1.05 eq, 0.031 mmol, 8.7 mg)14 was dried twice by evaporation of added dry pyridine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Et3N (8:2). Compound 8 (0.030 mmol, 22.3 mg) was added, then dry pyridine was added and evaporated twice, whereupon the mixture was further dried on a vacuum line for 50 min. The residue was dissolved in dry DCM:dry pyridine (9:1) and PivCl (1.6 eq, 0.049 mmol, 6 μL) was added after which stirring was continued for 30 min under argon atmosphere. A solution consisting of I2 (1.3 eq, 0.038 mmol, 9.6 mg), H2O (50 μL) and pyridine (0.5 mL) was added, the mixture was stirred for an additional 10 min and then the volatiles were removed under reduced pressure. After completion of the diester formation (TLC) 0.5 mL MeCN:Et3N (2:1) was added and reaction was left stirring overnight. The mixture was evaporated to dryness and the residue was dissolved in DCM and extracted with water (3 × 10 mL). The aqueous phase was concentrated and the crude phosphate 9 was dissolved in a saturated solution of ammonia in methanol (18%) (2 mL) and the reaction was left for 24 h at r.t. The volatiles were removed under reduced pressure. The residue was dissolved in diethyl ether and extracted with water (2 × 20 mL). The aqueous layer was then evaporated to dryness under reduced pressure. 1H NMR (400 MHz, D2O): δ 8.46 (1H, s), 8.26 (1H, s), 6.24 (1H, d, J = 5.0 Hz), 4.60–4.58 (1H, m), 4.37 (1H, bs), 4.25 (3H, m), 3.41–3.24 (4H, m), 3.17–3.11 (12H, m), 1.27–1.17 (20H, m). 13C NMR (100 MHz, D2O): δ 8.6 (6C), 16.9, 38.2, 47.1 (6C), 50.2, 52.4, 57.2, 69.7, 76.2, 83.5, 120.8, 122.4, 127.8, 129.2, 132.9, 172.8. 31P NMR (162.0 MHz, D2O): δ 0.07; MS (ESI) TOF-mass m/z 472.11 calculated for C14H19N9O8P, found 472.44, [M − H].
:Et3N (8:2). Compound 8 (0.030 mmol, 22.3 mg) was added, then dry pyridine was added and evaporated twice, whereupon the mixture was further dried on a vacuum line for 50 min. The residue was dissolved in dry DCM:dry pyridine (9:1) and PivCl (1.6 eq, 0.049 mmol, 6 μL) was added after which stirring was continued for 30 min under argon atmosphere. A solution consisting of I2 (1.3 eq, 0.038 mmol, 9.6 mg), H2O (50 μL) and pyridine (0.5 mL) was added, the mixture was stirred for an additional 10 min and then the volatiles were removed under reduced pressure. After completion of the diester formation (TLC) 0.5 mL MeCN:Et3N (2:1) was added and reaction was left stirring overnight. The mixture was evaporated to dryness and the residue was dissolved in DCM and extracted with water (3 × 10 mL). The aqueous phase was concentrated and the crude phosphate 9 was dissolved in a saturated solution of ammonia in methanol (18%) (2 mL) and the reaction was left for 24 h at r.t. The volatiles were removed under reduced pressure. The residue was dissolved in diethyl ether and extracted with water (2 × 20 mL). The aqueous layer was then evaporated to dryness under reduced pressure. 1H NMR (400 MHz, D2O): δ 8.46 (1H, s), 8.26 (1H, s), 6.24 (1H, d, J = 5.0 Hz), 4.60–4.58 (1H, m), 4.37 (1H, bs), 4.25 (3H, m), 3.41–3.24 (4H, m), 3.17–3.11 (12H, m), 1.27–1.17 (20H, m). 13C NMR (100 MHz, D2O): δ 8.6 (6C), 16.9, 38.2, 47.1 (6C), 50.2, 52.4, 57.2, 69.7, 76.2, 83.5, 120.8, 122.4, 127.8, 129.2, 132.9, 172.8. 31P NMR (162.0 MHz, D2O): δ 0.07; MS (ESI) TOF-mass m/z 472.11 calculated for C14H19N9O8P, found 472.44, [M − H].
5́-O-(4-Monomethoxytrityl)-2́-O-methyladenosine (11)
2́-O-Methyladenosine (3.2 mmol, 899 mg) was dried by evaporation of added dry pyridine (2×). The residue was dissolved in dry pyridine (33 mL) whereupon 4-monomethoxytrityl chloride (MMTr-Cl, 1.1 eq, 3.5 mmol, 1.1 g) was added. The mixture was stirred under argon atmosphere, at a temperature starting from 0 °C and that was allowed to rise to r.t overnight. The reaction was quenched after 24 h with sat. aqueous NaHCO3. The mixture was extracted with DCM (3 × 30 mL) and the collected organic fractions were dried over MgSO4. The mixture was filtered and the volatiles removed under reduced pressure. The crude product was purified with flash chromatography (silica gel, gradient elution dichloromethane : methanol (0–3% MeOH) Yield: 1.01 g, 57%. 1H NMR: (400 MHz, CDCl3): δ 8.02 (1H, s), 7.79 (1H, s), 7.35–7.06 (12H, m), 6.82–6.80 (2H, m), 5.83 (1H, m), 4.72 (1H, m), 4.57 (1H, bs), 4.35 (1H, s), 3.92 (1H, d), 3.73 (1H, d). 13C NMR: (100.6 MHz, CDCl3): δ = 1.1, 21.6, 29.8, 55.3, 63.4, 70.7, 71.2, 77.3, 82.6, 88.4, 89.8, 113.3, 122.7–130.3 (10 peaks), 136.1, 140.4, 145.0, 150.0, 151.8, 154.7, 158.5; MS (ESI) TOF-mass m/z 554.24, calculated for C31H32N5O5 observed mass: 553.95, [M+H].3́-O-(N-(2-Azidoethyl)carbamoylmethyl-2́-O-Methyladenosine 5′-phosphate (14)
Compound 11 (0.476 mmol, 263.8 mg) was dried by evaporation of added dry THF, and subsequent evacuation with a vacuum pump overnight. The nucleoside was dissolved in dry THF (20 mL) and t-BuOK (1.3 eq, 0.627 mmol, 70.4 mg) was added. After 40 min allyl bromoacetate (1.3 eq, 0.63 mmol, 113 mg) was added, and the stirring was continued for 6 h. The reaction mixture was concentrated to a quarter of the volume and first dry MeOH (1 mL) and then 2-azidoethylamine (4.3 mmol, 371.6 mg) was added. The mixture was left stirring overnight. The reaction was quenched with sat. aqueous NaHCO3, the organic phase was separated and the aqueous layer was extracted with DCM (3 × 30 mL). The combined organic phases were dried over MgSO4 and filtered. The volatiles were removed in vacuo. Crude compound 12 was subjected to deprotection with 80% AcOH in water for 2 h to give compound 13. The reaction mixture was concentrated to dryness and remaining acetic acide was removed by co-evaporation with toluene (2 × 10 mL) and DCM (2 × 10 mL). The crude was dried by evaporation of added dry pyridine (2 × 10 mL) dissolved in pyridine and added to ammonium (9H-fluoren-9-yl)methyl-H-phosphonate (0.0616 mmol, 17.9 mg) (pre-dried and converted to triethylammonium salt prior to the addition by twice evaporating added dry pyridine:Et3N (8:2)). The mixture of the two compounds was further dried by evaporation of added dry pyridine (2×) subsequent evacuation on a vacuum line for 2 h. The mixture was dissolved in 0.6 mL dry DCM:pyridine (9:1) and stirred under argon atmosphere, pivaloyl chloride (PvCl, 1.7 eq, 0.107 mmol, 12.9 mg) was added and the mixture was stirred for 30 min. A prepared solution of I2 (0.063 mmol, 16.1 mg), H2O (50 μL) and pyridine (0.5 mL) was then added to oxidize the H-phosphonate diester. To achieve the hydrolysis of the phosphate diester to monoester the mixture was first evaporated to dryness and then dissolved in MeCN:Et3N (2:1) and stirred overnight. The mixture was evaporated to dryness, dissolved in DCM and extracted with water (3 × 1 mL). The aqueous phase was concentrated under reduced pressure and purification of 14 was performed by RP-HPLC tR = 18.7 (method: 1 ml min−1, gradient 0–35% buffer B in A, during 30 min), yield: 8.6 mg. 1H NMR (400 MHz, D2O): δ 8.59 (1H, s), 8.20 (1H, s), 6.21 (1H, d, J = 6.2 Hz), 4.60 (2H, t, J = 4.8 Hz), 4.47 (1H, s), 4.23 (2H, ABq, H: J = 15.3 Hz, J = 23.3 Hz), 4.01 (1H, s), 3.47–3.41 (4H, m), 3.38 (3H, s), 3.15–3.10 (12H, m), 1.85 (2H, s), 1.22–1.15 (18H, m). 13C NMR (100 MHz, D2O): δ 8.6 (6C), 17.0, 38.8, 47.1 (6C), 50.4, 58.7, 64.6, 69.4, 78.5, 82.8, 83.1, 85.6, 119.4, 136.5, 138.7, 153.4, 156.1, 172.7. 31P NMR (162.0 MHz, D2O): δ 0.13; MS (ESI) TOF-mass m/z 486.13, calculated for C15H21N9O8P observed mass: 486.39, [M − H].
2′-O-Methyl-5′-(4-monomethoxytrityl)adenosine 3′- H-phosphonate triethylammonium salt (18)
Compound 11 (1 mmol, 550 mg) and triethylammonium (9H-fluoren-9-yl)methyl-H-phosphonate (1.1 eq, 1.1 mmol, 300 mg weighed as ammonium salt treated with Pyridine-Et3N as above) were made anhydrous by evaporation of added dry pyridine (2×), dissolved in 10 mL of DCM/pyridine (9:1, v/v) and PvCl (1.5 molar excess) was added. Reaction was complete (31P NMR, TLC) after 20 min. After this, solvents were evaporated under reduced pressure and the residue was dissolved in dichloromethane and washed with brine (2×). The organic layer was separated, dried over MgSO4 and concentrated. The remaining oily residue was dissolved in 10 mL of MeCN/Et3N (2:1, v/v) and kept for 20 min. at room temperature to effect a quantitative elimination of the (9H-fluoren-9-yl)methyl group. The reaction mixture was concentrated and product 18 was isolated by column chromatography using a stepwise gradient of water (0–15%) in 2-propanol containing Et3N (3% v/v). Yield: 650 mg, 91%. 1H NMR (400 MHz, CDCl3): δ 8.24 (1H, s), 7.98 (1H, s), 7.45–7.43 (4H, m), 7.32–7.19 (8H, m), 6.98 (1H, dd, J = 9.4 Hz, J = 626.0 Hz), 6.81–6.79 (2H, m), 6.21–6.19 (1H, m), 5.02 (1H, q, J = 4.7 Hz), 4.61 (1H, t, J = 5.1 Hz), 4.48–4.47 (1H, bs), 3.77 (3H, s), 3.54 (1H, s), 3.52–3.46 (2H, m), 3.00–2.94 (6H, m), 1.28–1.25 (9H, m). 13C NMR (100 MHz, CDCl3): δ 8.91 (3C), 45.5 (3C), 55.2, 58.6, 63.4, 71.4, 82.5, 83.6, 86.1, 86.9, 113.2 (2C), 120.0, 127.1 (2C), 127.9 (3C), 128.6 (3C), 130.5 (2C), 135.1, 138.9, 144.0 (2C), 150.0, 153.0, 155.6, 158.6. 31P NMR (162.0 MHz, CDCl3): δ 3.87 (dd, J = 9.4 Hz, J = 626.0 Hz); MS (ESI) TOF-mass m/z 616.20, calculated for C31H31N5O7P observed mass: 616.49, [M − H].
2′-O-Methyladenosine 3′-(17-azido-10-oxo-3,6,12,15-tetraoxa-9-azaheptadecyl)phosphate triethylammonium salt (20)
A: 2(2-Azidoethoxy)ethoxyacetic acid8 (0.5 mmol, 94 mg) 16 was dissolved in dry dichloromethane (3 mL) and first HBTU (1.05 eq, 0.53 mmol, 199 mg) and then NMM (10 eq, 5 mmol, 550 μL) was added and the reaction was stirred at r.t for 30 min. After this time 2-(2-(2-aminoethoxy)ethoxy)ethanol (1.1 eq, 0.55 mmol, 82 g) 15 was added to the activated acid and the reaction mixture was left stirring for 2 h. When the formation of 2-(2-(2-azidoethoxy)ethoxy)-N-(2-(2-(2-hydroxyethoxy)ethoxy)ethyl)acetamide 17 was completed (confirmed by ES-TOF MS) the crude product was used directly for the next reaction step. B: Nucleoside H-phosphonate 18 (1 eq, 0.33 mmol, 237 mg) was dried by evaporation of added dry pyridine (2×), dissolved in dichloromethane containing pyridine [10% (v/v)] (3 mL) and pivaloyl chloride (1.5 eq, 0.5 mmol, 62 μL) was added. When pivaloyl chloride assisted H-phosphonate activation15 was complete (5 min., 31P NMR), the reaction mixture was added to a solution of crude 17 in dichloromethane (ca. 1.5 eq, 0.5 mmol, 160 mg) and reaction was stirred at r.t for 20 min. To the generated H-phosphonate diester a solution of iodine (2 eq, 0.7 mmol, 167 mg) in 0.5 mL pyridine containing water [10% (v/v)] was added. After the reaction was complete (5 min., 31P NMR), solvents were removed by evaporation and the crude product was deprotected by treatment with 80% acetic acid at r.t overnight. The acid was removed by co-evaporation with toluene (2×). The product was dissolved in water and washed with dichloromethane (2×). The water was removed by evaporation under reduced pressure and the deprotected compound 20 was purified by column chromatography using a linear gradient of water (0–30%) in 2-propanol containing Et3N (5% v/v). Yield: 161 mg, 64%. 1H NMR: (400 MHz, CD3OD): δ 8.28 (1H, s), 7.09 (1H, s), 5.97 (1H, d, J = 5.9 Hz), 4.48 (1H, m), 4.37 (1H, bs), 3.95 (1H, m), 3.76 (2H, m), 3.58 (3H, s), 3.84–2.82 (22H, m), 3.10 (6H, q, J = 7.2 Hz), 1.20 (9H, t, J = 7.2 Hz). 13C NMR: (100.6 MHz, CD3OD): δ = 10.1, 38.0, 45.1, 49.9, 57.1, 61.3, 64.3, 69.7, 69.8, 70.1, 70.2, 70.3, 70.1, 70.2, 72.6, 81.7, 85.7, 126.4 139.9, 146.3, 148.2, 151.9, 174.9. 31P NMR (162 MHz, CD3OD): δ P 0.71; MS (ESI) TOF-mass m/z 662.23, calculated for C23H37N9O12P, observed mass: 661.83, [M − H].2′-O-methyl-5′-phosphoadenosine 3′-(17-azido-10-oxo-3,6,12,15-tetraoxa-9-azaheptadecyl)phosphate tris(triethylammonium) salt (21)
Compound 20 (1 eq, 0.1 mmol, 76 mg) and triethylammonium (9H-fluoren-9-yl)methyl -H-phosphonate (1.05 eq, 0.105 mmol, 30 mg weighed as ammonium salt and converted to triethylammonium salt as above) were made anhydrous by repeated evaporation of added pyridine. After this, the residue was dissolved in 1 mL of DCM/pyridine (9:1, v/v) and PvCl (1.5 eq, 0.15 mmol, 19 μL) was added. H-phosphonylation of nucleotide 20 was complete (31P NMR, TLC) after 40 min and then a solution of iodine (2 eq, 0.2 mmol, 51 mg) in 0.2 mL pyridine containing water [10% (v/v)] was added. After 10 min solvents were removed by evaporation and the crude product was dissolved in MeCN/Et3N (2:1, v/v) and left to react overnight at room temperature. After removing solvents by evaporation, the product was dissolved in water and washed with DCM (3×). The water phase was concentrated and compound 21 was purified by preparative RP HPLC using a gradient of buffer B in buffer A (0–35% over 42 min). Concentration of collected fractions gave pure product as tris(triethylammonium)salt. tR: 24 min. 1H NMR (400 MHz, D2O): δ 8.73–8.72 (1H, m), 8.20 (1H, s), 6.16 (1H, d, J = 7.0 Hz), 4.96 (1H, bs), 4.60 (1H, bs), 4.55 (1H, bs), 4.04–3.99 (6H, m), 3.72–3.55 (14H, m), 3.42–3.36 (5H, m), 3.15–3.10 (12H, m), 1.99–1.85 (2H, m), 1.22–1.19 (18H, m). 13C NMR (100 MHz, D2O): δ 7.4 (6C), 37.6, 45.8 (6C), 49.2, 57.2, 63.3, 64.0, 64.1, 67.9, 68.4, 68.6, 68.8, 69.2, 69.3, 69.4, 72.5, 81.3, 83.5, 84.0, 118.3, 139.2, 148.4, 152.1, 154.8, 171.7. 31P NMR (162.0 MHz, D2O): δ −0.38, 1.44; MS (ESI) TOF-mass m/z 742.20, calculated for C23H38N9O15P2, found: 742.03, [M − H].
2′-O-Methyl-5′-O-(4-monomethoxytrityl)uridine 3′-H-phosphonate triethylammonium salt (22)
2′-O-Methyluridine (7.8 mmol, 2 g) was dissolved in dry pyridine (10 mL) and concentrated to dryness and then subjected to evacuation with a vacuum pump and flushed with N2. The residue was dissolved in dry pyridine (70 mL) and monomethoxytrityl chloride was added (1.4 eq, 10.9 mmol, 3.35 g). The reaction was left stirring overnight at r.t. As the completion of reaction was confirmed the next day, by TLC analysis, diphenyl H-phosphonate was added (3 eq, 23.3 mmol, 4.5 mL) and stirring was continued overnight. After confirming with TLC that all monomethoxytritylated uridine was consumed, 5 mL of water and 10 mL of Et3N was added to the reaction which was left to stir overnight. The reaction was quenched after 24 h with sat. aqueous NaHCO3. The mixture was extracted with DCM (3 × 30 mL), dried over MgSO4, filtered and the volatiles removed under reduced pressure. Purification was done with flash chromatography (silica gel, gradient elution dichloromethane (containing 1% Et3N): methanol (0–10% MeOH) yield: 4.77 g, 88%. 1H NMR (400 MHz, CDCl3): δ 7.91–7.90 (1H, bs), 7.38–7.25 (12H, m), 6.98 (1H, dd, J = 8.1 Hz, J = 629.1 Hz), 6.85–6.84 (2H, bs), 6.08–6.06 (1H, m), 5.16–5.15 (1H, bs), 4.91 (1H, bs), 4.30 (1H, bs), 4.05 (1H, bs), 3.80–3.78 (3H, bs), 3.59–3.54 (5H, m), 3.02–2.91 (6H,m), 1.26–1.23 (9H,m). 13C NMR (100 MHz, CDCl3): δ 9.0 (3C), 45.6 (3C), 55.3, 58.4, 61.5, 70.0, 82.0, 83.2, 87.3, 102.1, 113.3 (2C), 127.3, 128.0 (2C), 128.59 (2C), 128.61 (4C), 130.6 (4C), 134.7, 139.9, 143.6, 143.9, 150.3, 158.8, 163.2, 31P NMR (162.0 MHz, CDCl3): δ 3.08 (dd, J = 8.1 Hz, J = 629.1 Hz); MS (ESI) TOF-mass m/z, 593.17, calculated for C30H30N2O9P, found: 593.35, [M − H].2́-O-(N-(2-azidoethyl)carbamoylmethyl-3́-O,N,N-tribenzoyl-adenosine 5′-(2′-O-methyluridine 3′-phosphate) (23)
Compounds 8 (1 eq, 0.05 mmol, 38 mg) and 22 (1.1 eq, 0.055 mmol, 39 mg) were dried by evaporation of added dry pyridine (2×), dissolved in dichloromethane containing pyridine [10% (v/v)] (1 mL). Pivaloyl chloride (1.5 eq, 0.075 mmol, 9.5 μL) was then added. When the formation of H-phosphonate diester was complete (10 min., 31P NMR), a solution of iodine (2 eq, 0.1 mmol, 25 mg) in 0.2 mL pyridine containing water [10% (v/v)] was added. After the reaction was complete (5 min., 31P NMR, TOF MS ES−), solvents were removed by evaporation and the crude product was deprotected by dissolving it in 80% acetic acid (2 mL) at r.t. overnight. The acid was removed by co-evaporation with toluene (2×). The product was purified by column chromatography using a linear gradient of methanol (0–30%) in dichloromethane containing Et3N (3% v/v). Yield: 48 mg, 85%,;1H NMR (400 MHz, CDCl3): δ 9.04 (1H, s), 8.67 (1H, s), 8.13–8.11 (1H, m), 7.98–7.96 (1H, m), 7.84 – 7.83 (4H, m), 7.69–7.63 (4H, m), 6.47 (1H, bs), 5.91–5.89 (1H, m), 5.82–5.81 (1H, m), 5.65 (1H, bs), 4.96 (1H, bs), 4.73 (1H, bs), 4.62 (1H, bs), 4.33 (1H, bs), 4.22 (1H, bs), 3.95–3.93 (3H, m), 3.59–3.54 (3H, m), 3.23–3.19 (4H, m), 2.77–2.74 (6H, m), 1.14–1.05 (11H, m). 13C NMR (100 MHz, CDCl3): δ 10.1 (3C), 38.2, 46.0 (3C), 50.3, 58.2, 60.1, 65.1, 72.7 (2C), 82.6 (2C), 83.2 (2C), 86.6, 88.1, 102.2, 126.7, 128.8 (4C), 129.4 (6C), 129.9 (3C), 133.1 (2C), 133.9, 134.1 (2C), 140.3, 144.0, 150.1 (2C), 151.7, 152.4, 163.5, 165.9, 168.7, 172.3 (2C). 31P NMR (162.0 MHz, CDCl3): δ −0.25, −0.61; MS (ESI) TOF-mass m/z, 1024.26, calculated for C45H43N11O16P, found: 1024.86, [M − H].2′-O-Methyl-5′-O-(4-monomethoxytrityl)N6- butyryladenosine 3′- H-phosphonate triethylammonium salt (24)
2′-O-Methyladenosine (7.1 mmol, 2 g) was dissolved in dry MeCN (10 mL) and concentrated to dryness. The flask was evacuated with a vacuum pump and flushed with N2. The residue was dissolved in dry pyridine (70 mL) and the reaction flask was placed on an ice bath. Trimethylsilyl chloride (4 eq, 28.4 mmol, 3.6 mL) was added with a syringe and after 15 min the bath was removed. The reaction was left to stir overnight. When completion of reaction was confirmed by TLC and MS analysis the following day, n-butyric anhydride (1.5 eq, 10.7 mmol, 1.74 mL) was added directly to the reaction mixture, which was left to stir for 3 h at r.t. After this time TLC analysis showed completion of reaction and the transient protection was removed by adding water (35 mL) and then leaving the reaction mixture over night at room temperature sat. NaHCO3 (aq) was then added and the reaction was extracted with DCM (3×). The organic layers were collected, dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The product (N6-Butyryl-2′-O-methyladenosine) was purified with flash chromatography (silica gel, gradient elution DCM: MeOH (0–10% MeOH)), yield: 1.8 g, 67%. 1H NMR (400 MHz, CD3OD): δ 8.53 (2H, s), 6.10 (1H, d, J = 5.1 Hz), 4.41 (1H, t, J = 4.6 Hz), 4.31 (1H, t, J = 5.1 Hz), 4.05 (1H, dd, H: J = 3.1 Hz, J = 6.8 Hz), 3.80 (1H, dd, H: J = 2.7 Hz, J = 12.4 Hz), 3.68 (1H, dd, H: J = 3.1 Hz, J = 12.4 Hz), 3.37 (3H, s), 2.53 (2H, t, J = 7.4 Hz), 1.73–1.64 (2H, m), 0.94 (3H, t, J = 7.4 Hz). 13C NMR (100 MHz, CD3OD): δ 14.4, 20.0, 40.4, 59.3, 63.1, 71.0, 85.3, 88.2, 89.2, 125.0, 144.7, 151.3, 153.1, 153.5, 174.8; MS (ESI) TOF-mass m/z, 352.16 calculated for: C15H22N5O5, found: 352.60 [M+H] 1.3g (3.7 mmol) of the intermediate product, N6-Butyryl-2′-O-methyladenosine) was dissolved in dry pyridine (10 mL) and concentrated to dryness. After evacuation on a vacuum pump and subsequent flushing with N2, the residue was dissolved in dry pyridine (37 mL) and 4-monomethoxytrityl chloride was added (1.1 eq, 4.7 mmol, 1.25 g). The reaction was left stirring overnight at r.t. When completion of reaction was confirmed the next day (by TLC analysis) diphenyl H-phosphonate was added (3 eq, 8.14 mmol, 1.57 mL) and stirring was continued overnight. After confirming with TLC that all monomethoxytritylated linker was consumed, 5 mL of water and 10 mL of Et3N was added to the reaction which was left to stir over night. The reaction was quenched after 24 h with sat. NaHCO3 (aq). The mixture was extracted with DCM (3 × 30 mL) and the combined organic phases were dried over MgSO4. The mixture was filtered and the volatiles removed under reduced pressure. Product 24 was purified with flash chromatography (silica gel, gradient elution dichloromethane (containing 1% Et3N)): methanol (0–5% MeOH) yield: 2.53 g, 87%. 1H NMR (400 MHz, CDCl3): δ 8.63–8.61 (2H, m), 7.44–7.24 (12H, m), 7.00 (1H, dd, J = 9.4 Hz, J = 626.7 Hz), 6.81–6.79 (2H, m), 6.25–6.23 (1H, bs), 5.05–5.01 (1H, bs), 4.63–4.62 (1H, bs), 4.50 (1H, bs), 3.79 (3H, d, J = 2.9 Hz), 3.55–3.48 (5H, m), 2.97–2.95 (6H, m), 2.85 (2H, t, J = 0.8 Hz), 1.88–1.77 (2H, m), 1.29–1.25 (9H, m), 1.07–1.03 (3H, m). 13C NMR (100 MHz, CDCl3): δ 8.1 (3C), 12.8, 17.4, 38.8, 44.7 (3C), 54.3, 57.7, 62.3, 70.4, 81.6, 83.1, 85.4, 86.1, 112.3, 121.2, 126.2 (2C), 126.6 (2C), 127.0 (4C), 127.6 (4C), 134.1, 143.0 (2C), 146.3, 148.1, 150.3, 151.6, 157.7, 172.0. 31P NMR (161.9 MHz, CDCl3): δ 3.08 (dd, J = 9.4 Hz, J = 626.7 Hz); MS (ESI) TOF-mass m/z, 686.24 calculated for: C35H39N5O7P, found: 686.60 [M − H].:1, v/v) and PvCl (1.5 eq, 0.18 mmol, 23 μL) was added. After 40 min. a solution of iodine (2 eq, 0.24 mmol, 61 mg) in 0.5 mL pyridine containing water [10% (v/v)] was added to the reaction mixture. After 5 min. solvents were removed by evaporation and the crude product was dissolved in MeCN/Et3N (2:1, v/v) and left to react overnight at room temperature. After removing solvents, the crude product 26 was dissolved in concentrated aqueous ammonia (3 mL) and the reaction was stirred overnight. Since TLC analysis showed full deprotection, solvents were evaporated and the product was dissolved in water and washed with dichloromethane (3×). The water phase was concentrated under reduced pressure and compound 5 was purified by RP HPLC using a gradient of buffer B in buffer A (tR = 23.8 min, 0–30 min 35% B). Concentration of collected fractions gave 5 as the tris(triethylammonium)salt. MS (ESI) TOF-mass m/z, 1136.22 calculated for: C35H47N16O22P3, found 1136.14, [M].
General method for m3G-capping of 5′-phosphates 10, 14, 21 and 5
The clickable m3G-CAP constructs 1, 2, 3 and 4 were synthesized by adaptation of the method described for capping of ONs.2,3 In summary N2,N2,N7-trimethylguanosine 5′-pyrophosphate imidazolide was added in excess (10–15 eq) to the respective 5′-phosphate derivatives (10, 14, 21 or 5), dissolved in N-methylmorpholine buffer (pH 7) containing equimolar amount of MnCl2 (in an amount equimolar to the trimethylguanosine pyrophosphate imidazolide) (200 μL). The reaction was left at 30 °C for 5–7 days and was monitored using RP HPLC. After no more imidazolide was detected (up to 50% conversion into the conjugate) the products were isolated using RP-HPLC with buffers A and B (see supplementary).Analytical data:
m3G-CAP-construct 1: TOF-mass m/z, [M]−1m/z calculated: 939.17, found: 938.82; tR = 27.1 min, (0–30 min 24% B)
m3G-CAP-construct 2: TOF-mass m/z, [M]−1m/z calculated: 953.19, found: 953.56; tR = 19.8 min, (0–30 min 35% B)
m3G-CAP-construct 3: TOF-mass m/z, [M]−1m/z calculated: 1210.27, found: 1210.68; tR = 19.2 min, (0–25 min 50% B)
m3G-CAP-construct 4: TOF-mass m/z, [M]−1m/z calculated: 1602,28 found: 1601.69; tR = 23.5 min, (0–30 min 35% B)
Synthesis of ONs containing the activated triple bond donor PATA linker.
The desired ON sequence containing the linker equipped with the activated p-(N-propynoylamino)toluic acid (PATA) triple bond donor was synthesized on solid support as previously described.8 The supported 5′-alkyne-ON was then placed in an Eppendorf tube with a screw cap and 1 mL of sat. methanolic ammonia was added to deprotect and to remove the ON from the support at r.t. overnight. For reactions in which a purified sequence was used, the purification was performed by RP-HPLC using a linear gradient of buffer B in A from 0 to 40% B in 40 min, detection at 260 nm. tR = 34.2 min (se supporting information for HPLC chromatogram); MS (ESI) TOF-mass m/z, calculated (M) 6175, found 6173.Conjugation of the “clickable” 2′-O-methylated trinucleotide pAUA (5) to 5′-alkyne-ONs.
34 nmol of crude, freeze-dried 5′-alkyne-ON were dissolved in an aqueous solution containing 3.35 eq. of the trinucleotide 5 (114 μl from a stock solution of 1 μmol mL−1) and transferred to a glass vial with a screw cap. Then 1 eq. of CuI in 5 μL of DMSO (from a stock solution of 1.2 mg mL−1 in DMSO) was added to the mixture followed by 8 eq. of diisopropyl ethyl amine (5 μL from a stock solution of 10 μL in 1 mL of H2O) and sodium ascorbate (3 eq, 5 μL from a stock solution of 4 mg mL−1 in H2O). The solution was gently agitated on a vortex at r.t overnight. Purification was done by RP-HPLC using a linear gradient of buffer B in A from 0 to 80% B in 25 min, detection at 260 nm. tR = 14.3 min; MS (ESI) TOF-mass m/z,, calculated (M) 7315, found 7350 (M+K).Conjugation of the “clickable” m3G-CAP constructs (1–4) to the 5′-alkyne-ON.
18mer-m3G-CAP 1. Conjugation between ON and m3G-CAP 1 was performed mixing 13.6 nmol of the HPLC purified 5′-alkyne-18merRNA(2′-OMe) dissolved in 50 μL of H2O and 0.5 eq of m3G-CAP 1 (in 20 μL of H2O). Then 1 eq of CuI in 5 μL of DMSO (from a stock solution of 0.52 mg mL−1) was added to the solution followed by 8 eq.of DIPEA (5 μL from a stock solution of 19 μL in 5 mL of H2O) and sodium ascorbate (3 eq., 5 μL from a stock solution of 1.6 mg mL−1 in H2O). The solution was gently agitated on a vortex at r.t overnight. Purification was done by RP-HPLC using a linear gradient of buffer B in A from 0 to 29% B in 45 min, detection at 260 nm. tR = 43.7 min;18mer-m3G-CAP 2–4. A crude solution of 5′-alkyne-18merRNA(2′-OMe) in H2O (26 nmol for reaction with cap 2 and 13 nmol for reaction with cap 3 and 4), was mixed with the desired m3G-CAP 2–4 (1.9, 2.8 and 1.25 eq respectively). Then 1 eq of CuI in 10 μL of DMSO was added to the solution followed by 8 eq of DIPEA to a final volume solution of 100 μL. An aliquot of each reaction was checked by HPLC and MS after 2 h and 1 extra eq of CuI was added to complete conversion. The solution was gently agitated on a vortex at r.t overnight. Purification was done by RP-HPLC for conjugates with caps 2 (tR = 28.6 min using a linear gradient from 0 to 55% B in 45 min with a flow of 4 mL min−1) and 3 (tR = 31.3 min using a linear gradient from 0 to 40% B in 40 min with a flow of 1 mL min−1) and with IE-HPLC for conjugates with cap 4 (tR = 24.5 min using a linear gradient of buffer C in D from 0 to 40% D in 40 min).
18mer-m3G-CAP1: calculated (M) 7120, (MAXENT) found 7124 (M).
18mer-m3G-CAP2: calculated (M) 7134, (MAXENT) found 7138 (M),
18mer-m3G-CAP3: calculated (M) 7405, (MAXENT) found 7404 (M),
18mer-m3G-CAP4: calculated (M) 7783, (MAXENT) found 7784 (M).
Results and discussion
Four different “clickable” m3G-CAP constructs (1–4, Fig. 1), all of them equipped with a handle (an azide group) for attachment to a cargo molecule, were designed and synthesized. We started from the most simplified m3G-CAP structure, which possesses a 2,2,7-trimethyl-guanosine connected via triphosphate bridge to an adenosine residue. The functional azide linker protrudes from the 2′-position of the adenosine nucleoside (structure 1, Fig. 1). Since the 2′-O-methyl position may have additional effect on the binding process with Snurportin1 m3G-CAP construct 2 was equipped with a functional linker at the 3′-position while the 2′-position is O-methylated, and thus a step closer to the native m3G-CAP (structure 2, Fig. 1). The m3G-CAP construct 3 is equipped with both a 2′-O-methylated adenosine residue and a 3′-phosphate as a part of a longer linker attachment. Finally the m3G-CAP construct 4 contains a 2,2,7-trimethylguanosine connected via triphosphate bridge to an AUA sequence, where the 5′-end A and the U are 2′-O-methylated (structure 4, Fig. 1). The 5′-phosphorylated trimer (structure 5, Fig. 1) can also be attached to cargos and used as a non-capped control of conjugates with 4 in further studies of nuclear transport.The synthesis of an azido containing adenosine building block was performed by alkylation of 5′-monomethoxytrityl protected adenosine followed by aminolysis. This two step one-pot procedure (potassium t-butoxide and allyl bromoacetate, followed by addition of 2-azidoethylamine) gives the required 2′-substituted product in yields of up to 65% of the isolated product after two steps.10 Using this method compound 7 (Scheme 1) was synthesized. Benzoylation and subsequent detritylation gave the tribenzoyl derivative 8, which was further used as building block for the synthesis of m3G-CAP constructs 1 and 4. For the synthesis of m3G-CAP construct 1 compound 8 is 5′-phosphorylated by means of robust tri-step one-pot phosphorylation method using (9H-fluoren-9-yl)methyl H-phosphonate14 with pivaloyl chloride, oxidation by iodine/pyridine/water and finally elimination of the fluorenylmethyl group with triethylamine. This resulted in the 5′-phosphorylated 2′-azidoethylcarbamoylmethyladenosine 9 which was then deprotected with methanolic ammonia to give compound 10 that was converted to the m3G-CAP construct 1 by reaction with m3G 5′-pyrophosphorylimidazolide.
 | ||
| Scheme 1 Synthesis of m3G-CAP constructs 1 (scheme A) and 2 (scheme B). i: THF, t-BuOK, 20 min, allyl bromoacetate r.t., 6 h, r.t.; ii: dry MeOH, 2-azidoethylamine, 24 h, r.t. iii: benzoyl chloride, pyridine, overnight, r.t.; iv: 80% acetic acid, r.t., 2 h; v: (9H-fluoren-9-yl)methyl H-phosphonate, DCM:Py 9:1, PvCl, r.t., 30 min; vi: I2, water, r.t., 10 min; vii: MeCN:Et3N 2:1, overnight, r.t.; viii: MeOH/NH3 sat., r.t. 50 h; ix: m3GppIm, MnCl2, N-methylmorpholine x HCl pH 7, 30 °C, 7 days; (Abbreviations: MeCN = acetonitrile, PvCl = pivaloyl chloride, Py = Pyridine, t-BuOK = potassium tertbutoxide, MeOH = methanol, DCM = dichloromethane, THF = tetrahydrofuran, NMM = N-methylmorpholine, m3GppIm = N2,N2,N7-trimethylguanosine 5′-pyrophosphorylimidazolide). | ||
Synthesis of m3G-construct 2 was based on 5′-Monomethoxytritylated 2′-O-methylated adenosine which was first alkylated using allyl bromoacetate in THF, and the formed allyl ester was then treated in situ with 2-azidoethylamine. Monomethoxytrityl removal gives compound 13 which was then 5′-phosphorylated to 14 and m3G-Capped (as described above for the 2-linked construct) to give 2.
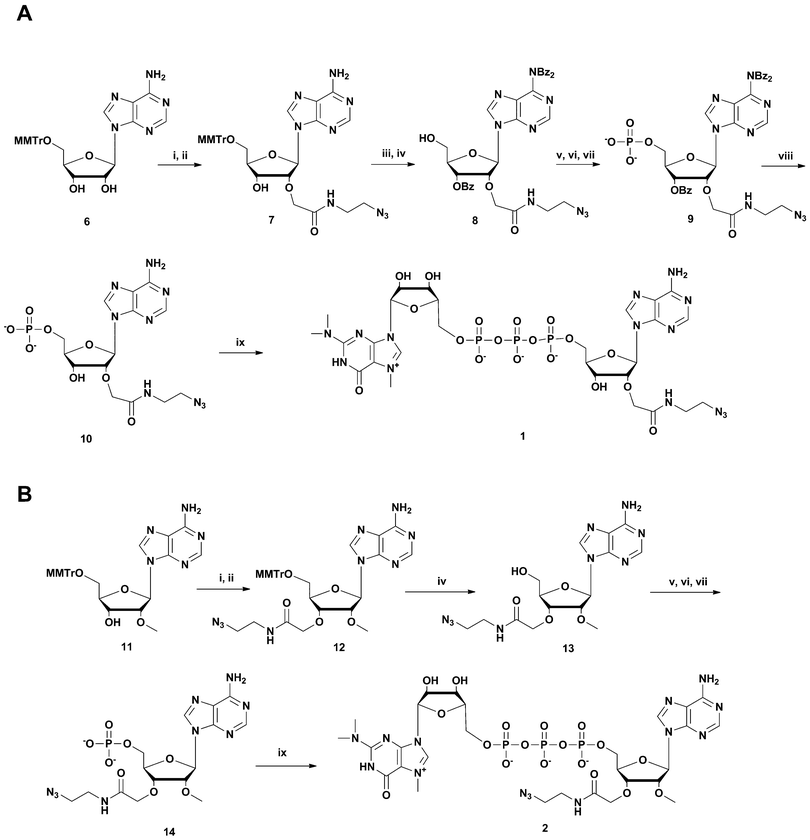
An alternative strategy was used for the synthesis of m3G-CAP construct 3 (Scheme 2). Aided by pivaloyl chloride 5′-O-MMTr-2′-O-methyladenosine 3′-H-phosphonate 18 was condensed with azidoalcohol 17 (made from (2(2-azidoethoxy)ethoxyacetic acid (16) activation with HBTU, and NMM for 30 min and subsequent addition of 2-(2-aminoethoxy) ethoxyethanol (15) Scheme 2, A) and then oxidized to 19. Subsequent detritylation and phosphorylation (as above) resulted in the azidolinked 2′-O-methyladenosine derivative 21, which was then capped with m3G pyrophosphorylimidazolide to give construct 3.
 | ||
| Scheme 2 Synthesis of m3G-CAP construct 3. A: i: HBTU, NMM, DCM, 2 h; B: i: DCM:Py 9:1, PvCl, 5 min; ii: compound 17, 20 min; iii: I2, water, r.t., 5 min; iv: 80% acetic acid, r.t., overnight; v: (9H-fluoren-9-yl)methyl H-phosphonate, DCM:Py 9:1, PvCl, r.t., 40 min; vi: I2, water, r.t., 10 min; vii: MeCN:Et3N 2:1, overnight, r.t.; viii: m3GppIm, MnCl2, NMM x HCl pH 7, 30 °C, 7 days; (Abbreviations: MeCN = acetonitrile, PvCl = pivaloyl chloride, Py = Pyridine, DCM = dichloromethane, HBTU = O-Benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate, NMM = N-methylmorpholine, DMF = dimethylformamide, m3GppIm = N2,N2,N7-trimethylguanosine 5′-pyrophosphorylimidazolide). | ||
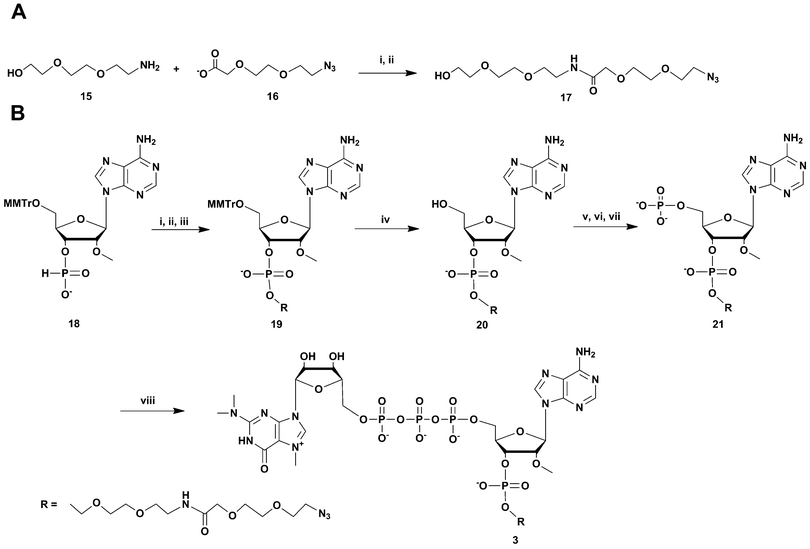
For the synthesis of the most UsnRNA-like clickable m3G-CAP construct 4 we employed the azido-linked adenosine 8 (Scheme 3) which was extended into a trimer using H-phosphonate chemistry16–18 and 5′-monomethoxytritylated H-phosphonates of 2′-O-methyluridine and N-nBu protected 2′-O-methyladenosine (compounds 22 and 24 respectively, Scheme 3). The monomers were synthesized in a one-pot procedure using diphenyl H-phosphonate19,20 as a phosphonylating reagent after 5′-monomethoxytritylation.
 | ||
| Scheme 3 Synthesis of m3G-CAP construct 4. i: DCM:Py 9:1, PvCl, compound 22, 10 min; ii: I2, water, r.t., 5 min; iii: 80% acetic acid, r.t., overnight; iv: DCM:Py 9:1, PvCl, compound 24, 30 min; v: I2, water, r.t., 10 min; vi: 80% acetic acid, r.t., overnight; vii: (9H-fluoren-9-yl)methyl H-phosphonate, DCM:Py 9:1, PvCl, r.t., 40 min; viii: I2, water, r.t., 10 min; ix: MeCN:Et3N 2:1, overnight, r.t.; x: NH3 aq. sat., r.t., overnight; xi: m3GppIm, MnCl2, NMM x HCl pH 7, 30 °C, 7 days; (Abbreviations: MeCN = acetonitrile, PvCl = pivaloyl chloride, Py = Pyridine, DCM = dichloromethane, NMM = N-methyl morpholine, m3GppIm = N2,N2,N7-trimethylguanosine 5′-pyrophosphorylimidazolide). | ||
H-Phosphonate 22 was coupled to 8 with pivaloyl chloride and the resulting dinucleoside H-phosphonate was subsequently oxidized with iodine-water. Removal of the 5′-monomethoxytrityl protection resulted in dinucleotide 23, which was further coupled with N6-butyryl-2′-O-methyladenosine H-phosphonate 24. This gave after 5′-monomethoxytrityl removal the trinucleotide 25. This clickable trinucleotide was further 5′-phosphorylated analogue using (9H-fluoren-9-yl)methyl H-phosphonate based phosphorylation (see above), which after ammonia deprotection gave the clickable 5′-phosphorylated trinucleotide 5 which was then purified by RP-HPLC. Treatment of 5 with N2,N2,N7-trimethylguanosine 5′-pyrophosphorylimidazolide in the presence of Mn2+ resulted in the clickable m3GpppA(OMe)U(OMe)A(linker) construct 4.
Having successfully synthesized the different clickable m3G-CAP nuclear localisation signals these could be generally conjugated to different molecules by use of “click chemistry”. One of the most immediate uses of the clickable m3G-CAPs is to attach these to therapeutic ONs that have their main site of action in the nucleus of cells, e.g. splice-switching ONs.21,22 From our experience in conjugating oligonucleotides to peptides by click chemistry, it seems that low concentration of the reacting biomolecules requires more active functional groups to achieve sufficient conversion. This can be achieved with an electron-withdrawing carbonyl group adjacent to the triple bond.8 The advantage of having an activated triple bond donor as a linker appeared even more important when developing a methodology for clicking of the m3G-CAPs as cap structures can, at least partially, decompose in the presence of copper ions,23,24 if conditions are too harsch.
Thus a method for attachment of m3G-CAPs 1–4 and 5 to ONs carrying a (N-Propynoylamino)-p-toluic acid (PATA) based alkyne linker was developed. The different “clickable” CAP-derivatives 1–4 were attached to a 2′-O-methylated 18-mer ON. The ON sequence has been used for splice correction in the HeLa-Luc 705 reporter cell line which contains a luciferase reporter construct that has the β-globin 705 intron inserted into the luciferase ORF.25 The following steps are subsequently performed to prepare alkyne-ONs (Fig. 2): a) commercially purchased 2′-O-methyl modified ON is 5′-detritylated using 2% DCA in DCM, b) the 5′-aminolinker8 is attached using H-phosphonate c) The 5′-amino linker on the ON is detritylated using 2% DCA in DCM, d) PATA is activated using HBTU and NMM in DMF and added to the ON on solid support, e) finally the ON with the linked triple bond donor is deprotected and cleaved from support using methanolic ammonia at r.t. during 16 h (overnight).
![Schematic representation of stepwise synthesis of ONs containing m3G-CAP analogues. i: 2-(N-(4-monomethoxytrityl)aminoethoxy)ethyl H-phosphonate, Py, PvCl, 20 min, r.t.; ii: I2, water, Py; iii: 2% DCA, DCM, 2 min; iv: pre-activation of (N-Propynoylamino)-p-toluic acid (PATA), HBTU, NMM, DMF, 0.5 h, r.t., v: addition of activated PATA to solid supported ON, 2 h; vi: MeOH/NH3 sat., 18 h, r.t., vii: CuI, DIPEA, overnight, water/DMSO 9/1 (dipolar cycloaddition [3+2] of ON-PATA with m3G-CAP); (Abbreviations: PvCl = pivaloyl chloride, Py = Pyridine, DCA = dichloroacetic acid, DIPEA = diisopropylethylamine, DMSO = dimethylsulfoxide, DCM = dichloromethane, MeOH = methanol, HBTU = N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate, NMM = N-methylmorpholine, DMF = dimethylformamide).](/image/article/2012/RA/c2ra22345g/c2ra22345g-f2.gif) | ||
| Fig. 2 Schematic representation of stepwise synthesis of ONs containing m3G-CAP analogues. i: 2-(N-(4-monomethoxytrityl)aminoethoxy)ethyl H-phosphonate, Py, PvCl, 20 min, r.t.; ii: I2, water, Py; iii: 2% DCA, DCM, 2 min; iv: pre-activation of (N-Propynoylamino)-p-toluic acid (PATA), HBTU, NMM, DMF, 0.5 h, r.t., v: addition of activated PATA to solid supported ON, 2 h; vi: MeOH/NH3 sat., 18 h, r.t., vii: CuI, DIPEA, overnight, water/DMSO 9/1 (dipolar cycloaddition [3+2] of ON-PATA with m3G-CAP); (Abbreviations: PvCl = pivaloyl chloride, Py = Pyridine, DCA = dichloroacetic acid, DIPEA = diisopropylethylamine, DMSO = dimethylsulfoxide, DCM = dichloromethane, MeOH = methanol, HBTU = N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate, NMM = N-methylmorpholine, DMF = dimethylformamide). | ||
This sequence of reaction provides 5′-alkyne linked ONs that then can be reacted in its crude form with the azide functionalized m3G-CAP constructs. It is important to note some specific observations. Since the m3G-CAP is susceptible to degradation under basic conditions (unpublished results), necessary for standard ON deprotection and cleavage from the support, we decided to carry out the final conjugation in solution. Deprotection of ONs carrying the activated triple bond donor PATA with concentrated aqueous ammonia at 50 °C leads to complete degradation of the triple bond (most likely due to Michael addition to the conjugated triple bond which was also supported by MS-analysis). Even mild deprotection using methanolic ammonia at room temperature has to be carefully monitored but could be successfully employed to achieve the deprotected 5′-PATA linked oligonucleotide. In addition, purification of the PATA-functionalized ON by RP HPLC with triethylammonium acetate buffer gave partial degradation of the triple bond linker, which led us to develop the alternative strategy of carrying out the conjugation on the crude alkyne-linked ON and only purify the final m3G-capped ON by HPLC. The methodology is most conveniently carried out with excess of the m3G-CAP construct since the product after m3G-CAP conjugation and the ON substrate had quite similar retention times. Thus, for the facilitation of the purification we wanted the alkyne-linked ON to be as fully consumed as possible and most of the ON was indeed converted (see Fig. 3 and supporting information for HPLC analysis). Thus, conjugation was efficient and the corresponding m3G-Cap derivatives, 18merRNA-CAP 1–4 as well as 18merRNA-AUA (via attachment of trinucleotide 5) were readily isolated.
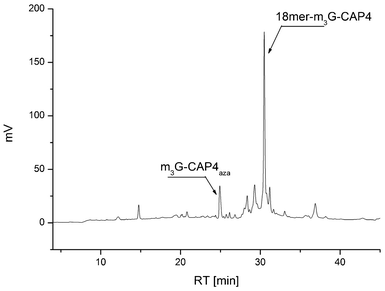 | ||
| Fig. 3 HPLC analysis of the crude product after click conjugation of clickable m3G-Cap 4 to 5′alkyne 18-merRNA to obtain the most complex construct, the 18merRNA-CAP 4. | ||
In conclusion, we report the synthesis of four different m3G-CAP constructs (1–4, Fig. 1) and their efficient conjugation to ONs using Cu (I) [3 + 2] catalysed cycloaddition to produce the corresponding ON conjugates (Fig. 2). This is enabled by azide functionalities of the m3G-CAP constructs, and conjugation in solution with the activated triple bond linker on the crude deprotected ON. The constructs are four different variants in order to be able to find out requirements for Snurportin1 recognition and nuclear transport of cargos with m3G-CAP signals, starting from the most simplified structure 18merRNA-CAP1 towards the more complex structure 18merRNA-CAP 4 that resembles the naturally transported UsnRNPs. The methodology also should allow for synthesis of clickable m3G-CAPs with triphosphate bridges modified for resistance towards enzymatic degradation and the conjugation of these to different cargos. Studies of the ability of the synthesized artificially linked m3G-CAP-ONs in promoting nuclear uptake is currently in progress and will be reported in the near future.
Acknowledgements
This work was supported by grants from The Swedish Science Research Council, Magnus Bergvalls Foundation, The Torsten and Ragnar Söderberg Foundation, the Duchenne Parent Project, The Swedish Foundation for Strategic Research, VINNOVA and the European Council (FP7:ITN-2008-238679).References
- S. Kitao, A. Segref, J. Kast, M. Wilm, I. W.Mattaj and M. Ohno, A compartmentalized phosphorylation/dephosphorylation system that regulates U snRNA export from the nucleus, Mol. Cell. Biol., 2008, 28, 487–497 CrossRef CAS.
- P. Moreno, M. Wenska, K. E. Ö. L. Wrange, R. Strömberg and E. Smith, A synthetic snRNA m3G-CAP enhances nuclear delivery of exogenous proteins and nucleic acid, Nucl. Acids Res., 2009, 1925–35 Search PubMed.
- H. Sawai, H. Wakai and A. Nakamura-Ozaki, Synthesis and reactions of nucleoside 5′-diphosphate imidazolide. A nonenzymatic capping agent for 5′-monophosphorylated oligoribonucleotides in aqueous solution, J. Org. Chem., 1999, 64, 5836–5840 CrossRef CAS.
- A. Strasser, A. Dickmanns, R. Lührmann and R. Ficner, Structural basis for m3G-cap-mediated nuclear import of spliceosomal UsnRNPs by Snurportin1, EMBO J., 2005, 6(24), 2235–2243 CrossRef.
- J. Huber, U. Cronshagen, M. Kadokura, C. Marshallsay, T. Wada, M. Sekine and R. Lührmann, Snurportin1, an m3G-cap-specific nuclear import receptor with a novel domain structure, EMBO J., 1998, 15, 4114–4126 CrossRef.
- R. Manetsch, A. Krasinski, Z. Radic, J. Raushel, P. Taylor, K. B. Sharpless and H.C. Kolb In situ click chemistry, enzyme inhibitors made to their own specifications, J. Am. Chem. Soc., 2004, 126, 12809–12818 CrossRef CAS.
- A. E. Speers, G. C. Adam and F. B. Cravatt, Activity-based protein profiling in vivo using a copper (I)-catalyzed azide-alkyne [3 + 2] cycloaddition, J. Am. Chem. Soc., 2003, 125, 4686–4687 CrossRef CAS.
- M. Wenska, M. Alvira, P. Steunenberg, M. Murtola and R. Strömberg, An activated triple bond linker enables ‘click’ attachment of peptides to oligonucleotides on solid support, Nucleic Acids Res., 2011, 39(20), 9047–9059 CrossRef CAS.
- J. D. Puglisi and I. Tinoco Jr., Methods Enzymol., 1989, 180, 304–25 CrossRef CAS.
- S. Milton, R. Stromberg and M. Wenska, One pot preparation of adenosine building blocks for click chemistry, Protoc. Exch., 2010 DOI:10.1038/nprot.2010.93.
- G. Gellerman, A. Elgavi, Y. Salitra and M. Kramer, Facile synthesis of orthogonally protected amino acid building blocks for combinatorial N-backbone cyclic peptide chemistry, J. Pept. Res., 2001, 57, 277–291 CrossRef CAS.
- T. Mayer and M. E. Maier, Design and synthesis of a tag-free chemical probe for photoaffinity labeling, Eur. J. Org. Chem., 2007, 4711–4720 CrossRef.
- P. Steunenberg and M. Wenska, Conversion of commercial peptides into “clickable” derivatives for labeling and conjugate synthesis, Protoc. Exch., 2010 DOI:10.1038/nprot.2010.94.
- J. Romanowska, A. Szymanska-Michalak, M. Pietkiewicz, M. Sobkowski, J. Boryski, J. Stawinski and A. Kraszewski, A new, efficient entry to non-lipophilic H-phosphonate mono-esters - preparation of anti-HIV nucleotide analogs, Lett. Org. Chem., 2009, 6, 496–499 CrossRef CAS.
- P. J. Garegg, T. Regberg, J. Stawinski and R. Strömberg, Nucleosides H-phosphonates. V. The mechanism of hydrogenphosphonate diester formation using acyl chlorides as coupling agents in oligonucleotide synthesis by the hydrogenphosphonate approach, Nucleosides, Nucleotides Nucleic Acids, 1987, 6, 655 CAS.
- P.J. Garegg, T. Regberg, J. Stawinski and R. Strömberg, Formation of inter-nucleotidic bonds via phosphonate intermediates, Chemica Scripta, 1985, 25, 280–282 CAS Garegg, P.J., Regberg, T., Stawinski, J. and Strömberg, R.1986.
- S. Sigurdsson and R. Strömberg, The H-phosphonate approach to oligonucleotide synthesis. An investigation on the mechanism of the coupling step, J. Chem. Soc., Perkin Trans., 2002, 2, 1682–1688 Search PubMed.
- T. Regberg, J. Stawinski and R. Strömberg, Nucleoside H-phosphonates. IX. Possible side-reactions during hydrogen phosphonate diester formation, Nucleosides Nucleotides, 1988, 7, 23–35 CAS.
- J. Stawinski and M. Thelin, Nucleoside H-phosphonates. XI. A cconvenient method for the preparation of Nucleoside H-phosphonates, Nucleosides Nucleotides, 1990, 9, 129–135 CAS.
- J. Stawinski and R. Strömberg, Deoxyribo- and Ribonucleoside H-phosphonates. Current Protocols in Nucleic Acid Chemistry 2.6.1-2.6.15, John Wiley & Sons, Inc., New York, 2010, pp 2001 Search PubMed.
- G. van Ommen-J., J. van Deutekom and A. Aartsma-Rus, The therapeutic potential of antisense-mediated exon skipping, Current Opinion in Molecular Therapeutics, 2008, 10, 140–149 Search PubMed.
- J. Bauman, N. Jearawiriyapaisarn and R. Kole, Therapeutic potential of splice-switching oligonucleotides, Oligonucleotides, 2009, 19, 1–13 CrossRef CAS.
- Z. Wieczorek, E. Darzynkiewicz, S. Kuusela and H. Lonnberg, The Cu2+-promoted cleavage of mRNA 5′-cap analogs: a kinetic study with P1-(7-methylguanosin-5′-yl) P3-(nucleosid-5′-yl) triphosphates and P1-(7-methylguanosin-5′-yl) P4-(guanosin-5′-yl) tetraphosphate, Nucleosides Nucleotides, 1999, 18, 11–21 CAS.
- E. Darzynkiewicz, J. Stepinski, S. M. Tahara, R. Stolarski, I. Ekiel, D. Haber, K. Neuvonen, P. Lehikoinen, I. Labadi and H. Lonnberg, Synthesis, conformation and hydrolytic stability of P1, P3-dinucleoside triphosphates related to mRNA 5′-cap, and comparative kinetic studies on their nucleoside and nucleoside monophosphate analogs, Nucleosides Nucleotides, 1990, 9, 599–618 CAS.
- S. Kang, R. Cho and R. Kole, Up-regulation of luciferase gene expression with antisense oligonucleotides: Implications and applications in functional assay development, Biochemistry, 1998, 37, 6235–6239 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: mass spectra of m3G-CAP constructs (1-4, and trimer 5) and their conjugates with 18-mer 2’-O-Methylated ON. HPLC analysis of the capping reaction that produces the clickable m3G-CAP constructs. HPLC profile of crude PATA-ON. Additional HPLC chromatograms of crude products after conjugation of the m3G-CAP constructs to the 18-mer RNA. Mass spectroscopy of PATA-ON conjugate, mass spectra of compounds 20 and 21. See DOI: 10.1039/c2ra22345g |
| This journal is © The Royal Society of Chemistry 2012 |