Fine-tuning the specificity of boronate affinity monoliths toward glycoproteins through pH manipulation
Yue Lu, Zijun Bie, Yunchun Liu and Zhen Liu*
State Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China. E-mail: zhenliu@nju.edu.cn; Fax: +86-25-8368-5639; Tel: +86-25-8368-5639
First published on 2nd October 2012
Abstract
Boronate affinity functionalized materials have recently drawn increasing attention due to their capability to selectively isolate and enrich glycoproteins and glycopeptides. As cheaper and more stable competitors to lectins, boronic acids are generally believed to yield a relatively wider spectrum specificity to glycoproteins. For better understanding and effective utilization of boronate affinity, it is necessary to establish if boronic acids exhibit lectin-like narrow specificity towards individual or a sub-class of glycoproteins. Here we report a pH manipulation strategy for fine-tuning the specificity of boronate affinity monoliths towards two sub-classes of glycoproteins, sialylated and nonsialylated glycoproteins. When the binding pH > the pKa of the boronic acid by one pH unit or more, the boronate affinity monolith preferentially binds to glycoproteins containing neutral sugars and excludes sialic acid containing glycoproteins due to electrostatic repulsion. When the binding pH < the pKa by one pH unit or more, the boronate affinity monolith binds to sialylated glycoproteins due to the exceptional binding affinity of the boronic acid towards sialic acid residues. The alternative specificity towards sialic acid and neutral sugar was first verified using an off-line combination of boronate affinity extraction with nano-ESI-Orbitrap MS/MS detection. The alternative specificity towards sialylated and nonsialylated glycoproteins was then demonstrated by means of off-line combination of boronate affinity extraction with MALDI-TOF MS. Finally, the developed approach was applied to the alternative extraction of intact sialylated and nonsialylated glycoproteins spiked in human serum.
Introduction
Protein glycosylation is a widely existing essential post-translational modification. In mammalian systems, more than 50% of the total proteins are glycosylated proteins. Glycoproteins play key roles in many biological processes, such as molecular recognition, inter- and intra-cell signaling, immune response, sperm–egg interaction, and regulation of development. The occurrence of many diseases is accompanied by glycosylation of related proteins. A variety of glycoproteins have been used as disease biomarkers for clinical diagnosis. For example, α-fetoprotein (AFP) and carcinoembryonic antigen (CEA) have been used in pre-clinical research and clinical diagnosis. Also, many other glycoproteins have been suggested as potential biomarkers for cancers,1 Alzheimer's disease,2 rheumatoid arthritis3 and chronic obstructive pulmonary disease.4 Due to the crucial role of glycoproteins in fundamental research and clinical applications, glycoproteomics has become an important frontier of proteomics studies.The first step of glycoproteomic analysis is the isolation of glycosylated proteins from the proteome. Glycoproteins can be captured in a variety of ways, such as solid-phase hydrazide capture,5 chemoenzymatic approach,6 hydrophilic interaction chromatography (HILIC),7–9 and lectin affinity chromatography10–13, which is one of the most popular techniques for the isolation of glycoproteins. Recently, boronate affinity chromatography (BAC)14–32 is becoming noteworthy for it serves as an effective extraction platform in the field of glycoproteomics. Particularly, boronate affinity functionalized materials (predominantly in microbead,14,21,22,26 nanoparticle16,19,23–25,27–29,32 and monolith17,18,31 format) are generally used for the extraction of glycoproteins and glycopeptides, and a powerful platform was built through their combination with MALDI-TOF MS.14,19–21,24,25,30 BAC appeared more than two decades ago.33 It is a useful tool for the isolation of carbohydrates, RNA, nucleosides, glycoproteins and glycopeptides. The principle relies on the chemistry through which boronic acids form covalent bonds with cis-diols to generate five or six-membered cyclic esters in a basic aqueous media when the environmental pH is greater than or equal to the pKa value of the boronic acids. The esters reversibly dissociate when the environmental pH is much lower than the pKa value. BAC has two advantages over lectin affinity chromatography. First, boronic acids are much cheaper and more stable. Additionally, the acidic desorbing solutions are compatible with MS detection. A unique characteristic of boronic acids compared with other ligands is their broad-spectrum affinity. In theory, boronic acids can bind to all kinds of glycoproteins, while different lectins exhibit different affinities to glycoproteins with different glycan structures.14 Up until now, most BAC-MALDI-TOF MS methods focused on the enrichment of glycopeptide and ignored the enrichment of intact glycoproteins and therefore it remains an open question whether boronic acids exhibit lectin-like affinity towards individual glycoproteins or a sub-class of glycoproteins.
A systematic investigation in a previous paper34 revealed that multiple intermolecular interactions can occur between analytes and boronate affinity monoliths under certain conditions, including boronate affinity, reverse-phase, cation-exchange and hydrogen bonding interactions. Even the steric hindrance of the porous monolith could have a great effect on the selectivity of boronate affinity monoliths.35 Such knowledge is helpful in choosing appropriate experimental conditions and designing new boronate affinity functionalized materials with desired properties and functions.17,27,35–39 Recently, we found that the affinity of boronate affinity monoliths to glycoproteins is strongly dependent on both the experimental conditions and the characteristics of the target glycoproteins.37 For example, the extraction of α1-acid glycoprotein requires a large abundance of salt in the sample solution to suppress electrostatic repulsion between the negatively charged protein and boronic acid. The finding suggests that boronic acids may exhibit discrimination effects for different glycoproteins.
As a sub-type of glycosylation, sialylation attaches terminal sialic acids to proteins or lipids. Sialic acids are a group of N- or O-substituted derivatives of neuraminic acid, though sialic acid is also used as the name for the most common member of this group, N-acetylneuraminic acid (Neu5Ac). Sialylation plays critical roles in the bioavailability, stability, metabolism, and immunogenicity of glycoproteins.40 Aberrant sialylation has been found to be correlated with diseases such as cancer metastatic progression41 and IgA nephropathy.42 Therefore, specific extraction of sialoglycoproteins is important not only for glycoproteomic research but also for diagnostic purposes. Larsen et al.43 presented a highly selective method utilizing TiO2 chromatography for purification and characterization of SA-containing glycopeptides. Zhao et al.44 developed a strategy for identifying sialylated glycoprotein markers in human cancer serum. However, none of these works was directly focused on the enrichment of intact sialoglycoproteins.
Herein, we present a simple and direct strategy for alternative extraction of sialylated or nonsialylated glycoproteins with boronate affinity monoliths. This strategy mainly relies on a finding by Otsuka and co-workers45 that sialic acids exhibit anomalously high binding constants with common boronic acids when the pH is lower than the pKa value of the boronic acid. This phenomenon was subsequently employed by Matsumoto and co-workers46 to develop a potentiometric method for the specific detection of sialic acids at the cell membrane. The principle of the proposed strategy is illustrated in Fig. 1, in which Neu5Ac and mannose are adopted as models for sialic acid and neutral sugar, respectively. The glycolyl side chain of Neu5Ac is a critical site for binding boronic acid, and the complex is stabilized through the coordination of the amide NH or CO located at the C-5 position of Neu5Ac, forming an intermolecular B–N or B–O linkage.45 In fact, intramolecular B–N and B–O linkages have been found to be effective strategies for the design and synthesis of boronic acids that can bind to cis-diol compounds at lowered pH.38,39,47,48
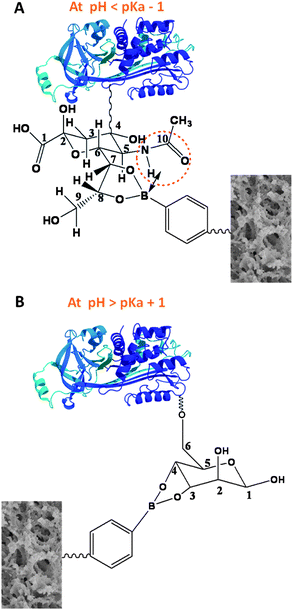 | ||
| Fig. 1 Principle of alternative binding of boronic acid with sialylated and nonsialylated glycoproteins at different pH. (A) When pH < pKa − 1, the boronic acid binds to the sialylated glycoproteins; (B) when pH > pKa + 1, the boronic acid binds to the nonsialylated glycoproteins. | ||
In this study, a hydrophilic boronate affinity monolithic capillary was employed as the extraction column, with 4-vinylphenylboronic acid (VPBA) as the functional monomer. VPBA is a common functional monomer for functionalized polymer synthesis. As phenylboronic acid has a pKa of 8.9,49 VPBA should have a pKa value very similar to this. In order to elucidate our strategy, we firstly applied the obtained boronate affinity monolithic capillary to the alternative extraction of monosaccharides, followed by nano-ESI-Orbitrap MS/MS detection. The alternative extraction of glycoproteins was achieved with monolithic capillary-based boronate affinity extraction followed by MALDI-TOF MS detection. Finally, the established method was applied to human serum samples. The model glycoprotein, either sialoglycoprotein or nonsialylated glycoprotein, spiked in the human serum, was alternatively extracted.
Experimental
Instrumentation
Nano-ESI-Orbitrap MS/MS experiments were performed on a LC-LTQ-Orbitrap XL system equipped with a Surveyor nano-LC system (Thermo Fisher Scientific, San Jose, CA). MALDI-TOF MS experiments were implemented on an Autoflex mass spectrometer (Brucker Daltonics, Germany). Scanning-electron microscopy (SEM) analyses were performed on a Hitachi FE-SEM S-4800 (Japan).Chemicals and materials
N,N-Methylenebisacrylamide (MBAA), VPBA and D-(+)-mannose were purchased from Alfa Aesar (Ward Hill, MA, USA). γ-Methacryloxypropyltrimethoxysilane (γ-MAPS), ribonuclease B (RNase B), ribonuclease A (RNase A), myoglobin, horseradish peroxidase (HRP), bovine serum albumin (BSA), siapinic acid (SA) and α-cyano-4-hydroxycinnamic acid (CHCA) were obtained from Sigma (St. Louis, MO, USA). Dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF), azobisisobutyronitrile (AIBN), acetic acid and ammonium formate were from Sinopharm Chemical Reagent (Shanghai, China). N-Acetylneuraminic acid was purchased from Maya Chemicals (Zhejiang, China). Capillaries of 100 μm i.d. were purchased from Yongnian Optic Fiber Plant (Hebei, China). The recombinant human erythropoietin (EPO) produced in CHO cell lines was from European Pharmacopoeia as Biological Reference Product (BRP) as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of α and β. Ultrapure water produced by a Milli-Q Advantage A10 system (Millipore, Bedford, MA, USA) was used to prepare all solutions in this study.
:1 mixture of α and β. Ultrapure water produced by a Milli-Q Advantage A10 system (Millipore, Bedford, MA, USA) was used to prepare all solutions in this study.Methods
Preparation of the monolithic capillary
Poly (VPBA-co-MBAA) monolithic capillaries were prepared according to a previously reported method.17 Briefly, the capillary was first pretreated with a vinyl silanizing agent to covalently anchor the polymer to the capillary wall. 1 mL DMSO, 0.5 mL DMF, and 0.1 mL water were mixed and then saturated with Na2HPO4. The saturated solution was mixed with 625 mg dodecanol as a porogenic agent. Then, 300 mg VPBA, 300 mg MBAA and 1 mg AIBN were added to the porogenic solution. The resultant solution was degassed with nitrogen for 10 min and injected into a pretreated 45 cm capillary to a certain length with a syringe. Both ends of the capillary were sealed with rubber, and the capillary was submerged in a water bath at 75 °C for 12 h. The copolymerized monolithic column was washed with methanol by an HPLC pump to flush out the residual reagents. The average size of the mesopores of the obtained monolithic capillaries was measured to be 11.2 nm.17 Such a pore size is suitable for application to medium size proteins. For application to larger proteins, the pore size needs to be enlarged, which can be realized through optimizing the ratio of the porogenic solvents.Alternative extraction of sialic acid and neutral sugar
For selective extraction of sialic acid, the mixture of Neu5Ac and mannose in a 1:1 molar ratio was dissolved in 100 mM phosphate buffer (pH 7.4). All procedures for the sample loading, washing and elution were performed by gas pressure as previously reported.50 Briefly, 10 μL of the sample was transferred to a 200 μL microcentrifuge tube which was placed in a high-pressure bomb connected to a nitrogen gas cylinder. A boronate affinity monolithic column of 100 μm i.d. and 8 cm length was inserted into the bomb. The sample was introduced by applying 0.1 MPa of nitrogen until all the sample was completely pushed into the capillary. 50 μL of 100 mM phosphate buffer (pH 7.4), which is the same as the loading buffer, was utilized for washing and 5 μL of 100 mM acetic acid (pH 2.7) for elution. To validate the effect of ionic strength, the same experiment was conducted in a 100 mM phosphate buffer containing 500 mM NaCl (pH 7.4). For selective extraction of neutral sugar, a sugar mixture of Neu5Ac and mannose in a 1:1 molar ratio was dissolved in 100 mM phosphate buffer (pH 10.0). 50 μL 100 mM phosphate buffer (pH 10.0), also the same buffer as the loading buffer, was utilized for washing and 5 μL of 100 mM acetic acid (pH 2.7) for elution. The obtained sample solutions were submitted for nano-ESI-Orbitrap MS/MS for individual detection.ESI-Orbitrap MS/MS analysis of monosaccharides
The flow rate of the mobile phase after splitting was 1 μL min−1. The mass spectrometric detections were set as follows: spray voltage, 1.7 eV; in each scan, a 100000 resolution full scan was performed in the Orbitrap; each full scan included one micro scan; full scan was performed over the range 100–450 m/z. In MS/MS experiments, a mass range of 0–500 m/z was scanned. It was performed through collision induced dissociation (CID) and the [M + NH4]+ ions were selected as parent ions.Preparation of EPO stock solution
Each EPO BRP sample vial contained about 250 μg EPO, 0.1 mg Tween 20, 30 mg trehalose, 3 mg arginine, 4.5 mg sodium chloride and 3.54 mg disodium hydrogen phosphate monohydrate, and it was adjusted to pH 7.00 with sodium hydroxide. To construct solutions for the EPO BRP, the content of each vial was dissolved in 0.25 mL of water. The low-molecular-mass excipients in all the samples were eliminated by ultrafiltration. Samples of certain volume were first diluted into 200 μL and then passed through an Amicon Ultra-0.5 (MWCO 10000 Da) cartridge (Millipore, Beverly, MA, USA), for 20 min at 14000×g and the retentate was washed three times with 180 μL water for 20 min. The retentate was recovered by inverting the cartridge and centrifuging at 2000×g for 3 min. To increase the recovery, this step was repeated three times by moving the collected liquid back to the cartridge. Finally, the volume of the liquid obtained was about 20 μL and the prepared stock solutions were preserved at 4 °C.Extraction of glycoproteins by boronate affinity monolith
RNase B, RNase A and myoglobin were mixed in a 1:1:1 molar ratio and diluted in 250 mM ammonium acetate buffer (pH 10.0). The extraction procedure was similar to the extraction of sialic acid and neutral sugar. The resultant solution was submitted for MALDI-TOF MS for detection.Alternative extraction of sialylated and nonsialylated glycoproteins
For specific extraction of sialylated glycoproteins, the protein mixture of EPO, BSA and HRP in a 1:1:1 molar ratio was dissolved in 100 mM phosphate buffer (pH 7.4) containing 500 mM NaCl before extraction. For specific extraction of nonsialylated glycoproteins, the protein mixture of EPO, BSA and HRP in a 1:1:1 molar ratio was dissolved in 100 mM phosphate buffer (pH 10.0) before extraction. The extraction procedure was similar to the extraction of sialic acid and neutral sugar. The resultant solutions were applied to MALDI-TOF MS for detection individually.Alternative extraction of sialylated and nonsialylated glycoproteins spiked in human serum
To further validate the extraction ability in biological samples, RNase B was directly spiked into 1 μL human serum which is diluted by 10 folds with 250 mM ammonium acetate buffer (pH 10.0). The final concentration of RNase B was 0.1 mg mL−1. 30 μL 250 mM ammonium acetate buffer (pH 10.0) was used for washing and 5 μL 100 mM acetic acid (pH 2.7) for elution. The obtained sample solution was applied to MALDI-TOF MS for detection.For specific extraction of sialoglycoproteins, the stock solution of EPO was directly spiked into 1 μL human serum which is diluted by 10 folds with 100 mM phosphate buffer (pH 7.4) containing 150 mM NaCl and 50 mM NaF. The final concentration of EPO was 0.1 mg mL−1. 30 μL 100 mM phosphate buffer (pH 7.4) containing 150 mM NaCl and 50 mM NaF was used for washing and 5 μL 100 mM acetic acid (pH 2.7) for elution.
MALDI-TOF MS analysis of glycoproteins
Positive ion spectra were recorded using stainless steel target plates with standard parameters, a nitrogen laser (λ = 337 nm) and 20 kV accelerating voltage. 250 ns and 350 ns pulse duration time were separately set for different samples. Saturated CHCA or 10 mg mL−1 SA in 0.1% trifluoroacetic acid:acetonitrile (70:30, v/v) was used as the matrix. After a 1.0 μL matrix solution was added to the eluate, the sample spots were allowed to dry. Each spectrum was recorded as the sum of 100–500 laser shots.Results and discussion
Specific extraction of glycoproteins
The general affinity towards glycoproteins is the precondition for any further specificity exploration. Thus, the specificity of the monolithic capillary was first investigated in terms of its affinity to RNase B against RNase A and myoglobin. Commercially available RNase B is a mixture of glycoforms that differ in the number and branching pattern of the mannose sugars on the nonreducing end of the saccharide moiety. It contains a single glycosylation site at Asn34 and the glycan varies from five to nine mannose residues. Due to the heterogeneity in the glycosylation, RNase B exhibits five glycosylated variants, with a molecular weight of 14901.2 Da for the most abundant isoform in the MALDI-TOF MS spectrum. RNase A (MW, 13682.5 Da) shares an identical polypeptide core but contains no carbohydrate. Myoglobin was another nonglycoprotein model (MW, 16961.7 Da). MALDI-TOF MS analysis of RNase B standard exhibited five major peaks, which stand for the five glycosylated isoforms (Fig. 2A). Meanwhile, a minor peak at ∼13700 Da was also observed, which may be attributed to in-source glycan loss of RNase B or an impurity in the original RNase B sample. MALDI-TOF MS analysis of a standard mixture containing RNase B, RNase A and myoglobin in a 1:1:1 molar ratio indicated the presence of the three model proteins (Fig. 2B). However, the signals for RNase B were much lower than for RNase A (13682.7 Da). This is due to the presence of the glycan in RNase B, which usually exhibits poor ionization efficiency. As comparison, for RNase B extracted by the boronate affinity monolithic capillary from the standard mixture, only the peaks standing for RNase B were observed in the spectrum, though in-source glycan loss was also present (Fig. 2C).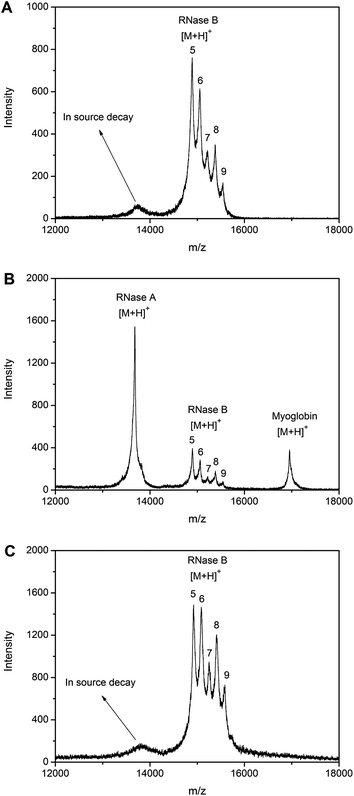 | ||
| Fig. 2 The specificity of the boronate affinity monolith to glycoproteins. MALDI-TOF MS mass spectra for (A) direct analysis of RNase B, (B) direct analysis of 1:1:1 RNase A, RNase B and myoglobin, (C) RNase B extracted by the boronate affinity monolith from the mixture. 5, 6, 7, 8 and 9 in the spectra represent for Man5, Man6, Man7, Man8 and Man9 glycoforms, respectively. | ||
It is interesting that the extraction efficiency for the five glycoforms was not identical. The amounts of Man6, Man7, Man8, and Man9 have increased relative to the amount of Man5. Although the same five glycoform peaks previously observed in Fig. 2A or Fig. 2B are present, the glycoprofile has changed. In comparison to the MALDI data, the intensity of glycoforms with higher mannose content (i.e., Man6–9) has increased relative to that of Man5, the glycoform with the lowest mannose content. The reason is the different content of mannose in the glycoforms; the higher the content of mannose, the stronger the interactions between the boronic acid ligands and the glycan, thus the higher the extraction efficiency.
These results demonstrate that the boronate affinity monolithic capillary exhibited excellent specificity to glycoproteins because of the interaction between glycan chains and boronic acids.
Alternative extraction of sialic acid or neutral sugar
To verify the principle for alternative extraction of the glycoproteins, we firstly tested the specificity toward sialic acids and neutral sugars, which serve as the binding sites of glycoproteins. N-Acetylneuraminic acid (Neu5Ac) was employed as a model sialic acid and mannose was used as a typical neutral sugar. Neu5Ac is the predominant sialic acid found in mammalian cells and also the major component of the glycan chains of sialylated glycoprotein, such as EPO, which is the model of sialylated glycoprotein in this paper. Mannose, which is one of the typical components of the glycan chains of glycoproteins, exists in RNase B and HRP, models for nonsialylated glycoproteins in this work. As a reference, the mixture of two sugars in a 1:1 molar ratio without extraction was directly submitted for nano-ESI-Orbitrap MS analysis. The peaks representing the [M + NH4]+ form of Neu5Ac and mannose ions were observed in the extracted ion chromatogram and the intensity of these two peaks were similar (Fig. 3). Then the mixtures containing these two sugars in a 1:1 molar ratio at pH 7.4 and 10.0 were examined. When the boronate affinity extraction was performed at physiological pH, Neu5Ac was predominant (Fig. 4). As a contrast, when the boronate affinity extraction was carried out at pH 10.0, most of the extracted substance was mannose (Fig. 5). The MS/MS spectra further verify the identities of these ions (Fig. 4B and 5B).![Nano-ESI-Orbitrap MS mass spectra for Neu5Ac and mannose standard. (A) Extracted ion chromatograms for mannose and Neu5Ac, (B) positive ion MS/MS spectra for mannose, generated from precursor ion [M + NH4]+ at 198.10, (C) positive ion MS/MS spectra for Neu5Ac generated from precursor ion [M + NH4]+ at 327.00.](/image/article/2013/AN/c2an36048a/c2an36048a-f3.gif) | ||
| Fig. 3 Nano-ESI-Orbitrap MS mass spectra for Neu5Ac and mannose standard. (A) Extracted ion chromatograms for mannose and Neu5Ac, (B) positive ion MS/MS spectra for mannose, generated from precursor ion [M + NH4]+ at 198.10, (C) positive ion MS/MS spectra for Neu5Ac generated from precursor ion [M + NH4]+ at 327.00. | ||
![The specificity of the boronate affinity monolith toward monosaccharide at physiological pH. (A) Extracted ion chromatogram for Neu5Ac extracted from the mixture containing Neu5AC and mannose in 100 mM phosphate buffer containing 500 mM NaCl (pH 7.4), (B) positive ion MS/MS spectra for Neu5Ac, generated from precursor ion [M + NH4]+ at 327.00.](/image/article/2013/AN/c2an36048a/c2an36048a-f4.gif) | ||
| Fig. 4 The specificity of the boronate affinity monolith toward monosaccharide at physiological pH. (A) Extracted ion chromatogram for Neu5Ac extracted from the mixture containing Neu5AC and mannose in 100 mM phosphate buffer containing 500 mM NaCl (pH 7.4), (B) positive ion MS/MS spectra for Neu5Ac, generated from precursor ion [M + NH4]+ at 327.00. | ||
![The specificity of the boronate affinity monolith toward monosaccharide at basic pH. (A) Extracted ion chromatogram for mannose extracted from the mixture containing Neu5Ac and mannose in 100 mM phosphate buffer (pH 10.0), (B) positive ion MS/MS spectra of mannose, generated from precursor ion [M + NH4]+ at 198.10.](/image/article/2013/AN/c2an36048a/c2an36048a-f5.gif) | ||
| Fig. 5 The specificity of the boronate affinity monolith toward monosaccharide at basic pH. (A) Extracted ion chromatogram for mannose extracted from the mixture containing Neu5Ac and mannose in 100 mM phosphate buffer (pH 10.0), (B) positive ion MS/MS spectra of mannose, generated from precursor ion [M + NH4]+ at 198.10. | ||
Effect of ionic strength
The ionic strength of the sample solution is another important factor that needs to be taken into account. It was observed that appropriate ionic strength is critical for specificity and affinity for sialic acids. When the ionic strength is not enough in the loading conditions, the affinity toward sialic acid was rather weak (Fig. 6). This is due to electrostatic repulsion between boronic acid and sialic acid, because under the pH used (7.4) boronic acid is partially negatively charged while sialic acid (pKa value, 2.6) is highly negatively charged. To suppress electrostatic repulsion, an adequate quantity of salt is necessary in the loading buffer. When 100 or 200 mM NaCl was added, neither sialic acid nor the sialylated glycoproteins could be effectively extracted. On the other hand, in the presence of 500 mM NaCl in the loading buffer, sialic acid and the sialylated glycoproteins could be specifically extracted. However, for different sialylated glycoproteins, the maximum salt content required may depend on the number of sialic acid moieties on the target molecule and the competing substance. The ionic strength is not only used for enhancing the interaction between sialic acids and boronic acids but also for suppressing non-specific adsorption, considering the fact that negatively charged boronic acids will attract positively charged substances. As the presence of excess salt in the samples is detrimental for MS detection, however, the concentration of salt in the samples should be kept as low as possible.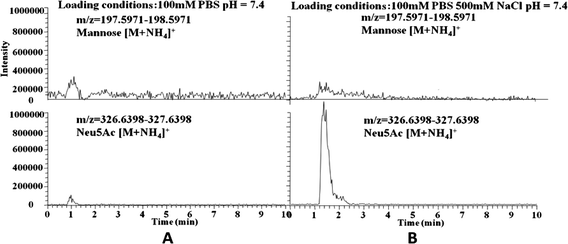 | ||
| Fig. 6 Effect of the ionic strength on the specific extraction of sialic acid. (A) Extracted ion chromatograms for mannose (top) and Neu5Ac (bottom) extracted from the mixture of mannose and Neu5AC in 100 mM phosphate buffer (pH 7.4), (B) extracted ion chromatograms for mannose (top) and Neu5Ac (bottom) extracted from the mixture of mannose and Neu5AC in 100 mM phosphate buffer containing 500 mM NaCl (pH 7.4). | ||
Alternative extraction of sialylated glycoproteins or nonsialylated glycoproteins
To verify the proposed strategy for alternative extraction of sialylated glycoproteins and nonsialylated glycoproteins, EPO was employed as a model sialoglycoprotein, while HRP and BSA were used as a typical nonsialylated glycoprotein and nonglycoprotein, respectively. EPO is a sialoglycoprotein hormone. Native and recombinant EPO can contain a maximum of 11 and 19 sialic acid residues, respectively.51 A mixture containing the three proteins in a 1:1:1 molar ratio was tested at pH 7.4 and 10.0. Direct MALDI TOF-MS analysis of the standard mixture exhibited four peaks, which represent HPR [M + 2H]2+, EPO [M + H]+, HRP [M + H]+ and BSA [M + H]+ (Fig. 7A). As opposed to RNase B, no glycoforms were observed for EPO and HRP. This was attributed to in-source glycan loss. When the extraction was carried out at pH 10.0, only HRP was observed (Fig. 7B). In contrast, when the extraction was performed at physiological pH, only EPO was extracted (Fig. 7C).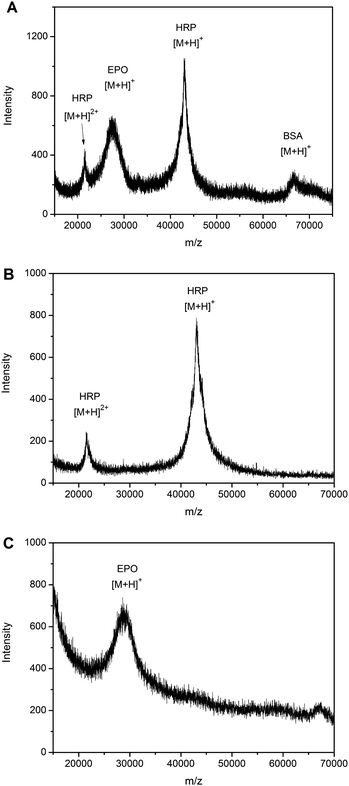 | ||
| Fig. 7 The alternative specificity of the boronate affinity monolith towards sialylated glycoproteins or nonsialylated glycoproteins at different pH. MALDI-TOF MS spectra for (A) direct analysis of a protein mixture containing a molar ratio 1:1:1 of EPO, BSA and HRP, (B) protein extracted from the mixture dissolved in 100 mM phosphate buffer containing 500 mM NaCl (pH 10.0), (C) protein extracted from the mixture dissolved in 100 mM phosphate buffer containing 500 mM NaCl (pH 7.4). | ||
Alternative extraction of sialylated glycoproteins or nonsialylated glycoproteins from complex samples
To demonstrate the feasibility of the proposed strategy for application to complex samples, human serum spiked with sialylated glycoprotein and nonsialylated glycoprotein was employed as a test sample, using RNase B and EPO as models for nonsialylated and sialylated glycoproteins, respectively. MALDI-TOF MS analysis of the eluate from RNase B-spiked human serum (pH adjusted to 10.0) indicated that RNase B and several other glycoproteins (located within the range 12.67–13.75 kDa) were extracted by the boronate affinity monolithic capillary (Fig. 8A). Although the identities of the peaks ranging from 12.67–13.75 kDa were unknown, they could probably be attributed to nonsialylated glycoproteins since the specificity under the pH conditions used has been confirmed. As a comparison, when the extraction was performed at pH 7.4, MALDI-TOF MS analysis of the eluate from EPO-spiked human serum (pH 7.4) indicated that EPO was extracted, along with two minor components located at ∼42 kDa and 66 kDa (Fig. 8B). The minor peak at 66 kDa is assigned to human serum albumin (HSA). Due to the abundance of HSA in human serum (the concentration of HSA is approximately 100 times the spiked model glycoprotein), nonspecific adsorption may have occurred. The 42 kDa can be assigned to α2 HS-glycoprotein,52 which is a sialic acid containing glycoprotein. However, because of the poor signal intensity, such an assignment needs further confirmation.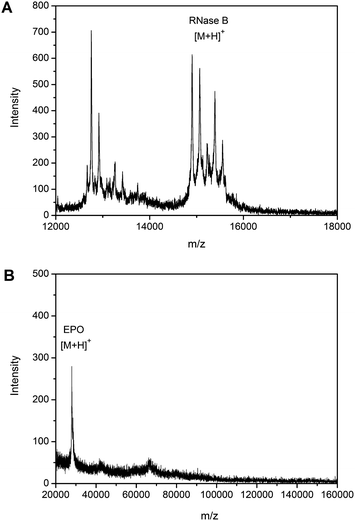 | ||
| Fig. 8 The alternative specificity of the boronate affinity monolith towards sialylated glycoproteins or nonsialylated glycoproteins spiked in human serum. MALDI-TOF MS spectra for (A) proteins extracted from RNase B-spiked serum in 250 mM ammonium acetate buffer (pH 10.0), (B) proteins extracted from EPO-spiked serum in 100 mM phosphate buffer containing 150 mM NaCl and 50 mM NaF (pH 7.4) (in comparison to Fig. 7C, less salt was added to the sample to improve the resolution). | ||
Conclusion
We have proposed a new strategy for alternative extraction of sialylated glycoproteins and nonsialylated glycoproteins by boronate affinity monolith through pH manipulation. An off-line combination of monolithic capillary-based boronate affinity extraction with MS detection was established for the extraction and identification of monosaccharide and intact glycoproteins. Under physiological pH, the boronate affinity monolithic column preferentially exhibited affinity to sialic acid rather than neutral sugar. On the other hand, under basic pH, the boronate affinity monolithic column preferentially exhibited affinity to neutral sugar rather than sialic acid. Based on this, we applied this strategy to the alternative extraction of intact glycoproteins with MALDI-TOF MS detection. As expected, the boronate monolithic column exhibited good specificity toward sialylated glycoproteins under physiological pH conditions while the nonsialylated glycoproteins were predominant under basic pH condition. Furthermore, this method was verified in a complex biological environment such as human serum. From another perspective, this study provides a deeper understanding of the boronate affinity mechanism. Boronate affinity materials may have different affinity towards each glycoform of individual glycoproteins and most importantly, the affinity varies dramatically for different types of glycoproteins. The affinity strongly depends on the nature of the glycoproteins and the experimental conditions, particularly the pH value. Therefore, when boronate affinity materials are used for comprehensive glycoproteomic analysis, such discrimination effect must be taken into account.Acknowledgements
We acknowledge the financial support of the General Grants (nos. 21075063 and 21275073) from the National Natural Science Foundation of China, the International Science & Technology Cooperation Grant (no. 2008DFA01910) from the Ministry of Science and Technology of China, the Fundamental Research Funds (no. 1093020506) and the New Century Excellent Talents Program (no. NCET-08-0270) from the Ministry of Education of China, and the General Grant (no. KB2011054) from the Natural Science Foundation of Jiangsu Province, China.Notes and references
- Y. Wang, S. Wu and W. S. Hancock, Glycobiology, 2006, 16, 514–523 CrossRef CAS.
- S. J. Van. Rensburg, P. Berman, F. Potocnik, P. MacGregor, D. Hon and N. De Villiers, Metab. Brain Dis., 2004, 19, 89–96 CrossRef CAS.
- T. Fournier, N. Medjoubi-N and D. Porquet, Biochim. Biophys. Acta, 2000, 1482, 157–171 CAS.
- U. Nihlen, P. Montnemery, L. H. Lindholm and C. G. Lofdahl, Scand. J. Clin. Lab. Invest., 1999, 17, 232–237 Search PubMed.
- H. Zhang, X. J. Li, D. B. Martin and R. Aebersold, Nat. Biotechnol., 2003, 21, 660–666 CrossRef CAS.
- N. Khidekel, S. B. Ficarro, E. C. Peters and L. C. Hsieh-Wilson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13132–13137 CrossRef CAS.
- M. Gilar, Y. Q. Yu, J. Ahn, H. W. Xie, H. H. Han, W. T. Ying and X. H. Qian, Anal. Biochem., 2011, 417, 80–88 Search PubMed.
- S. Mysling, G. Palmisano, P. Hojrup and M. Thaysen-Andersen, Anal. Chem., 2010, 82, 5598–5609 CrossRef.
- L. Yu, X. L. Li, Z. M. Guo, X. L. Zhang and X. M. Liang, Chem.–Eur. J., 2009, 15, 12618–12626 CrossRef CAS.
- Y. Mechref, M. Madera and M. V. Novotny, Methods Mol. Biol., 2008, 424, 373–396 Search PubMed.
- R. R. Drake, E. E. Schwegler, G. Malik, J. Diaz, T. Block, A. Mehta and O. J. Semmes, Mol. Cell. Proteomics, 2006, 5, 1957–1967 CrossRef CAS.
- H. Kaji, Y. Yamauchi, N. Takahashi and T. Isobe, Nat. Protoc., 2006, 1, 3019–3027 CAS.
- R. Qiu and F. E. Regnier, Anal. Chem., 2005, 77, 2802–2809 CrossRef CAS.
- K. Sparbier, T. Wenzel and M. Kostrzewa, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2006, 840, 29–36 CrossRef CAS.
- A. Monzo, M. Olajos, L. De Benedictis, Z. Rivera, G. K. Bonn and A. Guttman, Anal. Bioanal. Chem., 2008, 392, 195–201 Search PubMed.
- W. Zhou, N. Yao, G. P. Yao, C. H. Deng, X. M. Zhang and P. Y. Yang, Chem. Commun., 2008, 43, 5577–5579 Search PubMed.
- L. B. Ren, Y. C. Liu, M. M. Dong and Z. Liu, J. Chromatogr., A, 2009, 1216, 8421–8425 CrossRef CAS.
- M. Chen, Y. Lu, Q. Ma, L. Guo and Y. Q. Feng, Analyst, 2009, 134, 2158–2164 RSC.
- J. Tang, Y. C. Liu, D. W. Qi, G. P. Yao, C. H. Deng and X. M. Zhang, Proteomics, 2009, 9, 5046–5055 CrossRef CAS.
- K. K. Jang, Y. H. Kim, K. S. Lee, I. S. Choi, S. Kim and K. B. Lee, Rapid Commun. Mass Spectrom., 2009, 23, 3599–3602 Search PubMed.
- A. Takatsy, K. Boddi, L. Nagy, G. Nagy, S. Szabo, L. Marko, I. Wittmann, R. Ohmacht, T. Ringer, G. K. Bonn, D. Gjerde and Z. Szabo, Anal. Biochem., 2009, 393, 8–22 CrossRef CAS.
- B. Preinerstorfer, M. Laemmerhofer and W. Lindner, J. Sep. Sci., 2009, 32, 1673–1685 CrossRef CAS.
- L. J. Zhang, Y. W. Xu, H. L. Yao, L. Q. Xie, J. Yao, H. J. Lu and P. Y. Yang, Chem.–Eur. J., 2009, 15, 10158–10166 CrossRef CAS.
- W. W. Shen, C. N. Ma, S. F. Wang, H. M. Xiong, H. J. Lu and P. Y. Yang, Chem.–Asian J., 2010, 5, 1185–1191 Search PubMed.
- Y. W. Xu, L. J. Zhang, H. J. Lu and P. Y. Yang, Proteomics, 2010, 10, 1079–1086 CAS.
- D. W. Qi, H. Y. Zhang, J. Tang, C. H. Deng and X. M. Zhang, J. Phys. Chem. C, 2010, 114, 9221–9226 CrossRef CAS.
- L. Liang and Z. Liu, Chem. Commun., 2011, 47, 2255–2257 RSC.
- J. Qiu, Y. Zhang, H. J. Lu and P. Y. Yang, Acta Chim. Sin., 2011, 69, 2123–2129 Search PubMed.
- H. Y. Zhang, G. P. Yao, C. H. Deng, H. J. Lu and P. Y. Yang, Chin. J. Chem., 2011, 29, 835–839 Search PubMed.
- Y. Zhang, A. Giboulot, M. Zivy, B. Valot, E. Jamet and C. Albenne, Phytochemistry, 2011, 72, 1109–1123 Search PubMed.
- F. Yang, Z. A. Lin, X. W. He, L. X. Chen and Y. K. Zhang, J. Chromatogr., A, 2011, 1218, 9194–9201 CrossRef CAS.
- X. H. Zhang, X. W. He, L. X. Chen and Y. K. Zhang, J. Mater. Chem., 2012, 22, 16520–16526 RSC.
- R. Flueckiger, T. Woodtli and W. Berger, Diabetes, 1984, 33, 73–76 CrossRef CAS.
- L. B. Ren, Z. Liu, M. M. Dong, M. L. Ye and H. F. Zou, J. Chromatogr., A, 2009, 1216, 4768–4774 CrossRef CAS.
- Y. C. Liu, Y. Lu and Z. Liu, Chem. Sci., 2012, 3, 1467–1471 RSC.
- L. B. Ren, Z. Liu, Y. C. Liu, P. Dou and H. Y. Chen, Angew. Chem., Int. Ed., 2009, 48, 6704–6707 CrossRef CAS.
- Y. C. Liu, L. B. Ren and Z. Liu, Chem. Commun., 2011, 47, 5067–5069 RSC.
- H. Y. Li, Y. C. Liu, J. Liu and Z. Liu, Chem. Commun., 2011, 47, 8169–8171 RSC.
- H. Y. Li, H. Y. Wang, Y. C. Liu and Z. Liu, Chem. Commun., 2012, 48, 4115–4117 RSC.
- Y. Kaneko, F. Nimmerjahn and J. V. Ravetch, Science, 2006, 313, 670–673 CrossRef CAS.
- P. Thomas, Cancer J., 1996, 9, 1–10 Search PubMed.
- R. Coppo and A. Amore, Kidney Int., 2004, 65, 1544–1547 Search PubMed.
- M. R. Larsen, S. S. Jensen, L. A. Jakobsen and N. H. H. Heegaard, Mol. Cell. Proteomics, 2007, 6, 1778–1787 CrossRef CAS.
- J. Zhao, D. M. Simeone, D. Heidt, M. A. Anderson and D. M. Lubman, J. Proteome Res., 2006, 5, 1792–1802 CrossRef CAS.
- H. Otsuka, E. Uchimura, H. Koshino, T. Okano and K. Kataoka, J. Am. Chem. Soc., 2003, 125, 3493–3502 CrossRef CAS.
- A. Matsumoto, N. Sato, K. Kataoka and Y. J. Miyahara, J. Am. Chem. Soc., 2009, 131, 12022–12023 CrossRef CAS.
- G. Wulff, M. Lauer and H. Bohnke, Angew. Chem., Int. Ed., 1984, 23, 741–742 CrossRef.
- M. Dowlut and D. G. Hall, J. Am. Chem. Soc., 2006, 128, 4226–4227 CrossRef CAS.
- X. C. Liu, J. L. Hubbard and W. H. Scouten, J. Organomet. Chem., 1995, 493, 91–94 CrossRef CAS.
- M. M. Dong, M. H. Wu, F. J. Wang, H. Q. Qin, J. Dong, R. A. Wu, M. L. Ye, Z. Liu and H. F. Zou, Anal. Chem., 2010, 82, 2907–2915 CrossRef CAS.
- D. Choi, M. Kim and J. Park, J. Chromatogr., B: Biomed. Sci. Appl., 1996, 687, 189–199 CrossRef CAS.
- G. L. Hortin, Clin. Chem., 2006, 52, 1223–1237 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |