Nickel-catalyzed decyanation of inert carbon–cyano bonds†
Tuhin
Patra
,
Soumitra
Agasti
,
Akanksha
and
Debabrata
Maiti
*
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400076, India. E-mail: dmaiti@chem.iitb.ac.in
First published on 6th November 2012
Abstract
Nickel catalyzed decyanation of aryl and aliphatic cyanides with hydrosilane as the hydride source has been developed. This method is easy to handle, scalable and can be carried out without a glove box. The method has been applied in the cyanide directed functionalization reaction and α-substitution of benzyl cyanide.
Removal of a specific functional group after using its beneficial aspects plays an important role in synthetic chemistry.1 Although a wide variety of effective approaches now exists for a number of defunctionalization reactions,2 some of these have been only recently discovered and need further improvement. Synthetic methodology for the removal of the cyano group can be advantageous due to its ortho-directing ability and α-C–H acidity.3,4 The electron withdrawing character of a cyano group brings about challenging coupling reactions by making metal-mediated steps i.e. oxidative addition and reductive elimination faster than the unsubstituted analogues and thus a decyanation protocol will allow synthetic chemists to take advantage of the cyano group.1a,b
The high bond dissociation energy of the C–CN bond and limited successes of (aryl/alkyl) cyanides as the coupling partner compared to (aryl/alkyl) halides imposes a scientific challenge to develop mild and efficient methods for its reductive cleavage.5 Previous decyanation reactions relied on oxidative addition to the silyl hydride and then silicon-assisted carbon–cyano bond cleavage reactions.3a,c,h Another powerful approach could be via insertion of a transition metal into the C–CN bond and subsequent formation of the decyanated product with a hydride source (Scheme 1). In this context, we note that nickel mediated oxidative addition to the C–CN bond and related transformations are reported in the literature.1a,6 Herein, we report a general and mild Ni-catalyzed method for reductive cleavage of unactivated C–CN bonds with (Me2SiH)2O [tetramethyldisiloxane, TMDSO] as the hydride source.
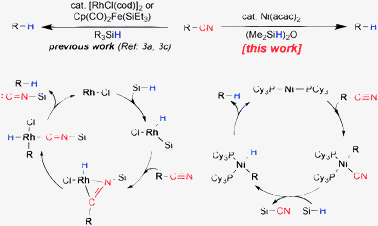 | ||
| Scheme 1 Catalytic decyanation reactions of R–CN. | ||
In order to realize the C–CN bond cleavage, we began our investigation by evaluating a variety of ligand systems for nickel in the presence of various hydride donors such as TMDSO, Et3SiH, iPr3SiH, and Ph2SiH2. We studied 2-cyanonaphthalene as the substrate in the presence of various nickel sources including Ni(COD)2 (COD, 1,5-cyclooctadiene), Ni(acac)2 (acac, acetylacetonate), NiCl2, Ni(OAc)2etc. Use of Lewis acid (AlMe3) can facilitate oxidative addition into the C–CN bond6 and was, therefore, found to be beneficial in terms of substrate scope and yields of the decyanation reactions.
These extensive experimentations showed that the Ni(acac)2 complex of PCy3 (or PCy3·HBF4 with equimolar K3PO4) in combination with TMDSO [(Me2SiH)2O] can catalyze the reductive decyanation of R–CN substrates efficiently.
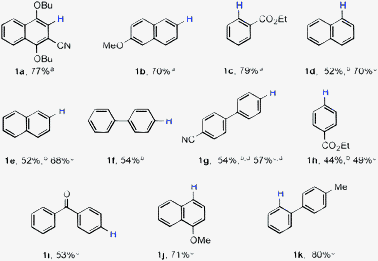
Encouraged by our initial findings, we set out to explore the substrate scope for this Ni(acac)2 catalyzed reductive decyanation reaction (Table 1). The aryl alkyl ether bonds (Ar–O–R; R = Me, nBu) were tolerated (1a, 1b and 1j) under our reaction conditions, thus exhibiting a preference for reductive cleavage of the C–CN bond over Ar–O–R.2b,c,7 A monodecyanated product was observed from substrates bearing dicyano groups (1a and 1g). Ester (1c and 1h) and keto (1i) groups were also tolerated. Although not essential, presence of an ortho-directing group or an ortho-substituent was found to be beneficial in achieving better yield of the desired decyanated product.

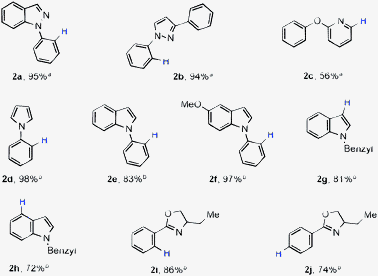
Having demonstrated the protocol on aryl derivatives, we next focused on heterocyclic cyano compounds. As expected, nickel catalyst can be employed in combination with a hydride source for effective decyanation of heterocyclic cyanides (Table 2). Substrates with two different heteroatoms can be employed successfully (2a, 2b, 2i and 2j) indicating lower Lewis acidity of the nickel catalyst. Notably, reductive decyanation reactions were efficient even with ortho-directing groups like pyrazole (2a and 2b), ester (1c) and oxazoline (2i and 2j).

Next we focused on catalytic decyanation of alkyl cyanides since such C–CN bond activations are problematic.8 We first tested the ability of the nickel-catalyzed method to transform benzylic C–CN bonds (Table 3) into C–H bonds (3a–3e). 2-(4-Hydroxyphenyl)acetonitrile produced the expected product in preparatively useful yields (3a). Benzylic cyanide with a long chain alkyl substituent also afforded the corresponding decyanated product (3c).
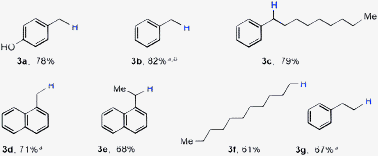
Synthetic transformations involving alkyl groups (RalkylX) are considered challenging due to their propensity to undergo side reactions from alkyl-metal intermediates to β-hydride elimination.8 Consistent with our expectations, aliphatic nitriles (3f and 3g) were successfully decyanated under the present conditions.
Isolation of the deuterated product by using Ph2SiD2 indicated that hydrosilane is the source of hydrogen in the resulting decyanated product (Scheme 2). Selective incorporation of the deuterium atom in unbiased arenes can thus easily be done under neutral conditions.
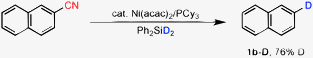 | ||
| Scheme 2 Deuterium labeling. | ||
A stable trans-[Ni(PCy3)2(CN)2] was isolated and characterized by X-ray crystallography9 (Fig. 1) from various decyanation reactions (e.g. entries 1f, 1g, 1i and 3f). Attempted decyanation reactions of 1-cyanonaphthalene by using trans-[Ni(PCy3)2(CN)2] as the catalyst [instead of Ni(acac)2] did not produce any of the desired decyanated product. Therefore, we suspected that the quick formation of this nickel complex, trans-[Ni(PCy3)2(CN)2], is possibly contributing to catalyst deactivation.
![ORTEP diagram of trans-[Ni(PCy3)2(CN)2]. Hydrogen atoms are omitted for clarity.](/image/article/2013/CC/c2cc36883h/c2cc36883h-f1.gif) | ||
| Fig. 1 ORTEP diagram of trans-[Ni(PCy3)2(CN)2]. Hydrogen atoms are omitted for clarity. | ||
We next set out to determine the realtive ease of nickel catalyzed defunctionalization reactions with aryl–CN, –SMe, –OMe and –OAr functional groups under the present reaction conditions (Table 4). We have carried out competition experiments with 2-cyanonaphthalene and 2-X-naphthalene (X = OMe, OAr and SMe) where two substrates were reacted in one flask. We observed that reduction of the aromatic C–CN bond occurs faster than ArOAr and ArOMe. Further investigations showed that removal of –SMe is faster than that of the –CN group.2a Thus our present study along with the recent literature2a–c,7 enabled us to find a reactivity pattern which is summarized below:
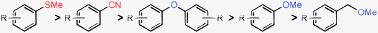
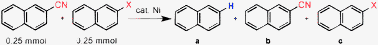
| X | Temp (°C) | a (mmol) | b (mmol) | c (mmol) |
|---|---|---|---|---|
| SMe | 90 | 0.29 | 0.20 | 0.005 |
| 130 | 0.495 | — | — | |
| O-p-tol | 130 | 0.18 | 0.04 | 0.24 |
| OMe | 130 | 0.175 | 0.06 | 0.245 |
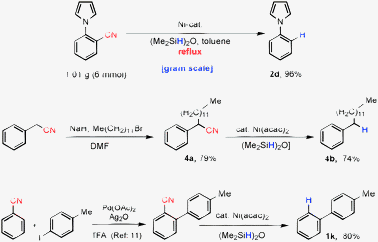
The decyanation reaction proved to be scalable, with 2-(1H-pyrrol-1-yl)benzonitrile producing 96% isolated yield of the expected product (Scheme 3, 2d). Benzyl cyanide can be alkylated upon NaH treatment. Subsequent decyanation of the resulting product will therefore allow use of benzyl cyanide as a benzyl anion equivalent (Scheme 3, 4a). A cyano group is also ortho directing10 and therefore direct C–H arylation at the ortho-position and successive decyanation would allow efficient synthesis of biaryls (Scheme 3, 1k). These two-step methods (Scheme 3) demonstrate the power of this decyanation strategy to synthesize useful molecules.
 | ||
| Scheme 3 Synthetic applicability. | ||
In summary, we have developed a general protocol for nickel-mediated decyanation of aryl and alkyl cyanides. A wide range of nitriles with varying electronic and steric substituents were successfully decyanated as well as the substrates with β-hydrogen. Synthetic advantages of the cyano group including ortho-directing ability, α-C–H acidity and electron-withdrawing ability can thus temporarily be used. The Ni-catalyzed decyanation reaction is expected to be applicable in synthetic chemistry due to its wide availability and easy to handle protocol. Studies are ongoing in our research group to develop related synthetic transformations and to elucidate mechanistic details for the reductive decyanation of the C–CN bond.
This activity is supported by BRNS (2011/20/37C/13/BRNS), India. Financial support received from UGC-India (fellowship to T.P.) and IIT Bombay (fellowship to S.A. and Akanksha) is gratefully acknowledged. X-ray structural study was carried out at the National Single Crystal Diffractometer Facility, IIT Bombay.
Notes and references
- (a) Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, Germany, 2nd edn, 2004 Search PubMed; (b) J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, CA Search PubMed; (c) J. March, Advanced Organic Chemistry, John Wiley & Sons, New York, 3rd edn, 1985 Search PubMed; (d) T. Patra, S. Manna and D. Maiti, Angew. Chem., Int. Ed., 2011, 50, 12140–12142 CrossRef CAS.
- (a) N. Barbero and R. Martin, Org. Lett., 2012, 14, 796–799 CrossRef CAS; (b) A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS; (c) P. Alvarez-Bercedo and R. Martin, J. Am. Chem. Soc., 2010, 132, 17352–17353 CrossRef CAS; (d) W. Hartwig, Tetrahedron, 1983, 39, 2609–2645 CrossRef CAS; (e) N. Chatani, H. Tatamidani, Y. Ie, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2001, 123, 4849–4850 CrossRef CAS; (f) T. C. Fessard, H. Motoyoshi and E. M. Carreira, Angew. Chem., Int. Ed., 2007, 46, 2078–2081 CrossRef CAS; (g) J. Tsuji and T. Ohno, Synthesis, 1969, 157–169 CrossRef CAS; (h) C. R. Hauser and B. E. Hudson, Jr., Org. React., 1942, 1, 266 Search PubMed; (i) A. Modak, A. Deb, T. Patra, S. Rana, S. Maity and D. Maiti, Chem. Commun., 2012, 48, 4253–4255 RSC; (j) Akanksha and D. Maiti, Green Chem., 2012, 14, 2314–2320 RSC.
- (a) M. Tobisu, R. Nakamura, Y. Kita and N. Chatani, J. Am. Chem. Soc., 2009, 131, 3174 CrossRef CAS; (b) M. Tobisu, R. Nakamura, Y. Kita and N. Chatani, Bull. Korean Chem. Soc., 2010, 31, 582–587 CrossRef CAS; (c) H. Nakazawa, K. Kamata and M. Itazaki, Chem. Commun., 2005, 4004–4006 RSC; (d) H. Nakazawa, M. Itazaki, K. Kamata and K. Ueda, Chem.–Asian J., 2007, 2, 882–888 CrossRef CAS; (e) K. Fukurnoto, T. Oya, M. Itazaki and H. Nakazawa, J. Am. Chem. Soc., 2009, 131, 38–39 CrossRef; (f) M. Tobisu, Y. Kita and N. Chatani, J. Am. Chem. Soc., 2006, 128, 8152–8153 CrossRef CAS; (g) K. Fukumoto, A. A. Dahy, T. Oya, K. Hayasaka, M. Itazaki, N. Koga and H. Nakazawa, Organometallics, 2012, 31, 787–790 CrossRef CAS; (h) M. Tobisu, Y. Kita, Y. Ano and N. Chatani, J. Am. Chem. Soc., 2008, 130, 15982–15989 CrossRef CAS.
- J. Y. Corey, Chem. Rev., 2011, 111, 863–1071 CrossRef CAS.
- (a) J. M. Mattalia, C. Marchi-Delapierre, H. Hazimeh and M. Chanon, ARKIVOC, 2006, 90–118 CAS; (b) T. A. Atesin, T. Li, S. Lachaize, W. W. Brennessel, J. J. Garcia and W. D. Jones, J. Am. Chem. Soc., 2007, 129, 7562–7569 CrossRef CAS.
- (a) Y. Nakao and T. Hiyama, Pure Appl. Chem., 2008, 80, 1097–1107 CrossRef CAS; (b) M. Tobisu and N. Chatani, Chem. Soc. Rev., 2008, 37, 300–307 RSC; (c) J. J. Garcia, N. M. Brunkan and W. D. Jones, J. Am. Chem. Soc., 2002, 124, 9547–9555 CrossRef CAS; (d) Y. Nakao, A. Yada, S. Ebata and T. Hiyama, J. Am. Chem. Soc., 2007, 129, 2428–2429 CrossRef CAS; (e) J. A. Miller and J. W. Dankwardt, Tetrahedron Lett., 2003, 44, 1907–1910 CrossRef CAS; (f) Y. Hirata, A. Yada, E. Morita, Y. Nakao, T. Hiyama, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2010, 132, 10070–10077 CrossRef CAS; (g) Y. Hirata, T. Yukawa, N. Kashihara, Y. Nakao and T. Hiyama, J. Am. Chem. Soc., 2009, 131, 10964–10973 CrossRef CAS; (h) Y. Nakao, A. Yada and T. Hiyama, J. Am. Chem. Soc., 2010, 132, 10024–10026 CrossRef CAS; (i) A. Yada, T. Yukawa, Y. Nakao and T. Hiyama, Chem. Commun., 2009, 3931–3933 RSC; (j) N. M. Brunkan, D. M. Brestensky and W. D. Jones, J. Am. Chem. Soc., 2004, 126, 3627–3641 CrossRef CAS; (k) Y. Nakao, S. Ebata, A. Yada, T. Hiyama, M. Ikawa and S. Ogoshi, J. Am. Chem. Soc., 2008, 130, 12874–12875 CrossRef CAS; (l) M. P. Watson and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 12594–12595 CrossRef CAS.
- M. Tobisu, K. Yamakawa, T. Shimasaki and N. Chatani, Chem. Commun., 2011, 47, 2946–2948 RSC.
- (a) X. L. Hu, Chem. Sci., 2011, 2, 1867–1886 RSC; (b) A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674–688 CrossRef CAS; (c) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656–2670 CrossRef CAS.
- (a) B. H. Xia, C. M. Che, D. L. Phillips, K. H. Leung and K. K. Cheung, Inorg. Chem., 2002, 41, 3866–3875 CrossRef CAS; (b) T. Schaub, C. Doring and U. Radius, Dalton Trans., 2007, 1993–2002 RSC.
- W. Li, Z. P. Xu, P. P. Sun, X. Q. Jiang and M. Fang, Org. Lett., 2011, 13, 1286–1289 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed description of experimental procedures, characterization of all compounds. See DOI: 10.1039/c2cc36883h |
| This journal is © The Royal Society of Chemistry 2013 |