A luminescent hydrogel based on a new Au(I) complex†
Raquel
Gavara
a,
Jordi
Llorca
b,
João Carlos
Lima
a and
Laura
Rodríguez
*c
aREQUIMTE, Departamento de Química, CQFB, Universidade Nova de Lisboa, Monte de Caparica, Portugal
bInstitut de Tècniques Energètiques i Centre de Recerca en NanoEnginyeria, Universitat Politècnica de Catalunya, Diagonal 647, 08028 Barcelona, Spain
cDepartament de Química Inorgànica, Universitat de Barcelona, Barcelona, Spain. E-mail: laura.rodriguez@qi.ub.es; Fax: +34 934907725; Tel: +34 934039130
First published on 2nd November 2012
Abstract
The reaction of the water soluble phosphine 1,3,5-triaza-7-phosphaadamantane (PTA) with [Au(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–C5H4N)]n yields the highly luminescent water soluble [(PTA)Au(4-pyridylethynyl)] complex. A detailed analysis of the compound shows the formation of gel structure giving rise to very long fibers, being the first example reported with such a simple structure.
C–C5H4N)]n yields the highly luminescent water soluble [(PTA)Au(4-pyridylethynyl)] complex. A detailed analysis of the compound shows the formation of gel structure giving rise to very long fibers, being the first example reported with such a simple structure.
Gels are well known materials with everyday applications and their designation is based on visual observation of their flow characteristics (a crude rheological measurement).1 They are generally comprised of at least two components: a sample-spanning, fibrous solid network and a solvent or liquid component that is retained within the solid by physical effects such as surface tension.2 They are termed organogels whenever the fluid component is an organic compound and hydrogels when it is water.1 They present highly anisotropic 3D structure in solution, often through non-covalent interactions, which are commonly van der Waals forces, hydrogen bonding, electrostatic attraction and π–π stacking interactions.3 Their diversity and remarkable properties make supramolecular gels interesting candidates for a number of high-technology applications.4–8 Gels derived from the non-covalent self-assembly of small molecules are called low molecular weight gelators (LMWGs). Though gel formation by organic molecules has been widely reported, metallogels have only been a subject of study in the last few years. Metallogelators may consist of coordination polymers or discrete metal complexes that form supramolecular assemblies or aggregates. The presence of metal ions may also modify the physical properties of the gels.9 A particular kind of metallogelators are organometallic gelators which can display metal–metal interactions that influence their properties.3,10,11 These interactions could also be responsible for the gelation process. Some well-known gelators include bis(ureas) and amides,12 nucleobase-derived compounds,13 fatty acids, steroids, anthryl derivatives, amino acid derivatives, dendrimers,14 phthalocyanines and porphyrins.15 In general, however, most of these groups are derivatized with long alkyl chains to favour gelation. Nevertheless, this process becomes unpredictable in some cases. In this work, we report, to the best of our knowledge, the formation of the first example of a luminescent organometallic gold(I) hydrogelator obtained from a discrete molecule, that is, [(PTA)Au(4-pyridylethynyl)] (PTA = 1,3,5-triaza-7-phosphaadamantane). The complex does not contain any of the aforementioned groups that induce gelation. Moreover, the analogous derivative with a 2-pyridyl unit was previously reported in the literature16 as a non-water soluble complex. This fact definitively proves that small changes in the structure could induce unexpected rheological properties. The compound was synthesized following a similar method previously reported in the literature17 and characterized by different techniques (see ESI†).
The corresponding 1H-NMR spectrum shows the characteristic Hα and Hβ protons of the pyridine together with the bridging N–CH2–P (equivalent protons, since this group lies in the plane of the molecule) and the N–CH2–N (AB system) of the PTA phosphine (Fig. S1, ESI†). The presence of a singlet instead of a doublet for the N–CH2–P group (no coupling with the phosphorus atom) has been observed in other metallic PTA derivatives16,18–20 and verified by the 2D 1H-13C NMR spectrum.21 ESI-MS experiment displays the molecular peak (Fig. S2, ESI†). Interestingly, the compound immediately gelified in water (Fig. S3, ESI†) even at very low concentration (0.015–0.05% weight). To the best of our knowledge, this is the lowest concentration reported in the literature for the formation of a gel. The 1H-NMR spectrum of the gelified solution shows very broad peaks, especially for pyridine protons, which almost disappear (Fig. S4, ESI†). The broadening of the peaks could also be observed for the sample recorded in CDCl3 at higher concentrations (Fig. S5, ESI†). These facts are in agreement with an aggregation process, responsible for the formation of the gel.22 The larger effect observed for Hα and Hβ pyridine protons (mainly in water) indicates the involvement of the pyridyl unit in this process. Thus, π–π interactions between these groups are expected to occur in this system. This fact supports previous research done with similar derivatives, where excitonic splitting resulting from interactions involving the CC-py units (together with Au⋯Au interactions) was detected.17 The 2D-NOESY NMR spectrum is a very important tool to obtain additional information about molecular organization during aggregation (Fig. 1). It shows the existence of interactions between pyridyl units and PTA protons, which is supported by the trace data retrieved for N–CH2–N phosphine protons (Fig. S6, ESI†). This particular point is crucial since NOESY spectra are generally used to detect protons that are close together within a molecule (not necessarily directly connected). In this case, information has been gleaned about intermolecular interactions, and thus, about their environment since protons are very far apart and the rigidity of the molecule prevents their interaction in an intramolecular way. Accordingly, we suggest that the disposition of the molecules could be as exemplified in Fig. S7 (ESI†). That is, head to head 1D chains based on π–π stacking interactions between the Au–CCpy units (observed by NMR and broadening of the UV/Vis absorption bands, indicative of exciton splitting, see below)17 that associate in head to tail intermolecular conformation (based on the NOESY coupling).
![NOESY spectrum of [(PTA)Au(CC–C5H4N)] in CDCl3.](/image/article/2013/CC/c2cc37262b/c2cc37262b-f1.gif) | ||
| Fig. 1 NOESY spectrum of [(PTA)Au(CC–C5H4N)] in CDCl3. | ||
Characterization of both the solid and the xerogel was carried out by powder DRX and they display indistinguishable peaks, which means that both have the same crystalline unit cell (Fig. S8, ESI†). Hence, the low concentration of fibers present in the samples does not allow us to identify them by this technique. The most intense DRX peaks are observed below 35° and most of them between 26.4° and 29.0° which correspond to a d spacing between 3.1–3.4 Å, very close to typical π–π stacking interactions.23 This supports our previous findings observed by NMR and absorption spectra.
The electronic absorption spectra both in water and dichloromethane display intense high-energy absorption bands in the range of 240–285 nm (Table S1, ESI†) assigned to PTA and well-resolved intraligand (IL) π → π* (CCpy) transitions (Fig. S9, ESI†).17,24–29 The latter becomes broader in water with respect to dichloromethane although broadening increases with concentration in both solvents. Moreover, a new broad band at ca. 350 nm is observed in this medium which has been assigned to a σ*Au–Au → π* transition30 (Fig. S10, ESI†). These facts were previously attributed to π–π interactions involving CCpy units favoured by the formation of aurophilic interactions,17 and so, they seem to be more easily established in water, as observed by NMR and are in agreement with the lower extinction coefficient values calculated in water in comparison with dichloromethane (Table S1, ESI†). The compound displays luminescence both in solution and in solid state. The emission spectra recorded in solution upon excitation of the samples at 280 nm at room temperature present a vibronically resolved band centred at ∼420 nm, which demonstrates the involvement of ligand character in their emission origin (Fig. S11, ESI†). Excitation spectra collected at the maxima reproduce the absorption band (Fig. S12, ESI†). Thus, our experimental results and the comparison with previously published data led us to assign this high-energy emission band to an intraligand 3[π–π*(alkynyl)] origin.17,25,27 Excitation of the samples at λexc = 320 nm and low temperature (77 K) gives rise to a new broad band at longer wavelengths (ca. 500 nm) superimposed to the well resolved emission band observed at 298 K (Fig. 2). This broad band could be attributed to the π–π excimeric 3IL emission because of the possible π–π stacking of the ethynylpyridyl ligand in the solid state, as suggested very recently by Yam and co-workers.31 Excitation collected at the broad band emission wavelength (77 K) displays two different superimposed bands (Fig. 2). The highest energy band (at 280 nm) matches the absorption spectrum and is attributed to the intraligand transition assigned above. The lowest energy band (360 nm) has been attributed to the previously related σ*Au–Au → π* transition.30
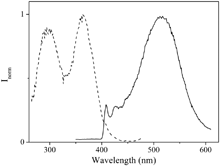 | ||
| Fig. 2 Normalized excitation (λem = 530 nm, left) and emission (λexc = 320 nm, right) spectra in CH2Cl2 recorded at 77 K. | ||
The emission spectrum in the solid state displays the same broad red shifted band (centred at 535 nm) recorded at low temperature (Fig. S13, ESI†). The excitation spectrum is also in agreement with the same assignment since it shows the same broad band at 360 nm. The large Stokes shift and the recorded monoexponential luminescence decay times in the order of microseconds (2.5 μs for 2 × 10−6 M solution in water, 25 °C) are indicative of a triplet emissive state. Quantum yields measured in an integrating sphere both in solution (H2O, 25 °C) and in solid state are ca. 0.1 and 0.15, respectively, which are very high values for this kind of complexes and at least ten times higher than those recorded for similar compounds.30
Optical microscopy analysis of the air-dried gel (xerogel) indicates clear formation of organized solid structures (after the gelation process) that were not observed previously in the powder (Fig. S14, ESI†). The formation of very long structures, up to a few millimetres, which is due either to the interaction between smaller aggregates or to the formation of long empty fibers (Fig. 3, left), could be observed. Smaller aggregates are also observed (Fig. S15, ESI†). Observation of the samples under a fluorescence optical microscope with a 395–440 nm filter shows us that the fibers emit in the blue region while the emission of the smaller crystallites is red shifted (in agreement with the emission spectra obtained in the solid state, Fig. 4). We were able to separately record the emission spectra of both fibers and crystallites using a micro-spectrofluorimeter. As depicted in Fig. 4, fibers are responsible for the emission centred at ∼420 nm while the broad red-shifted emission band (green solid emission) corresponds to the solid crystallite particles. Other types of aggregates are also present, e.g. dendritic growth (Fig. S16, ESI†). It is worth noting that for such a small molecule, the formation of these kinds of millimetre sized structures is extremely unusual and unexpected. Images taken using a 510–560 nm filter only show emission of the small particles while that corresponding to the fibers disappears (Fig. S17, ESI†).
 | ||
| Fig. 3 Optical microscopy (left) and SEM (right) images of the xerogel. | ||
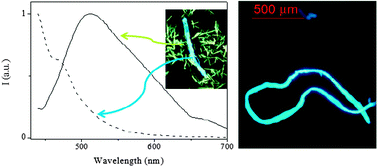 | ||
| Fig. 4 Emission spectra of the xerogel using emission microscopy spectroscopy (left, λexc = 390 nm): fibers (dashed line) and solid (solid line); optical fluorescence microscopy image of the fibers (right). | ||
Scanning electron microscopy (SEM) was used to gain insight into the structure of the gel (Fig. 3 right and Fig. S18, ESI†) and the recorded images are in agreement with previous data. Thus, there exist different kinds of structures such as long stacking derivatives, dendritic growth geometries as well as very long fibers (up to 2–3 mm). The latter correspond to those with blue emission recorded by fluorescence microscopy, while smaller and stacking crystallites should correspond to the green luminescent species. The information retrieved from microscopy has been complemented with optical interferometry to measure the 3D size of the structures. As shown in Fig. S19 (ESI†), the height of the assemblies is about 2–3.5 μm (i.e. diameters between 1–1.8 μm).
The thermal stability of the gels has been also studied both by electronic absorption and 1H-NMR spectra at different temperatures giving rise to a melting point at ca. 50 °C (see ESI†).
To conclude, we would like to point out that, as far as we know, this is the first reported luminescent gel obtained with such a simple structure, the [(PTA)Au(4-pyridylethynyl)] molecule. We have demonstrated that very small modifications of the structure, e.g. the position of the pyridyl unit, can induce unexpected and interesting rheological properties in water. All these data are supported by a wide range of different techniques (optical and fluorescence microscopy, SEM, luminescence, NMR and powder DRX). Some experiments are in progress in order to gain information about the photophysical properties of the different aggregation motifs, the gelation process and the potential applications as therapeutics and molecular recognition in water.32
The support and sponsorship provided by COST Action CM1005 is acknowledged. The authors are also grateful to the Ministerio de Ciencia e Innovación of Spain (Project CTQ2012-31335) and Fundação para a Ciência e Tecnologia of Portugal (PTDC/QUI-QUI/112597/2009). The authors also would like to thank Dr Maria João Melo from the Universidade Nova de Lisboa for the help with microspectrofluorimetry experiments and Dr Francisco Cárdenas and Dr Eurico Cabrita from the NMR Unities of the Faculties of Chemistry-Universitat de Barcelona and Universidade Nova de Lisboa, respectively, for fruitful help with some NMR experiments. The NMR spectrometers are part of the Portuguese National NMR Network (RNRMN) and are funded by Fundação para a Ciência e a Tecnologia (FCT). J. Ll. is grateful to the ICREA Academia program. R. G. thanks FCT for the post-doctoral grant (SFRH/BPD/44639/2008).
Notes and references
- J. W. Steed, Chem. Commun., 2011, 47, 1379 RSC.
- J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686 RSC.
- S.-T. Lam, G. Wang and V. W.-W. Yam, Organometallics, 2008, 27, 4545 CrossRef CAS.
- S. Banerjee, R. K. Das and U. Maitra, J. Mater. Chem., 2009, 19, 6649 RSC.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002 CrossRef CAS.
- A. Ajayaghosh, V. K. Praveen, C. Vijayakumar and S. J. George, Angew. Chem., Int. Ed., 2007, 119, 6376 CrossRef.
- K. Peng, I. Tomatsu and A. Kros, Chem. Commun., 2010, 46, 4094 RSC.
- Z. Dzolic, M. Cametti, A. Dalla Cort, L. Mandolini and M. Zinic, Chem. Commun., 2007, 3535 RSC.
- T. H. T. Hsu, J. J. Naidu, B.-J. Yang, M.-Y. Jang and I. J. B. Lin, Inorg. Chem., 2012, 51, 98 CrossRef CAS.
- C. A. Strassert, C.-H. Chien, M. D. Galvez Lopez, D. Kourkoulos, D. Hertel, K. Meerholz and L. De Cola, Angew. Chem., Int. Ed., 2011, 50, 946 CrossRef CAS.
- A. Kishimura, T. Yamashita and T. Aida, J. Am. Chem. Soc., 2005, 127, 179 CrossRef CAS.
- F. Fages, F. Vögtle and M. Žinic, Top. Curr. Chem., 2005, 256, 77 CrossRef CAS.
- K. Araki and I. Yoshikawa, Top. Curr. Chem., 2005, 256, 133 CrossRef CAS.
- D. K. Smith, Adv. Mater., 2006, 18, 2773 CrossRef CAS.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201 CrossRef CAS.
- E. Vergara, E. Cerrada, A. Casini, O. Zava, M. Laguna and P. J. Dyson, Organometallics, 2010, 29, 2596 CrossRef CAS.
- M. Ferrer, A. Gutiérrez, L. Rodríguez, O. Rossell, J. C. Lima, M. Font-Bardía and X. Solans, Eur. J. Inorg. Chem., 2008, 2899 CrossRef CAS.
- F. Mohr, S. Sanz, E. R. T. Tieknik and M. Laguna, Organometallics, 2006, 25, 3084 CrossRef CAS.
- D. Dolfen, K. Schottler, S.-M. Vaiahdi, M. A. Jakupec, B. K. Keppler, E. R. T. Tieknik and F. Mohr, J. Inorg. Biochem., 2008, 102, 2067 CrossRef CAS.
- R. Wanke, P. Smoleński, Guedes da Silva, L. M. D. R. S. Martins and A. J. L. Pombeiro, Inorg. Chem., 2008, 47, 10158 CrossRef CAS.
- M. Sternberg, C. H. Suresh and F. Mohr, Organometallics, 2010, 29, 3922 CrossRef CAS.
- Aggregation decreases the free movement of the molecule in the solution, giving rise to broader peaks in NMR.
- J. A. Foster and J. W. Steed, Angew. Chem., Int. Ed., 2010, 49, 6718 CrossRef CAS.
- K. L. Cheung, S. K. Yip and V. W.-W. Yam, J. Organomet. Chem., 2004, 689, 4451 CrossRef CAS.
- H. Y. Chao, W. Lu, Y. Q. Li, M. C. W. Chan, C. M. Che, K. K. Cheung and N. Y. Zhu, J. Am. Chem. Soc., 2002, 124, 14696 CrossRef CAS.
- V. W.-W. Yam, K. K. W. Lo and K. M. C. Wong, J. Organomet. Chem., 1999, 578, 3 CrossRef CAS.
- V. W.-W. Yam, K. L. Cheung, S. K. Yip and K. K. Cheung, J. Organomet. Chem., 2003, 681, 196 CrossRef CAS.
- H. de la Riva, M. Nieuwhuyzen, C. M. Fierro, P. R. Raithby, L. Male and M. C. Lagunas, Inorg. Chem., 2006, 45, 1418 CrossRef CAS.
- E. C. Constable, C. E. Housecroft, M. K. Kocik and J. A. Zampese, Polyhedron, 2011, 30, 2704 CrossRef CAS.
- L. Rodríguez, M. Ferrer, R. Crehuet, J. Anglada and J. C. Lima, Inorg. Chem., 2012, 51, 7636 CrossRef.
- V. K.-M. Au, W. H. Lam, W.-T. Wong and V. W.-W. Yam, Inorg. Chem., 2012, 51, 7537 CrossRef CAS.
- J. C. Lima and L. Rodríguez, Chem. Soc. Rev., 2011, 40, 5442 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, 1H-NMR and ESI-MS(+) spectra; Photophysical characterization of the aggregates and solid; Characterization of the xerogel by DRX, Optical Microscopy, Fluorescence Optical Microscopy, SEM images, Optical Interferometry. Stability experiments of the gel. See DOI: 10.1039/c2cc37262b. |
| This journal is © The Royal Society of Chemistry 2013 |