A homoleptic tetravalent cerium silylamide†
Alan R.
Crozier
a,
Andre M.
Bienfait
a,
Cäcilia
Maichle-Mössmer
b,
Karl W.
Törnroos
a and
Reiner
Anwander
*b
aDepartment of Chemistry, University of Bergen, Allégaten 41, N-5007, Bergen, Norway
bInstitut für Anorganische Chemie, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, D-72076, Tübingen, Germany. E-mail: reiner.anwander@uni-tuebingen.de; Fax: +49 7071 292436
First published on 5th November 2012
Abstract
Treatment of Ce[N(SiHMe2)2]3(thf)2 with the chlorinating agents PhICl2, Ph3CCl or C2Cl6 gave the homoleptic Ce(IV) silylamide Ce[N(SiHMe2)2]4. When performed in the absence of donating (solvent) molecules, the trivalent cluster Ce5[N(SiHMe2)2]8Cl7 was isolated.
Silylamido groups are a well-established ligand set in many s-, d- and f-block metal complexes, first and foremost the ubiquitous monovalent hexamethyldisilazide N(SiMe3)2.1 Less frequently, but increasingly employed is the tetramethyl derivative N(SiHMe2)2 featuring the Si–H moiety as a spectroscopic probe and revealing superior reactivity in sterically demanding ligand exchange reactions.2 Particularly, the trivalent rare-earth metal derivatives, Ln[N(SiHMe2)2]3(thf)x (Ln = Sc: x = 1, Ln = Y, La–Lu: x = 2), have emerged as prominent synthesis precursors according to amine and amide elimination protocols.3,4 Although only a few heteroleptic tetravalent cerium amide complexes have been reported,5–9 including Lappert's Ce[N(SiMe3)2]3Cl,7a silylamide ligands seem to provide a stabilising environment for Ce(IV) centres. Strikingly, Lappert et al. also described the X-ray structure analysis of the only homoleptic Ce(IV) amide complex, Ce(NCy2)4.10 This black bis(cyclohexyl)amide complex was obtained by air oxidation/redistribution of the corresponding Ce(III) complexes. In this report, we wish to illustrate the synthesis of the homoleptic Ce(IV) silylamide complex Ce[N(SiHMe2)2]4 as well as the isolation of the trivalent complex Ce5[N(SiHMe2)2]8Cl7.
Based on the successful synthesis of the black tetravalent (C5H5)3CeCl, utilising PhI(III)Cl2 as the oxidant for Ce(C5H5)3,11 we now treated Ce[N(SiHMe2)2]3(thf)2 (1) with 0.5 equivalents of the same chlorinating agent in THF at ambient temperature (Scheme 1, (i)). An instant reaction was indicated by a colour change from pale yellow to deep red. Removal of the solvent produced an oily residue. Upon concentrating a hexane solution of this residue and cooling to −35 °C dark red crystals were produced. DRIFT spectroscopy of these crystals revealed the presence of the bis(dimethylsilyl)amido ligand in distinct environments. This can be derived from two well-resolved peaks for the Si–H stretching vibrations at 2100 and 2010 cm−1, the latter being indicative of significant Ce(IV)–SiH β-agostic interactions in the solid state (Fig. S1, ESI†). 1H NMR spectroscopy of this compound 2, in [D6]benzene, showed a doublet at 0.34 and a septet at 6.01 ppm with a ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as expected for N(SiHMe2)2 (Fig. S2, ESI†), whereas the 29Si NMR spectrum (δ = −23.25 ppm, 1JSiH = 167.73 Hz) points to the presence of very weak secondary Ce(IV)–SiH interactions in solution.3 However, CHN elemental analysis of 2 was not in agreement with the envisaged chlorinated reaction product Ce[N(SiHMe2)2]3Cl and so an X-ray diffraction study was warranted.
:1 as expected for N(SiHMe2)2 (Fig. S2, ESI†), whereas the 29Si NMR spectrum (δ = −23.25 ppm, 1JSiH = 167.73 Hz) points to the presence of very weak secondary Ce(IV)–SiH interactions in solution.3 However, CHN elemental analysis of 2 was not in agreement with the envisaged chlorinated reaction product Ce[N(SiHMe2)2]3Cl and so an X-ray diffraction study was warranted.
![Synthesis of compounds 2 and 3 from Ce[N(SiHMe2)2]3(thf)x (1: x = 2; 1a: x = 0) and PhICl2 at ambient temperature in (i) THF and (ii) toluene.](/image/article/2013/CC/c2cc37404h/c2cc37404h-s1.gif) | ||
| Scheme 1 Synthesis of compounds 2 and 3 from Ce[N(SiHMe2)2]3(thf)x (1: x = 2; 1a: x = 0) and PhICl2 at ambient temperature in (i) THF and (ii) toluene. | ||
Surprisingly, the homoleptic compound Ce[N(SiHMe2)2]4 (2) was revealed‡ (Fig. 1). The molecular structure shows a Ce(IV) centre adopting a distorted tetrahedral geometry with N–Ce–N angles in the range from 98.35(4)° to 116.33(4)° placing 2 in the middle of known redox-stable complexes Hf[N(SiHMe2)2]4 (102°–105°)12 and U[N(SiHMe2)2]4 (99°–126°).13
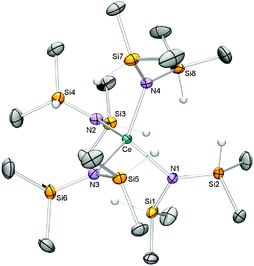 | ||
| Fig. 1 Molecular structure of 2 (ellipsoids set to 50%). All hydrogen atoms except Si–H have been omitted for clarity. Selected interatomic distances (Å) and angles (°): Ce(1)–N(1) 2.2438(11), Ce(1)–N(2) 2.2378(11), Ce(1)–N(3) 2.2488(11), Ce(1)–N(4) 2.2574(11), Ce(1)–Si(1) 3.2297(4), Ce(1)–Si(2) 3.4773(4), Ce(1)–Si(3) 3.6245(4), Ce(1)–Si(4) 3.1814(4), Ce(1)–Si(5) 3.1714(4), Ce(1)–Si(6) 3.5234(4), Ce(1)–Si(7) 3.5524(4), Ce(1)–Si(8) 3.1919(4), Ce(1)–N(1)–Si(1) 108.68(5), Ce(1)–N(1)–Si(2) 122.41(6), Ce(1)–N(2)–Si(3) 125.73(6), Ce(1)–N(2)–Si(4) 106.60(5), Ce(1)–N(3)–Si(5) 105.64(5), Ce(1)–N(3)–Si(6) 124.74(6), Ce(1)–N(4)–Si(7) 126.08(6), Ce(1)–N(4)–Si(8) 106.23(5), N(1)–Ce(1)–N(2) 113.89(4), N(1)–Ce(1)–N(3) 99.47(4), N(1)–Ce(1)–N(4) 116.33(4), N(2)–Ce(1)–N(3) 114.03(4), N(2)–Ce(1)–N(4) 98.35(4), N(3)–Ce(1)–N(4) 115.70(4). | ||
The average Ce–N bond length for 2 is 2.247 Å comparing similarly to other known tetravalent silylamide complexes (Ce[N(SiMe3)2]3Cl: 2.217 Å,7a Ce(L)[N(SiMe3)2]2Cl: 2.258 Å (L = OCMe2CH2(CNCH2CH2NDipp); Dipp = C6H3iPr2-2,6)8 and {Ce[N(SiMe3)2]2(μ-O)}2: 2.245 Å)9 as well as to Ce(NCy2)4 (2.242 Å).10 For further comparison, trivalent [Na(thf)4(Et2O)][Ce{N(SiMe3)2}4], which features the only other structurally authenticated cerium tetrakis silylamide, exhibits Ce(III)–N bond lengths in the range from 2.434(6) to 2.448(6) Å.14 The Ce(IV) ion in 2 appears to be coordinatively saturated due to the absence of donating THF in the structure despite it being used as the reaction solvent. Ce(IV)–SiH β-agostic interactions exclude THF donation (also seen for Sm(II))15 with SiH bonds from each silylamido ligand contributing to the coordination sphere. Ce–H contacts range from 2.81 Å–3.53 Å (av. 3.08 Å) and Ce–Si av. 3.170 Å.
When sounding out less labourious chlorinating reagents, we found that Ph3CCl and C2Cl6 produce compound 2 as well and in better yields (PhICl2 oxidation: crystallised yield 20%). Trityl chloride has previously been shown to quantitatively convert Ce[N(SiMe3)2]3 into Ce[N(SiMe3)2]3Cl,8 while hexachloroethane was mainly used for preparing CeCl3(thf)2 from metal powder.16 Although the Ce[N(SiHMe2)2]3(thf)2/C2Cl6 reaction, producing C2Cl4 as the major co-product, proceeds at a slower rate (crystallized yield 45%) than the Ph3CCl-based one (60% yield calculated on the basis of NMR data, cf., Fig. S3, ESI†), product separation via crystallisation is more straightforward.
Interested in the role THF played in the oxidation of 1, the reaction was repeated using donor-free starting material [Ce{N(SiHMe2)2}3]2 (1a) with toluene as the solvent (Scheme 1, (ii)). A similar colour change was observed as above, however, FTIR spectroscopy of the red residue showed a shift in the SiH peak centred at 2067 cm−1 with a significantly more detailed β-H agostic interaction shoulder. 1H NMR spectroscopy produced broader peaks indicating the likelihood of a trivalent complex. Cooling a concentrated hexane solution of the residue to −35 °C gave colourless crystals that were used in an X-ray diffraction experiment. Fig. 2 shows a representation of the molecular structure [{Ce[N(SiHMe2)2]}4{Ce[N(SiHMe2)2]2}(μ2-Cl)2(μ3-Cl)4{μ2-N(SiHMe2)2}2] (3).§. To our knowledge complex 3 is the first donor-free rare-earth metal(III) complex featuring amido and halo ligands exclusively. It is noteworthy that trivalent cerium complexes and in particular [Ce{N(SiMe3)2}2(μ2-Cl)2(thf)]2 occurred as significant reaction products during the synthesis of Ce[N(SiMe3)2]3Cl from Ce[N(SiMe3)2]3 and TeCl4.7¶
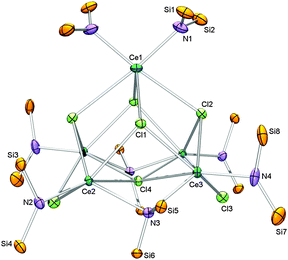 | ||
| Fig. 2 Molecular structure of 3 (ellipsoids set to 50%). All hydrogen and carbon atoms have been omitted for clarity. The molecule has a 2-fold axis ($) along space diagonal Ce(1)–Cl(4). Selected interatomic distances (Å) and angles (°): Ce(1)–N(1) 2.305(2), Ce(2)–N(2) 2.280(2), Ce(2)–N(3) 2.628(2), Ce(3)–N(3)$ 2.566(2), Ce(3)–N(4) 2.283(2), Ce(1)–Cl(1) 2.9438(4), Ce(1)–Cl(2) 3.1411(5), Ce(2)–Cl(1) 2.9476(4), Ce(2)–Cl(2) 2.8729(4), Ce(2)–Cl(3) 2.9129(4), Ce(2)–Cl(4) 2.9410(2), Ce(3)–Cl(1)$ 2.9185(4), Ce(3)–Cl(2) 2.8729(4), Ce(3)–Cl(3) 2.8483(5), Ce(3)–Cl(4) 3.0550(2), Ce(2)–Ce(3)$ 4.0708(2), Ce(1)–Si(1) 3.2284(6), Ce(2)–Si(3) 3.1570(5), Ce(2)–Si(6) 3.2412(5), Ce(3)–Si(5)$ 3.2472(5), Ce(3)–Si(8) 3.1429(6), Ce(1)–N(1)–Si(1) 106.7(1), Ce(1)–N(1)–Si(2) 117.3(1), Ce(2)–N(2)–Si(3) 103.91(8), Ce(2)–N(2)–Si(4) 128.06(8), Ce(2)–N(3)–Si(5) 115.90(7), Ce(2)–N(3)–Si(6) 93.87(6), Ce(3)–N(3)–Si(5) 96.30(7), Ce(3)–N(3)–Si(6) 121.97(7), Ce(3)–N(4)–Si(7) 120.2(1), Ce(3)–N(4)–Si(8) 103.50(9), N(1)–Ce(1)–N(1)$ 96.4(1), N(2)–Ce(2)–N(3) 108.61(5), N(3)$–Ce(3)–N(4) 115.5(6). | ||
Compound 3 is a pentanuclear Ce(III) complex with each metal centre adopting a six-coordinate arrangement in a distorted octahedral geometry. Two metal coordination environments exist, Ce(1) consists of two terminal amido groups and four (μ3) chlorine atoms. In the second Ce(2) and Ce(3) are each surrounded by a terminal and a μ2-amido group. Additionally, four chlorine atoms (1 μ2 Cl(3); 2 μ3 Cl(1 and 2); 1 μ4 Cl(4)) bind to each metal centre. The terminal Ce–N bond lengths (av. 2.285 Å) are similar to the terminal Nd–N(silylamido) distances in the neodymium-centred complex [{Nd{N(SiHMe2)2}(thf)}2(Nd{N(SiHMe2)2}2)(μ2-Cl)2(μ3-Cl)2{μ-N(SiHMe2)2}] (A, av. 2.296 Å).17
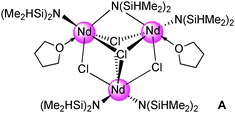
As expected the Ce–Cl bond lengths are in the order μ2 (av. 2.957 Å) < μ3 (av. 2.968 Å) < μ4 (av. 3.004 Å). Both SiH moieties belonging to the bridging bis(dimethylsilyl)amido ligands are involved in agostic interactions with short Ce–H contacts (av. 2.638 Å). The terminal amido groups exhibit longer metal–hydrogen distances with asymmetrical Ce–H contacts averaging 2.786 Å and 3.392 Å, respectively.
In summary, the bis(dimethylsilyl)amido ligand facilitates an oxidation/redistribution sequence and the formation of homoleptic Ce[N(SiHMe2)2]4via reaction of Ce[N(SiHMe2)2](thf)2 with chlorinating reagents. This is in contrast to the generation of heteroleptic complexes Ce[N(SiMe3)2]3X (X = Cl, Br) in the presence of a sterically more demanding silylamido ligand. In the absence of donating solvent molecules (here THF) reduction/redistribution reactions feature prominent side-reactions as shown for the formation of the Ce(III) silylamide-chloride complex Ce5[N(SiHMe2)2]8Cl7. Unsurprisingly, such donor-free dimethylsilylamide complexes show significant Ce(IV)–SiH β-agostic interactions in the solid state.
This work was funded by the University of Bergen (program Nanoscience@UiB) and the Meltzer Foundation. We are grateful to Prof. N. Å. Frøystein for technical assistance with the 29Si NMR spectroscopy.
Notes and references
- Metal And Metalloid Amides, ed. M. F. Lappert, P. P. Power, A. R. Sanger and R. C. Srivastava, Ellis Horwood, Chichester, 1980 Search PubMed.
- (a) W. A. Herrmann, R. Anwander, F. C. Munck, W. Scherer, V. Dufaud, N. W. Huber and G. R. J. Artus, Z. Naturforsch., B: Chem. Sci., 1994, 49, 1789–1797 CAS; (b) R. Anwander, O. Runte, J. Eppinger, G. Gerstberger, E. Herdtweck and M. Spiegler, J. Chem. Soc., Dalton Trans., 1998, 847–858 RSC; (c) W. A. Herrmann, F. C. Munck, G. R. J. Artus, O. Runte and R. Anwander, Organometallics, 1997, 16, 682–688 CrossRef CAS; (d) C. Meermann, G. Gerstberger, M. Spiegler, K. W. Törnroos and R. Anwander, Eur. J. Inorg. Chem., 2008, 2014–2023 CrossRef CAS.
- For initial silylamine elimination reactions, see: (a) O. Runte, T. Priermeier and R. Anwander, Chem. Commun., 1996, 1385–1386 RSC; (b) J. Eppinger, M. Spiegler, W. Hieringer, W. A. Herrmann and R. Anwander, J. Am. Chem. Soc., 2000, 122, 3080–3096 CrossRef CAS.
- For initial silylamide elimination reactions, see: (a) I. Nagl, M. Widenmeyer, E. Herdtweck, G. Raudaschl-Sieber and R. Anwander, Microporous Mesoporous Mater., 2001, 44–45, 311–319 CrossRef CAS; (b) M. G. Klimpel, R. Anwander, M. Tafipolsky and W. Scherer, Organometallics, 2001, 20, 3983–3992 CrossRef CAS.
- A. F. England, The Thorium Benzyne and the Cerium(IV)–Nitrogen Bond, PhD thesis, Massachusetts Institute of Technology, 1995 Search PubMed.
- C. Morton, N. W. Alcock, M. R. Lees, I. J. Munslow, C. J. Sanders and P. Scott, J. Am. Chem. Soc., 1999, 121, 11255–11256 CrossRef CAS.
- (a) O. Eisenstein, P. B. Hitchcock, A. G. Hulkes, M. F. Lappert and L. Maron, Chem. Commun., 2001, 1560–1561 RSC; (b) P. B. Hitchcock, A. G. Hulkes and M. F. Lappert, Inorg. Chem., 2004, 43, 1031–1038 CrossRef CAS.
- P. L. Arnold, Z. R. Turner, N. Kaltsoyannis, P. Pelekanaki, R. M. Bellabarba and R. P. Tooze, Chem.–Eur. J., 2010, 16, 9623–9629 CrossRef CAS.
- M. P. Coles, P. B. Hitchcock, A. V. Khvostov, M. F. Lappert, Z. Li and A. V. Protchenko, Dalton Trans., 2010, 39, 6780–6788 RSC.
- P. B. Hitchcock, M. F. Lappert and A. V. Protchenko, Chem. Commun., 2006, 3546–3548 RSC.
- P. Dröse, A. R. Crozier, S. Lashkari, J. Gottfriedsen, S. Blaurock, C. G. Hrib, C. Maichle-Mössmer, C. Schädle, R. Anwander and F. T. Edelmann, J. Am. Chem. Soc., 2010, 132, 14046–14047 CrossRef.
- E. d. L. Jiménez, S. Javed and D. M. Hoffman, Inorg. Chim. Acta, 2009, 362, 385–388 CrossRef.
- S. M. Mansell, B. F. Perandones and P. L. Arnold, J. Organomet. Chem., 2010, 695, 2814–2821 CrossRef CAS.
- W. J. Evans, D. S. Lee, D. B. Rego, J. M. Perotti, S. A. Kozimor, E. K. Moore and J. W. Ziller, J Am. Chem. Soc., 2004, 126, 14574–14582 CrossRef CAS.
- I. Nagl, W. Scherer, M. Tafipolsky and R. Anwander, Eur. J. Inorg. Chem., 1999, 1405–1407 CrossRef CAS.
- (a) G. B. Deacon, T. Feng, S. Nickel, B. W. Skelton and A. H. White, J. Chem. Soc., Chem. Commun., 1993, 1328–1329 RSC; (b) G. B. Deacon, T. Feng, P. C. Junk, B. W. Skelton, A. N. Sobolev and A. H. White, Aust. J. Chem., 1998, 51, 75–89 CrossRef CAS.
- C. Meermann, K. W. Törnroos, W. Nerdal and R. Anwander, Angew. Chem., Int. Ed., 2007, 46, 6508–6513 CrossRef CAS.
- A. Baranowski, D. Plachta, L. Skulski and M. Klimaszewska, J. Chem. Res. (S), 2000, 435–437 CrossRef CAS.
- H. F. Yuen and T. J. Marks, Organometallics, 2008, 27, 155–158 CrossRef CAS.
- Bruker, SMART, SAINT, SADABS and XPREP. Area detector control and data integration and reduction software, Bruker Analytical X-ray Instruments Inc., Madison, WI, USA, 2008 Search PubMed.
- G. M. Sheldrick, Acta. Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, NMR and FTIR data. CCDC 904595 (2) and 904596 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2cc37404h |
| ‡ Crystal data for 2: C16H56CeN4Si8, M = 669.49, monoclinic, a = 18.3037(9) Å, b = 11.1158(5) Å, c = 18.8563(9) Å, β = 107.559(1)°, V = 3657.8(3) Å3, T = 103(2) K, space group P21/c, Z = 4, 65647 reflections measured, 12241 independent reflections (Rint = 0.0443). R1 values = 0.0243 (I > 2σ(I)), wR(F2) = 0.0662 (all data). |
| § Crystal data for 3: C32H112Ce5Cl7N8Si16, M = 2007.49, monoclinic, a = 17.2810(6) Å, b = 25.5225(9) Å, c = 19.8095(7) Å, β = 100.2028(4)°, V = 8598.9(5) Å3, T = 100(2) K, space group C2/c, Z = 4, 71792 reflections measured, 12787 independent reflections (Rint = 0.0292). R1 values = 0.0210 (I > 2σ(I)). wR(F2) values = 0.0549 (all data). Data collection was done on a Bruker AXS Ultra TXS rotating anode CCD instrument using an Oxford Cryosystems series 700 N2 cryostat. Data collection and reduction were done using the Bruker AXS APEX2 suite programs.20 Structure solution and refinement were done using programs SHELXS and SHELXL, respectively.21 CCDC 904595 (2) and 904596 (3). |
| ¶ Synthesis of Ce[N(SiHMe2)2]42: a solution of 1 (0.266 g, 0.390 mmol) in THF (10 ml) was added to a solution of PhICl218 (0.053 g, 0.193 mmol) in THF (2 ml) in one portion in the dark. A colour change from yellow to red was observed immediately. This solution was allowed to stir for 1 h. The volatiles were removed via vacuum leaving an oily solid. To this, hexane (2 ml) was added and upon subsequent cooling to −35 °C dark red crystals suitable for X-ray diffraction were produced (yield 20%). In pure form, 2 can be sublimed at 80 °C/1.2 × 10−4 mbar. IR (DRIFT): vmax = 2953 m, 2900 w, 2102 m, 2009 m, 1942 vw sh, 1419 vw, 1248 s, 1020 s, 939 s, 889 vs, 841 s, 795 s, 768 m, 746 vw, 679 m, 631 w, 586 m, 409 w cm−1. 1H NMR (400 MHz, [D6]benzene, 20 °C): σ = 0.34 (d, J = 7.4 Hz, 48H, Si(H)Me2), 6.01 (septet, J = 2.9 Hz, 8H, Si(Me2)H). Anal. Calc. for C16H56CeN4Si8: C, 28.71; H, 8.43; N, 8.37%. Found: C, 28.30; H, 7.88; N, 6.43%. Alternative synthesis of Ce[N(SiHMe2)2]42: a solution of trityl chloride (0.099 g, 0.35 mmol) in toluene (3 ml) was slowly added to a solution of 1 (0.241 g, 0.35 mmol) in toluene (3 ml). A colour change from yellow to red was observed immediately. This solution was allowed to stir for 0.5 h. The volatiles were removed via vacuum leaving a dark red solid (0.327 g). To this, toluene (0.5 ml) was added and upon subsequent cooling to −40 °C dark red crystals of 2 suitable for X-ray diffraction were produced (along with colourless crystals of Gomberg's dimer). Yield calculated from NMR data (based on the Ce-content): 60%. Second alternative synthesis of Ce[N(SiHMe2)2]42: a solution of hexachloroethane (0.045 g, 0.19 mmol) in toluene (2 ml) was slowly added to a solution of 1 (0.257 g, 0.38 mmol) in toluene (5 ml). A colour change from yellow via orange and bright red to dark red occurred within 18 h, after which the volatiles were removed via vacuum leaving dark red sticky crystals (0.240 g). To this, toluene (0.5 ml) was added and upon subsequent cooling to −40 °C dark red crystals of 2 suitable for X-ray diffraction were produced (yield of two crops combined: 0.116 g, 0.17 mmol, 45%). Synthesis of Ce[N(SiHMe2)2]8Cl73: a solution of 1a19 (0.488 g, 0.910 mmol) in toluene (10 ml) was added to a solution of PhICl2 (0.125 g, 0.454 mmol) in toluene (2 ml) in one portion in the dark. A colour change from yellow to red was observed immediately. This solution was allowed to stir for 1 h. The volatiles were removed via vacuum leaving an oily solid. To this, hexane (2 ml) was added and upon subsequent cooling to −35 °C colourless crystals suitable for X-ray diffraction were produced (yield 38%). IR (DRIFT): vmax = 2952 m, 2898 w, 2123 m sh, 2066 m, 2008 m sh, 1959 m sh, 1910 m sh, 1418 w, 1296 w sh, 1254 m, 1057 m sh, 1024 m sh, 1008 m sh, 959 m, 893 s, 837 m, 795 m, 769 m, 687 m, 591 m, 565 w, 410 m cm−1. 1H NMR (400 MHz, [D6]benzene, 20 °C): δ = 0.12, 2.11, 4.71. Anal. Calc. for C32H112Ce5Cl7N8Si16: C, 19.15; H, 5.63; N, 5.59%. Found: C, 17.53; H, 4.44; N, 4.57%. |
| This journal is © The Royal Society of Chemistry 2013 |