Vapor-phase crystallization route to oxidized Cu foils in air as anode materials for lithium-ion batteries†
Kunfeng
Chen
ab,
Shuyan
Song
b and
Dongfeng
Xue
*ab
aSchool of Chemical Engineering, Dalian University of Technology, Dalian, 116024, China
bState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Renmin Street No. 5625, Changchun, 130022, China. E-mail: dongfeng@ciac.jl.cn
First published on 11th October 2012
Abstract
This paper describes a facile vapor-phase strategy for the crystallization of copper oxide on Cu foil substrate in air and the further fabrication of integrated CuxO–Cu electrodes with CuxO readily on the Cu foil current collector, without using conductive carbon and binder. The oxidation behavior of Cu foil in air was identified by XRD, SEM and electrochemical measurements. In the whole temperature range from 200 to 800 °C, Cu2O is the dominant product of the as-oxidized Cu, and CuO is grown on the surface of the Cu2O layer. The phase composition and thickness of oxidation products can be controlled by the reaction temperature and time. Both thermodynamic data and crystallography of the as-oxidized Cu products indicate that the Cu/Cu2O/CuO tri-layered structures can be conveniently fabricated according to the oxidation sequence Cu → Cu2O → CuO. The integrated CuxO–Cu electrodes can show better cycling stability and higher capability than those CuxO–C blend electrodes. The present work provides a new avenue for the large-scale configuration of integrated metal oxide materials as anode materials for lithium-ion batteries.
1. Introduction
Oxide materials have attracted the most intense technological interest, such as catalysis, sensing, biomedicine, solar cells and lithium-ion batteries, because they exhibit an ever growing range of structural motifs, and are the key components of many hybrids and composites.1–6 3d transition metal oxides with large rechargeable capacities are potential candidates for the anode materials of lithium-ion batteries.3,4,7,8 Among transition metal oxides, cuprous oxide (Cu2O) and cupric oxide (CuO) are undoubtedly an attractive choice, since they are abundant, environmentally benign and inexpensive in comparison to other investigated systems.8–11 However, the pure CuO and Cu2O electrodes suffer from poor cyclability and low capacity, which is mainly caused by their low conductivity and by the large volume change during the charge–discharge cycles.12,13 In an attempt to solve this problem, powdered CuxO were blended with conductive carbon–polymer binders to coat the metal collector.7,13 The blends often suffered a macro scale phase separation of the CuxO materials, which caused the poor cyclability.13 In addition, the nanocomposites with carbon nanotubes or graphenes have been investigated to attain homogeneous mixing and stable cycling performance.13,14 For example, the CuO–CNT nanocomposite in which the mesoporous CuO particles were threaded by CNTs were designed to significantly improve their cycling performance and the rate capabilities in lithium-ion batteries, owing to the synergic effect of the morphological stability of the CuO particles and high conductivity by CNTs.13 Although improved electrochemical performances have been reported, the charge transfer between CuxO and current collector is still limited.7 The synthesis of anodes with CuxO directly connected to the Cu foil can enhance the conductivity since the intimate contact with the current collector facilitates electron transfer processes, and avoids the use of binders and conductive carbon.7,15 Recently, hierarchically mesoporous nanosheet-assembled gear-like pillar array CuO directly grown at the Cu foil show excellent cycling stability and high-rate capability.15 The CuxO–TiO2 (x = 1, 2) thin film lithium batteries synthesized on polycrystalline Ti substrates by chemical vapor deposition (CVD) approach also exhibit very attractive high-rate capability and good stability.7 Vapor-phase oxidation of copper substrate is a facile and convenient method for the synthesis of copper oxide, because it includes only two starting materials, metal Cu and oxygen (air). However, the use of vapor-phase thermal oxidation crystallized CuxO on Cu foils as anode materials for lithium-ion batteries is not reported.Here, we crystallized copper oxide on Cu foil by a facile thermal oxidation of metal Cu in air at different temperatures. Through the temperature range, Cu2O was first formed by direct oxidation of Cu. Then, Cu atoms diffuse through the Cu2O layer and react with ambient oxygen, this results in the formation of CuO on the surface. Therefore, Cu/Cu2O/CuO tri-layered structures were fabricated. Besides enabling us to avoid the use of binders and conductivity carbon, this approach resulted in an optimal adhesion between CuxO and Cu foil, which improves the cycling stability and capability of CuxO–Cu electrodes. Notably, the vapor phase strategy proposed herein enabled a simultaneous tailoring of the system phase composition and thickness of the oxide layer by controlling the reaction temperature and time. This work is expected to open a new, simple guidance for the general synthesis of Cu/Cu2O/CuO tri-layered structures according to the intrinsic oxidation sequence, Cu → Cu2O → CuO. Although representing a fundamental study, the results obtained in the present work can provide a valuable avenue in view of further optimization of standard batteries.
2. Experimental
2.1. Synthesis and characterization
Prior to the synthesis, high-purity copper substrates (99.99% purity) were carefully cleaned with absolute alcohol and deionized water, respectively, in an ultrasound bath to remove surface impurities. In a typical procedure, the Cu foil was placed in an alumina boat and heated to the set-point temperature (at ambient pressure) in a tube furnace. The Cu sample was heated to the desired temperature at 5 °C min−1 in air. After the Cu sample was oxidized for different temperatures and durations, it was cooled down to room temperature. When the heating temperature was above 400 °C, the oxide layer was easy peeled from Cu substrates. Surface morphology and chemical composition of the oxidized samples are examined using a Field-emission scanning electron microscope (FESEM, Hitachi-S4800) and Powder X-ray diffractometer (XRD, Rigaku-D/max 2500 V).2.2. Electrochemical measurements
Two kinds of working electrodes were prepared: first, CuxO grown on the Cu foil was used directly as the working electrode without binders and conductivity agents. Second, CuxO–C blend electrodes were prepared from a mixture of sample, acetylene black, and polyvinylidene fluoride (PVDF) in a weight ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10. The mixtures were slurried with N-methyl-2-pyrrolidone, pasted onto the copper foils, and then cut into discs. Lithium was used for the counter electrodes and reference electrodes, Celgard 2400 were used as separators, and the electrolyte was 1 M LiPF6 in ethylene carbonate/dimethyl carbonate/diethyl carbonate (EC/DMC/DEC, 1:1:1 vol%). A galvanostatic cycling test of the assembled half-cells was conducted on a LAND CT2001A system (Wuhan Landian) at a discharge–charge rate of 100 mA g−1 and in the voltage range of 0.01–3.0 V (vs. Li+/Li).
:10:10. The mixtures were slurried with N-methyl-2-pyrrolidone, pasted onto the copper foils, and then cut into discs. Lithium was used for the counter electrodes and reference electrodes, Celgard 2400 were used as separators, and the electrolyte was 1 M LiPF6 in ethylene carbonate/dimethyl carbonate/diethyl carbonate (EC/DMC/DEC, 1:1:1 vol%). A galvanostatic cycling test of the assembled half-cells was conducted on a LAND CT2001A system (Wuhan Landian) at a discharge–charge rate of 100 mA g−1 and in the voltage range of 0.01–3.0 V (vs. Li+/Li).
3. Results and discussion
When Cu foil is heated in air, both Cu2O and CuO can be formed. In many cases, the major product is Cu2O, and CuO is formed slowly only through a second step of oxidation.16 The reactions involved in the entire synthesis can be summarized as the following:| 4Cu + O2 → 2Cu2O | (1) |
| 2Cu2O + O2 → 4CuO | (2) |
Fig. 1a shows the oxidation processes of Cu foil heated in air. In the typical experiment, Cu foil was heated in air at temperatures from 200 to 800 °C for different oxidation times. Cu2O was first formed at the interface of Cu foil and air, and CuO was formed at the top surface of the Cu2O layer. The transformation from Cu to Cu2O is more facile than that from Cu to CuO based on their crystal structures. It notes that Cu and Cu2O share a similar cubic structure, Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, a = 3.615 Å for Cu and Pnm, a = 4.267 Å for Cu2O, while CuO has a low-symmetry monoclinic structure (C2/c, a = 4.684 Å, b = 3.423 Å, c = 5.129 Å, β =99.55°).17–19 The crystal structure of Cu2O is cuprite, in which oxygen resides in the tetrahedral sites (1/4, 1/4, 1/4) and (3/4, 3/4, 3/4) of fcc (face-cubic centered) Cu lattice (Fig. 1b). The oxidation can be qualitatively interpreted as an oxygen diffusion and lattice expansion process, where Cu2O can be formed by the diffusion of the O atom into the tetrahedral sites of metal Cu (Fig. 1b). However, the formation of monoclinic CuO from fcc Cu is required for atom rearrangement and lattice/unit cell reconstruction. Therefore, Cu2O is initially the main product formed by Cu oxidation in air or oxygen atmosphere, which have been proved by many studies.16,20–24 Recently, it was proved that the resulting Cu–O tetrahedrons along the domain boundary at the onset of the bulk oxidation of Cu(100), strikingly resemble that of the bulk oxide phase of Cu2O.20 Namely, the Cu2O is formed first as a major product, and subsequently CuO is formed with Cu2O serving as a precursor. Thus, the oxidation sequence is Cu → Cu2O → CuO. When Cu foil is heated at high temperature, Cu2O layers are formed on the Cu foil which act as an interface between the Cu and atmospheric oxygen. The outward diffusion of copper atoms is faster compared to the inward diffusion of oxygen.21 Cu atoms diffuse through the Cu2O layer and react with ambient oxygen, this results in the formation of CuO on the surface (Fig. 1a). It is reported that the rate-determining step is concluded to be the outward diffusion of copper atoms in Cu2O in the oxidation of Cu.22
m, a = 3.615 Å for Cu and Pnm, a = 4.267 Å for Cu2O, while CuO has a low-symmetry monoclinic structure (C2/c, a = 4.684 Å, b = 3.423 Å, c = 5.129 Å, β =99.55°).17–19 The crystal structure of Cu2O is cuprite, in which oxygen resides in the tetrahedral sites (1/4, 1/4, 1/4) and (3/4, 3/4, 3/4) of fcc (face-cubic centered) Cu lattice (Fig. 1b). The oxidation can be qualitatively interpreted as an oxygen diffusion and lattice expansion process, where Cu2O can be formed by the diffusion of the O atom into the tetrahedral sites of metal Cu (Fig. 1b). However, the formation of monoclinic CuO from fcc Cu is required for atom rearrangement and lattice/unit cell reconstruction. Therefore, Cu2O is initially the main product formed by Cu oxidation in air or oxygen atmosphere, which have been proved by many studies.16,20–24 Recently, it was proved that the resulting Cu–O tetrahedrons along the domain boundary at the onset of the bulk oxidation of Cu(100), strikingly resemble that of the bulk oxide phase of Cu2O.20 Namely, the Cu2O is formed first as a major product, and subsequently CuO is formed with Cu2O serving as a precursor. Thus, the oxidation sequence is Cu → Cu2O → CuO. When Cu foil is heated at high temperature, Cu2O layers are formed on the Cu foil which act as an interface between the Cu and atmospheric oxygen. The outward diffusion of copper atoms is faster compared to the inward diffusion of oxygen.21 Cu atoms diffuse through the Cu2O layer and react with ambient oxygen, this results in the formation of CuO on the surface (Fig. 1a). It is reported that the rate-determining step is concluded to be the outward diffusion of copper atoms in Cu2O in the oxidation of Cu.22
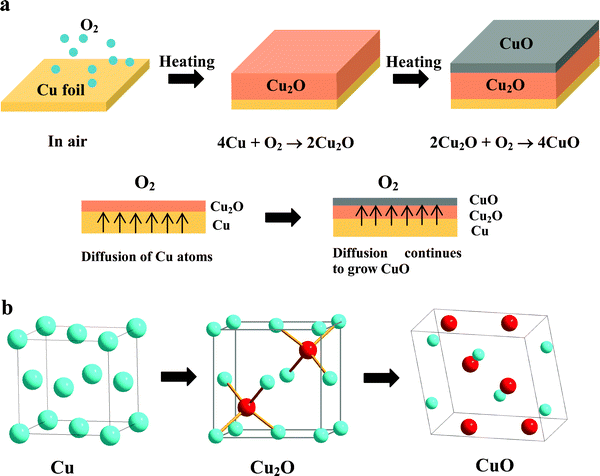 | ||
| Fig. 1 (a) Growth mechanism for copper oxides. (b) Comparison of the fcc structure of Cu, the cubic structure of Cu2O and the monoclinic structure of CuO. Oxygen atoms have to reside in the tetrahedral sites of fcc Cu to form the Cu2O structure. | ||
This speculation was confirmed by the XRD patterns shown in Fig. 2, which were taken from CuxO–Cu foil heated at 200 and 300 °C (Fig. 2a and Fig. S1, ESI†), and the separated oxide layer from Cu foil at above 400 °C (Fig. 2c). XRD patterns of these samples show that all of the peaks match well with Bragg reflections of the standard Cu2O (JCPDS 5-667) and CuO (JCPDS 5-661) as shown in Fig. 2 except the Cu substrate (JCPDS 3-1005). As the temperature dropped below 300 °C and the heating time was less than 3 h, only the Cu2O phase was present. The peak intensities of Cu2O increased with the temperature increase from 200 to 300 °C. When the oxidation temperature was 200 °C, only a very small peak of Cu2O (111) was detected (Fig. 2a). CuO was formed after the reaction at 300 °C for 6 h (Fig. S1, ESI†). As the temperature was increased to 700 °C, the peak intensities of Cu2O were significantly increased, while CuO peaks showed little increase (Fig. 2c). It is reported that for oxidation of Cu under ambient oxygen pressures at which only Cu2O is thermodynamically stable, a single Cu2O layer is formed. At oxygen pressures above the dissociation pressure of CuO, a CuO layer will be formed on the Cu2O layer.22 At higher temperatures and longer reaction durations, CuO can reach its dissociation pressure and the crystallization of CuO can be achieved.
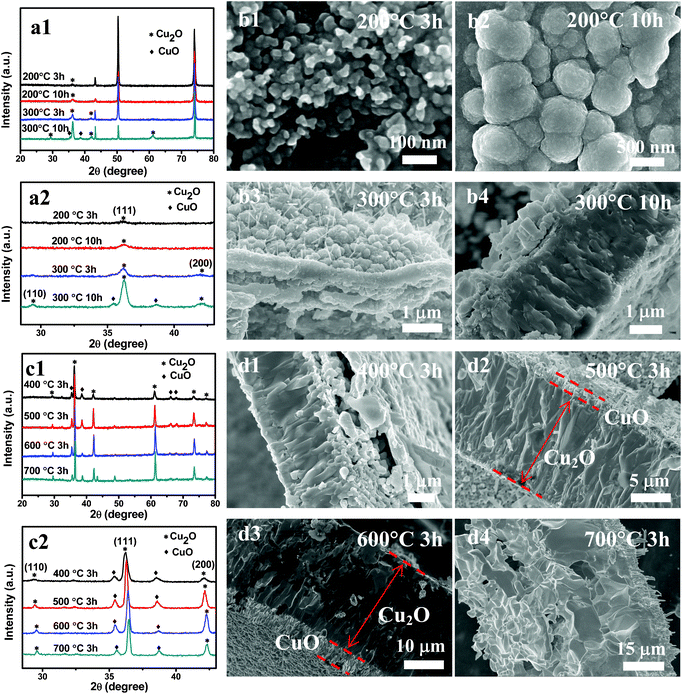 | ||
| Fig. 2 XRD patterns and SEM images of the samples synthesized at different temperatures and reaction times (indicated in the graphs). (a) Samples included both oxide layer and copper substrates. (c) Samples were only the oxide layer separated from the Cu foil. (b and d) Cross-sectional SEM images show the Cu2O and CuO layers. | ||
When the temperature was 200 °C, the Cu foil turned gold-yellow in color, showing the presence of a thin Cu2O film, which was in accordance with XRD patterns (Fig. 2). Optically the surface of the Cu substrate turned black in color after the oxidation treatment at above 300 °C, suggesting that the surface oxide is CuO, which is known to be black in color. However, XRD patterns show the majority of oxidation products are Cu2O, with the presence of a small amount of CuO. We believe that a thin CuO layer was formed at the surface of the Cu2O layer. After grinding the oxide layer separate from the Cu foil, yellow powder (Cu2O) was present instead of black powder, which further proved a thin layer of CuO was formed on top of the Cu2O layer (Fig. 3). It is believed that the X-ray patterns, coupled with observed color changes during the reaction, provide substantial evidence for the oxidation sequence, Cu → Cu2O → CuO, and Cu2O is the major product and CuO is formed on the surface of Cu2O.
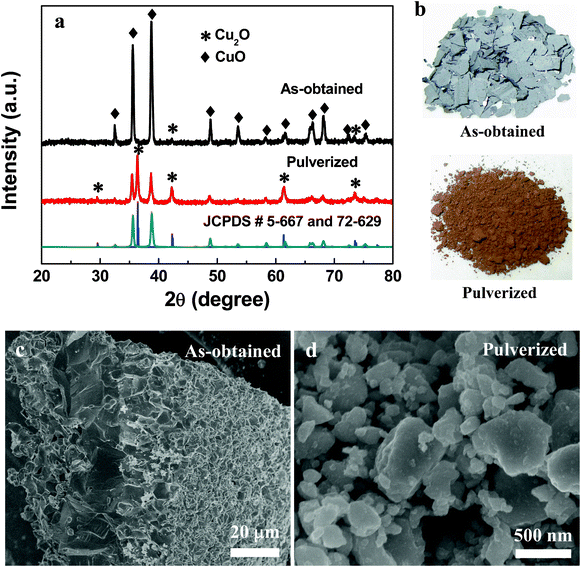 | ||
| Fig. 3 XRD patterns (a), photographs (b) and SEM (c, d) of as-obtained and pulverized sample obtained at 800 °C for 2 h. | ||
In the growth of bulk oxide during metal oxidation, the relief of stresses may occur by fracture in the oxide and/or in the underlying metals, or by separation of the oxide–metal interface.25 In addition, there exists a large volume (∼60%) increase accompanied with the conversion of Cu to Cu2O. When temperatures were at above 400 °C, copper oxide was easy peeled from the Cu foil owing to geometrically induced stresses that result from volume change. The microstructure of oxide is analyzed by SEM. Fig. 2 shows cross-sectional morphologies of the oxide layer, from which a two-layered structure can be identified: a thick bottom layer that lies directly above the Cu substrate and a thin top layer. According to XRD and the color change of products, the thick bottom layer was the Cu2O phase, while the thin top layer was the CuO phase. The Cu2O layer is composed of coarse columnar grains perpendicular to the Cu base whereas the CuO layer consists of much finer grains. The Cu2O/CuO two-layered structures exist in all samples obtained at different temperatures, showing the same oxidation route from Cu to Cu2O, and finally to CuO (Fig. 2).
The diffraction peaks from Cu disappeared with the reaction at 800 °C for 2 h (Fig. 3a). This finding testifies that the complete oxidation of Cu into copper oxide can be realized at temperatures of 800 °C. One intriguing phenomenon was that it shows different XRD patterns after grinding the same sample obtained at 800 °C (Fig. 3a). The as-obtained samples show black color and CuO is the main phase with a little amount of Cu2O (Fig. 3a and b). However, the Cu2O phase is the major product in the pulverized sample with the appearance of yellow color (Fig. 3a and b). Fig. 3c and d show that the as-obtained product mainly includes large layered bulk, while nanoparticles are the main form of the pulverized product. The results further confirm the CuO layer was grown on the surface of the thick Cu2O layer. After grinding, the Cu2O was exposed and XRD patterns show the Cu2O peaks.
The reaction from Cu2O to CuO occurring at high temperature, can be explained according to thermodynamic data. Since the change in entropy (ΔS) for reaction 2 has a negative sign, the change in free energy (ΔG) for this reaction will change sign (from negative to positive) when the temperature is sufficiently high.16 On the basis of the standard thermodynamic data from the Handbook, this transition temperature was estimated to be around 964 °C.16 This number agreed reasonably well with the temperature (800 °C) observed in the present study. Cu2O is the stable phase at temperatures of <800 °C and the occurrence of reaction 1 is energy preferential. Coupled with crystal structure, thermodynamic data and experimental results, synthesis of the Cu/Cu2O/CuO tri-layered structure can be easily achieved by the vapor strategy of heating Cu in air.
Fig. 4 shows the change in thickness of the oxide layer upon oxidation temperature. The thickness of Cu2O is increased linearly upon temperature, while the thickness of CuO shows little increase (Fig. 4). At higher reaction temperatures, the diffusion rates of both the O2 molecules and the Cu atoms are increased, according to Arrhenius-type equation, D = D0e−Q/RT, where D is the diffusivity, D0 is a proportionality constant independent of temperature, Q is the activation energy of diffusion species, and R is the molar gas constant.25 More oxygen molecules and the Cu atoms can diffuse to the surface and conduct the oxidation reaction at higher temperature. Thus, the thickness of both Cu2O and CuO increases with increase of temperature. After the formation of Cu2O on the Cu substrate, an interfacial strain field was formed owing to the large lattice mismatch at the Cu2O–Cu interface, which can hinder the interfacial diffusion of oxygen. In addition, the thermal expansion coefficient of Cu2O (1.9 × 10−6 K−1) is much smaller than that of Cu (17 × 10−6 K−1).25 With the increase of oxidation temperature, the lattice mismatch is reduced and the interfacial strain is decreased, which permits oxygen diffusion to occur more rapidly. Thus the enhanced kinetics of oxygen interfacial diffusion contributes significantly to thicken the copper oxide layers. The oxidation of copper is expected as a function of temperature and time. The thickness of oxide can be calculated by using the following equation:26
| doxide(t) = Aexp(−Q/RT) × t1/2 + d0 | (3) |
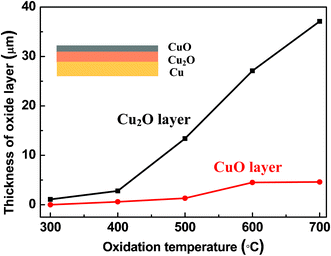 | ||
| Fig. 4 Thickness of oxide layers upon different oxidation temperatures. Inset shows Cu/Cu2O/CuO tri-layered structure. The reaction time was set as 3 h. | ||
To confirm the oxidation process, the Cu substrates oxidized at low temperature (200 and 300 °C) were used to observe their initial growth morphology. As illustrated in Fig. 2b, the oxide surface consists of small sized grains (∼30 nm) for 3 h reaction and large size of grains (∼400–600 nm) for 10 h reaction. It has been demonstrated by previous researchers that Cu2O nanoislands first nucleate at the initial oxidation stage, and then the islands grow and coalescence into larger ones.25 This process can be significantly influenced by the oxidation temperature. High temperature can make more Cu diffuse to the surface and conduct the oxidation reaction, which make the Cu2O grow faster and the formation of thicker Cu2O layers including large coarse columnar grains. At low temperature, lattice diffusion is slow because of the contribution of the grain boundary diffusion, which results in a thin oxide layer.22 Surface morphologies of oxide are shown in Fig. 5. Owing to the existence of the intrinsic growth stress, Cu2O and CuO layers were separated (Fig. 2 and 5a and b). The interface of Cu2O and CuO layers includes many porous grains (Fig. 5d). It is shown that the volume change associated with the solid-state transformation at the CuO–Cu2O interface produces compressive stresses. The surface of the CuO layer consists of considerably large solid grains compared to small grains on the top surface of the Cu2O layer (Fig. 5b). Small amounts of CuO nanorods also formed on the surface of the CuO layer with a temperature window between 300 and 800 °C (Fig. 5e and f). Recently, CuO nanorods have been synthesized by heating copper substrates in air or oxygen gas.16,24,27 The growth continues in the presence of oxygen, which results in the promotion of an outward diffusion of copper atoms, thus leading to the formation of CuO nanorods.21
 | ||
| Fig. 5 (a–d) SEM micrographs of copper oxide layers oxidized at 400 °C for 3 h. (e, f) Nanorods were grown from the upper surface layer of the CuO layer obtained at 500 °C for 3 h. The inset is a schematic illustration of the interface of the Cu2O and CuO layer. | ||
To test the applicability of the present systems as anodes in thin film batteries, their properties with respect to Li insertion–extraction were investigated. The CuxO–Cu thin film, which was synthesized at 200 and 300 °C, was directly used in electrodes. Fig. 6a and Fig. S2, ESI,† show the galvanostatic discharge–charge curves of CuxO–Cu thin film electrodes at a rate of 100 mA g−1. The first discharge and charge capacities were 703.2 and 463 mA h g−1, respectively. The initial irreversible capacity loss might be assigned to interfacial lithium-storage, the formation of a solid electrolyte interface (SEI) layer and the electrolyte decomposition.7,15,28 For comparison, separated CuxO from Cu foil, obtained with the temperatures higher than 400 °C, were blended with carbon and binder as electrodes (Fig. S3, ESI†). Fig. 6b and Fig. S4–S7, ESI,† show the discharge–charge curves of all electrodes. The open circuit voltage (∼2.8 V) decreased sharply to reach the discharge plateau in the range of 1.0–1.4 V (vs. Li+/Li) at the first cycle; and the discharge voltage plateau became more and more sloping with the increase of cycle number.12,15 CuxO–Cu thin film electrodes show higher discharge and charge capacities than CuxO–C blend electrodes between the first and 50th cycles.
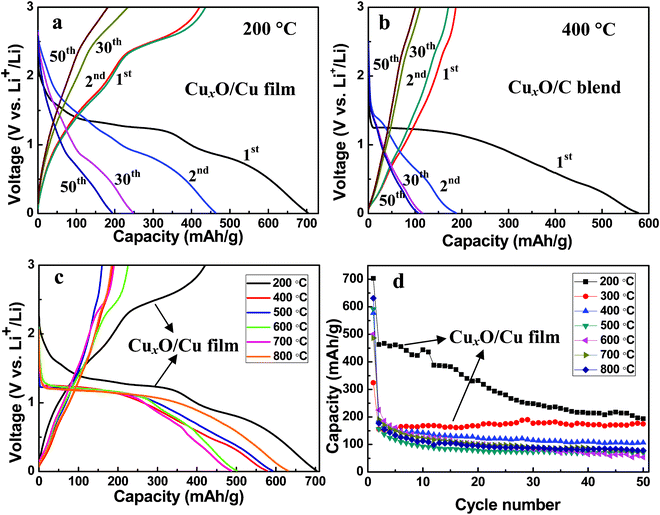 | ||
| Fig. 6 Electrochemical properties of the CuxO–Cu thin film electrode and the CuxO–C blend electrode. (a) Voltage profiles of the CuxO–Cu thin film electrode. (b) Voltage profiles of the CuxO–C blend electrode. (c) The initial discharge–charge curves and (d) cycling performances of the electrodes made of copper oxide at a rate of 100 mA g−1. | ||
Upon discharging, copper oxide was transformed into nanometric Cu particles as following:7
| Cu2O + 2Li+ + 2e− ↔ Li2O + 2Cu | (4) |
| CuO + 2Li+ + 2e− ↔ Li2O + Cu | (5) |
It is reported that the initial charge–discharge curves of the CuO anode have three obvious sloping voltage ranges, 2.3–1.3, 1.3–1.0, and 1.0–0.01 V versus Li+/Li. Fig. 6a–c only show two sloping voltage ranges, 1.4–1.0, and 1.0–0.01 V versus Li+/Li, which can be interpreted as Cu2O decomposition into Cu and Li2O, and the growth of the organic layers of the electrolytes, as discussed in the literature.4 Therefore, the discharge–charge curves also prove that Cu2O is the major oxidation product and the amount of CuO in the oxidation products is small. Thus, the charge–discharge curves did not reflect the character of CuO.
Fig. 6c show the first discharge and charge curves of electrodes. The initial discharge capacities are 703.2, 577.3, 591.7, 501.0, 487.1, 631.0 mA h g−1 for 200, 400, 500, 600, 700, and 800 °C, respectively. The better electrochemical performance of CuxO–Cu thin film electrodes is ascribed to the intimate contact with Cu current collector. The large first discharge capacity can be related to the formation of a SEI layer.7,15,28 The cycle performances of these oxide electrodes obtained at different oxidation temperatures are shown in Fig. 6d and specific discharge capacities of copper oxide electrodes are shown in Table 1. The electrochemical performances of the separated oxidation products show the comparable discharge–charge capacities and cyclic performances. Therefore, the microstructure and morphology of these separated oxidation products have no clear influence on their electrochemical performances, which is due to the fabricating method of CuxO–C blend electrodes via grinding active materials. After a few cycles, the CuxO–C blend electrode showed a significant capacity decrease, while the CuxO–Cu thin film electrode showed a stable cycling performance during 50 cycles (Fig. 6d). Normally, the significant capacity fading of CuxO–C blend electrodes is caused by the aggregation of the CuxO after repeated cycles, resulting in the reduction of electronic conductivity of CuxO.13 The integrated CuxO–Cu electrodes can facilitate electron transfer between active materials and current collectors.15,28 Therefore, such an integration infrastructure can show better cycling stability and higher capability than the CuxO–C blend electrodes.
| Sample | Discharge capacity/mA h g−1 | ||||
|---|---|---|---|---|---|
| 1st | 2nd | 3rd | 30th | 50th | |
| 200 °C | 703.2 | 463.0 | 467.1 | 248.0 | 193.9 |
| 300 °C | 323.9 | 155.5 | 152.8 | 176.7 | 174.9 |
| 400 °C | 577.3 | 187.5 | 167.3 | 115.3 | 106.4 |
| 500 °C | 591.7 | 156.1 | 136.7 | 72.6 | 70.2 |
| 600 °C | 501.0 | 225.6 | 185.3 | 82.5 | 53.9 |
| 700 °C | 487.1 | 190.3 | 171.3 | 90.4 | 76.7 |
| 800 °C | 631.0 | 177.1 | 154.6 | 88.7 | 78.2 |
As shown in Table 1, CuxO–Cu thin films synthesized at 300 °C can show better cycling stability than those synthesized at 200 °C, while the capacity of CuxO–Cu thin films synthesized at 200 °C are higher. High temperature favors the growth of thicker oxide layers. Therefore, the electrochemical performances of these copper oxide films are strongly dependent on their film thickness. During the charging process, when the Cu substrate acted as an electron drain, the inner layers were the first ones undergoing oxidation and the subsequent electron conduction from the outermost region was limited by the Cu → CuxO conversion.7,15
The current design of integrated Cu/Cu2O/CuO thin films can be good candidates for lithium-ion battery anode materials. However, owing to the existance of growth strain between Cu and oxides at high temperature, oxide layers can be separately peeled from the Cu substrate. Therefore, the selection of suitable oxidation temperature is dramatically important. The present results indicate that the vapor-phase strategy is facile and effective to crystallize copper oxides on the Cu foil, providing a new avenue for the large-scale configuration of integrated metal oxide electrodes with unique electrochemical properties.
4. Conclusions
In summary, a facile vapor-phase strategy was proposed to crystallize copper oxides on the Cu foil substrate in air. The novel CuxO–Cu electrodes can be integrated with CuxO directly on the Cu foil current collector, without using any conductive carbon and binder. The Cu/Cu2O/CuO tri-layered structure of the oxidized products validate the oxidation route from Cu to Cu2O, and finally to CuO. The driving force for our current oxidation sequence by simply heating Cu foil in air, Cu → Cu2O → CuO, can be ascribed to their similar cubic crystal structure of Cu and Cu2O, while the formation of CuO initially from Cu demands the stage of atom rearrangement and unit-cell restructuring. When the oxidation temperature is higher than 400 °C, the oxidized products can be peeled from the Cu foil substrate owing to the growth strain between oxide layers and Cu substrate. The as-obtained and pulverized oxidation products show completely different XRD patterns, which clearly confirm the Cu/Cu2O/CuO tri-layered structure during oxidizing Cu foil. In addition, the film thickness of these oxidized products can be controlled by selecting different oxidation temperatures and times. The integrated CuxO–Cu electrodes can show better cycling stability and higher capability than those CuxO–C blend electrodes, because they can facilitate the electron transfer processes between active materials and current collectors. More generally, the present work can serve as a successful example of the synthesis of integrated metal oxide electrodes, which also provides a novel strategy to improve the electrochemical performances of anode oxide materials.Acknowledgements
Financial support from the National Natural Science Foundation of China (grant nos. 50872016, 20973033 and 51125009), the National Natural Science Foundation for Creative Research Group (grant no. 20921002) and the Hundred Talents Program of the Chinese Academy of Sciences is acknowledged.References
- G. R. Patzke, Y. Zhou, R. Kontic and F. Conrad, Angew. Chem., Int. Ed., 2011, 50, 826 CrossRef CAS.
- J. Liu and D. Xue, Adv. Mater., 2008, 20, 2622 CrossRef CAS.
- J. Liu, H. Xia, D. Xue and L. Lu, J. Am. Chem. Soc., 2009, 131, 12086 CrossRef CAS.
- J. C. Park, J. Kim, H. Kwon and H. Song, Adv. Mater., 2009, 21, 803 CrossRef CAS.
- J. Wu and D. Xue, CrystEngComm, 2011, 13, 1966 RSC.
- Y. Zhang, C. Sun, P. Lu, K. Li, S. Song and D. Xue, CrystEngComm, 2012, 14, 5892 RSC.
- D. Barreca, G. Carraro, A. Gasparotto, C. Maccato, M. Cruz-Yusta, J. L. Gómez-Camer, J. Morales, C. Sada and L. Sánchez, ACS Appl. Mater. Interfaces, 2012, 4, 3610 CAS.
- J. Liu, H. Xia, L. Lu and D. Xue, J. Mater. Chem., 2010, 20, 1506 RSC.
- K. Chen and D. Xue, CrystEngComm, 2012 10.1039/c2ce26084k.
- J. Xu and D. Xue, Acta Mater., 2007, 55, 2397 CrossRef CAS.
- K. Chen and D. Xue, Nanosci. Nanotechnol. Lett., 2012, 4, 1 CrossRef CAS.
- W. Kang, F. Liu, Y. Su, D. Wang and Q. Shen, CrystEngComm, 2011, 13, 4174 RSC.
- S. Ko, J. Lee, H. S. Yang, S. Park and U. Jeong, Adv. Mater., 2012, 24, 4451 CrossRef CAS.
- J. Liu and D. Xue, Nanoscale Res. Lett., 2010, 5, 1525 CrossRef CAS.
- X. Chen, N. Q. Zhang and K. N. Sun, J. Phys. Chem. C, 2012, 116, 21224 CAS.
- X. Jiang, T. Herricks and Y. Xia, Nano Lett., 2002, 2, 1333 CrossRef CAS.
- M. Yin, C. Wu, Y. Lou, C. Burda, J. T. Koberstein, Y. Zhu and S. O'Brien, J. Am. Chem. Soc., 2005, 127, 9506 CrossRef CAS.
- X. Zhao, Z. Bao, C. Sun and D. Xue, J. Cryst. Growth, 2009, 311, 711 CrossRef CAS.
- K. Chen, Y. Si and D. Xue, Mod. Phys. Lett. B, 2009, 23, 3753 CrossRef CAS.
- L. Li, X. Mi, Y. Shi and G. Zhou, Phys. Rev. Lett., 2012, 108, 176101 CrossRef.
- S. L. Shinde and K. K. Nanda, RSC Adv., 2012, 2, 3647 RSC.
- Y. Zhu, K. Mimura, J. W. Lim, M. Isshiki and Q. Jiang, Metall. Mater. Trans. A, 2006, 37A, 1231 CAS.
- G. Zhou, W. Slaughter and J. C. Yang, Phys. Rev. Lett., 2005, 94, 246101 CrossRef.
- L. Yuan, Y. Wang, R. Mema and G. Zhou, Acta Mater., 2011, 59, 2491 CrossRef CAS.
- Z. Huang, Y. Lu, H. Qin, B. Yang and X. Hu, Adv. Eng. Mater., 2012, 14, 491 CrossRef CAS.
- I. C. Cheng and A. M. Hodge, Adv. Eng. Mater., 2012, 14, 219 CrossRef CAS.
- S. M. Cai, T. Matsushita, H. Fujii, K. Shirai, T. Nonomura, H. Tatsuoka, C. W. Hsu, Y. J. Wu and L. J. Chou, e-J. Surf. Sci. Nanotechnol., 2012, 10, 175 CrossRef CAS.
- F. Liu, S. Song, D. Xue and H. Zhang, Adv. Mater., 2012, 24, 1089 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ce26544c |
| This journal is © The Royal Society of Chemistry 2013 |