Synthesis, spectroscopic and structural characterization of the first phenyl bis-cyanoximes: non-chelating extended ionisable building block ligands for new MOFs†
Scott
Curtis
,
Olesya
Ilkun
,
Amy
Brown
,
Svitlana
Silchenko
and
Nikolay
Gerasimchuk
*
Department of Chemistry, Missouri State University, Temple Hall 456, Springfield, MO, USA. E-mail: NNGerasimchuk@missouristate.edu; Fax: +1 4178365507; Tel: +1 4178365165
First published on 16th October 2012
Abstract
Two new isomeric bis-cyanoximes were synthesized and characterized by a variety of spectroscopic methods including UV-visible, infrared, NMR, pKa measurements and X-ray analysis. These synthesized compounds represent the first non-chelating bis-cyanoximes that can function as building blocks for new MOF-like structures.
Introduction
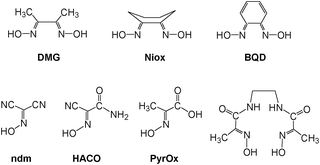
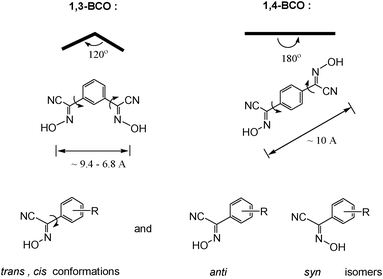
Bifunctional ionizable ligands possess a wide array of potential applications in modern inorganic and materials chemistry. The presence of ionizable groups combined with bifunctionality elicits the formation of coordination polymers that demonstrate a large variety of different structures in the complexes formed.1 Thus, there are 1D, 2D, and 3D coordination polymers which have, for example, applications in electronic devices2 and luminescent materials.3 The most interesting are 3D coordination polymers that arise from bifunctional ionizable ligands which are frequently called Metal–Organic Frameworks (MOFs). They adopt unusual lattice architectures, possess a large surface area, along with high porosity, and, as such, are currently being researched for their ability to trap gases like CO2,4 SO2,4d NOx,4e and H2.4a The last gas is thought to be the key for “hydrogen economy”.4fThe vast majority of bifunctional ionizable ligands that are being studied currently are dicarboxylic acids of both aliphatic5 and aromatic origin.4b,6a Oximes, and especially dioximes, represent a class of excellent ambidentate ligands for analytical6b and coordination chemistry.6c Oximes offer N− or O− atoms for coordination, thus acting as bidentate chelating agents, forming stable complexes with numerous metal ions.6c Examples of some most commonly used oximes and dioximes are dimethylglyoxime (DMG), 1,2-cycloxanedionedioxime (niox), 1,2-benzoquinonedioxime (BQD), isomeric 1,2-nitrosonaphtoles and nitroso-R-salt, nitrosodicyanmethnide (ndm), 2-cyano-2-oximinoacetamide (HACO), pyruvate oxime (PyOx) and its conjugates, etc. (Scheme 1). Nevertheless, prior to this study no non-chelating dioximes were known in general. As a result, no preparations, properties or subsequent studies of 3D coordination polymers and MOFs based on non-chelating dioximes have been reported yet. In this work we present the synthesis and some properties of the two novel oxime-based bifunctional ligands shown in Scheme 2. These compounds can act as bridges rigid at the core, but flexible at the cyanoxime fragments, which is due to their ability to adopt numerous conformations (Scheme 2; ESI† Fig. S1 and S2). Yet these dioximes represent ionizable spacers of 120° or 180° topologies that are able to form and support interesting crystal lattice architectures that can accommodate small guest molecules of volatile organic compounds, solvents or gases.
 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
Experimental
General considerations: chemicals and methods
Starting compounds such as phenylacetonitriles, sodium metal, sodium nitrite, sulfuric acid and hydrochloric acid (Aldrich, Fluka) and HPLC-grade organic solvents (Spectrum Chemicals, J. T. Baker) were used without additional purification. Melting points or decomposition temperatures for the synthesized compounds were determined using the Mel-Temp apparatus (Thomas Hoover). TLC for the ligands and their precursors was carried out on silica-coated aluminium-backed plates with a fluorescent indicator. The mobile phase was 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 = chloroform:methanol. Elemental analyses on C, H, N content were performed at the Atlantic Microlab (Norcross, GA).
:1 = chloroform:methanol. Elemental analyses on C, H, N content were performed at the Atlantic Microlab (Norcross, GA).
The synthesized, new, organic ligands were characterized at 296 K using UV-visible spectroscopy on an HP 8354 spectrophotometer (200–1100 nm; using 1 mm and 10 mm quartz cuvettes from Starna, Inc.), IR spectra (Bruker FT-IR spectrophotometer in ATR mode), and 1H, 13C{1H} NMR spectroscopy (solutions in DMSO-d6; TMS was an internal standard; Varian INova 400 MHz spectrometer; T = 23 °C unless otherwise specified).
The pKa studies were carried out using a Sirius Analytical Instruments automated titration station (Sussex, UK) equipped with a temperature-controlled bath. Since protonated cyanoximes HL are poorly soluble in water, all measurements were conducted in mixed solvent systems using DMSO as a solubilizing co-solvent. Measurements consisted of the three-step multi-stage titration in water–DMSO mixtures from 10 wt% to 20 wt% with ionic strength adjusted to 0.165 with KCl.7c,8a The values were extrapolated to "zero" DMSO content to obtain an aqueous pKa value using the Yasuda–Shedlovsky procedure. The pH in titration experiments ranged from 3 to 11.
Suitable single crystals of the isomeric bis-cyanoximes 1,3-BCO and 1,4-BCO were examined using a Bruker APEX 2 diffractometer (Mo Kα, λ = 0.71073 Å; a highly-oriented graphite monochromator; ω-scanning mode) equipped with a SMART CCD area detector. Intensities were integrated from 4 series of 364 exposures, each covering 0.5°, with the total data set being a sphere.13 The space group determination was done with the aid of XPREP software.14 Numerical absorption corrections were applied based on crystal face indexing obtained using images recorded by the video-microscope camera with the help of the SADABS program that was included in the Bruker AXS software package.15 The structures were solved by direct methods and refined by least-squares on weighted F2 values for all reflections using the SHELXTL program.
Synthesis of ligands and crystals growth
Preparation of target bis-cyanoximes was carried out according to Scheme 3 where respective monoximes represent intermediate products (ESI† Table S3 and Fig. S4 and S5). Typical synthesis is described below.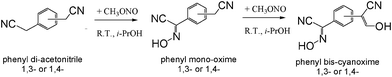 | ||
| Scheme 3 | ||
Thus, 2 g (12.8 mmol) of 1,3-phenylenediacetonitrile (or 1,4-phenylenediacetonitrile) was dissolved in 150 ml of i-PrOH at ambient conditions. 0.588 g of thinly sliced metallic sodium (25.5 mmol) was dissolved in the same solvent and added to the above solution under stirring and N2 protection. The base is needed for the activation of the methylene group in phenyl-bis-acetonitriles. A slow flow of gaseous methylnitrite CH3ONO was bubbled through the reaction mixture at room temperature under intense stirring for 10–15 minutes.9 The reaction mixture changed to a bright-yellow color and a thick precipitate began to form. The solid was collected by filtration and allowed to air dry. The precipitate was then re-dissolved in 50 ml H2O and acidified to a pH ∼3, with 25% by volume HCl added dropwise. The final product 1,3-BCO (or 1,4-BCO) was collected by gravity filtration, washed with water and then dried in a desiccator. The yield for 1,3-BCO was 34%; m.p. = 193 °C (with decomposition); Rf = 0.42 using 9:1 chloroform–methanol mobile phase. Analysis for C10H6N4O2·0.5 H2O calculated (found, %): C 53.81 (54.54), H 3.16 (3.16), N 25.10 (24.76). IR spectrum (cm−1, tentative assignments): 1021 ν(N–O), 3293 ν(O–H), 1700 ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1403, 1254, 1232 ν(CC), 2260 ν(C
N), 1403, 1254, 1232 ν(CC), 2260 ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N). The NMR 1H spectrum (complicated, due to presence of two isomers, ESI† Fig. S7; δ ppm): 14.07 (minor narrow singlet oxime), 13.96 (main narrow singlet), 8.00 (multiplet, 1H, between two cyanoxime groups), 7.83–7.80 (multiplet, 2H, at ortho-positions to cyanoxime groups), 7.66–7.61 (multiplet, 1H, at meta-position to cyanoxime groups). 13C{1H} NMR spectrum, δ ppm (double set of signals due to presence of two isomers; tentative assignments, ESI† Fig. S8), main component: 110.29 (CN group), 122.45, 127.98, 131.24 (oxime carbon), 130.84, 130.90; minor component: 115.77 (CN group), 126.03, 128.55, 133.09 (oxime carbon), 130.56, 128.65. For 1,3-BCO the UV spectra, λmax, nm (ε, cm−1 M−1): in methanol −258 nm (2600), ethanol −259 nm (6840), n-propanol −260 nm (4160).
N). The NMR 1H spectrum (complicated, due to presence of two isomers, ESI† Fig. S7; δ ppm): 14.07 (minor narrow singlet oxime), 13.96 (main narrow singlet), 8.00 (multiplet, 1H, between two cyanoxime groups), 7.83–7.80 (multiplet, 2H, at ortho-positions to cyanoxime groups), 7.66–7.61 (multiplet, 1H, at meta-position to cyanoxime groups). 13C{1H} NMR spectrum, δ ppm (double set of signals due to presence of two isomers; tentative assignments, ESI† Fig. S8), main component: 110.29 (CN group), 122.45, 127.98, 131.24 (oxime carbon), 130.84, 130.90; minor component: 115.77 (CN group), 126.03, 128.55, 133.09 (oxime carbon), 130.56, 128.65. For 1,3-BCO the UV spectra, λmax, nm (ε, cm−1 M−1): in methanol −258 nm (2600), ethanol −259 nm (6840), n-propanol −260 nm (4160).
Preparation of 1,4-BCO was carried out in a similar to 1,3-BCO. The yield for 1,4-BCO was 47%; m.p. = 197 °C (with decomposition); Rf = 0.26. Analysis for C10H6N4O2·H2O calculated (found, %): C 51.73 (52.28), H 3.47 (3.57), N 24.13 (23.36). IR spectrum (cm−1): 3508 ν(O–H), 3324 ν(C–H), 2239 ν(CN), 1624 (ν(CN), 1516, 1421, 1409 ν(CC), 973 ν(N–O), 468 (δ (CC)). NMR 1H spectrum (δ ppm): 7.82 (singlet, 2H phenyl group), 13.98 (H, oxime, singlet); 13C{1H} NMR spectrum (δ ppm): 110.30 (CN-group), 131.90 (oxime carbon), 130.98 (ipso-carbon), 126.78 (phenyl group carbon). For 1,4-BCO the UV spectra, λmax, nm (ε, cm−1 M−1): methanol −297 nm (3480), ethanol −300 nm (5370), n-propanol −302 nm (3870).
According to analytical and spectroscopic data, both compounds were obtained as pure, “one TLC spot” compounds suitable for further syntheses of a variety of complex compounds.
Both isomeric bis-cyanoximes demonstrate a great possibility for adoption of a variety of conformations that arises from syn/anti geometrical isomers (ESI† Fig. S1 and S2), resulting in difficulties and lengthy times required for the growth of crystals suitable for the X-ray analysis. All appropriate single crystals were obtained from the mixed water–acetonitrile, or DMF, or DMSO solutions within months of waiting. We explain this fact as a kinetic factor involved in the crystal packing and lattice formation.
Results and discussion
Properties of new bis-cyanoximes in solution
The synthesized novel bis-cyanoximes represent weak acids in solution and show a stepwise dissociation as depicted below for 1,3-BCO: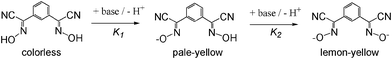
For 1,3-BCO, the measured ionization constants were: pKa1 = 6.89 ± 0.03 and pKa2 = 7.52 ± 0.10, while for 1,4-BCO they were pKa1 = 6.24 ± 0.06 and pKa2 = 7.67 ± 0.07.
The deprotonation of studied bis-cyanoximes leads to a color gain that is due to weak n → π* transitions in the nitroso chromophore in the visible region in the UV-visible spectra16 at ∼400–450 nm (ε ∼60–70). This solvent-dependent transition represents negative solvatochromism7a,12 (ESI† Fig. S9). A similar behavior was previously observed for the NO2− anion,7b nitrosodicyanomethanide11,12 ONC(CN)2−, and other cyanoximes.7c It is interesting to note that 1,4-BCO2− demonstrated the weakest solvochromism of all the known cyanoximes that have been previously studied. This is due to the symmetric positions of the cyanoxime groups in the ligand, leading to an insignificant change in the polarity of the dianion in the excited state compared to the ground state.
The 13C{1H} NMR spectrum of pure, one-spot on TLC sample of 1,3-BCO in DMSO-d6 demonstrates a double set of signals. They have minor and major components reflected in their different intensity and appear due to the presence of two geometrical syn /anti isomers (ESI† Fig. S1). Heating of the sample to 95 °C affords a simplified spectrum that indicates conversion of the mixture to the most stable, presumably anti isomer. Similar behaviour was earlier observed for several mono-cyanoximes.10
To the contrary, both 1H and 13C{1H} NMR spectra of 1,4-BCO at room temperature contain a few signals consistent with high symmetry (C2h) of the molecule in solution and absence of geometrical isomers and conformers (ESI† Fig. S2). Thus, the proton NMR spectrum consists of only two single lines that belong to all equivalent protons of the phenyl group (7.82 ppm) and to oxime hydrogens (13.98 ppm). The 13C{1H} NMR spectrum contains only 4 lines.
Solid-state structures
Both bis-cyanoximes that were studied in this work possess great structural diversity owing to their flexibility to adopt trans and cis conformations, as well as the presence of syn and anti geometrical isomers as shown in Scheme 2. Thus, the rotation around the C–C bond between the cyanoxime fragment and the phenyl group causes trans and cis conformations, while the temperature-dependent syn/anti isomerism,8 that is typical for oximes, contributes to further variety of possible geometries (Scheme 2), leading to 9 structures for 1,3-BCO and 6 structures for 1,4-BCO (ESI† Fig. S1 and S2). Moreover, the rich stereochemistry of bis-cyanoximes explains the difficulty encountered during the crystal growth that was reflected in a prolonged time for crystals to appear and their overall quality and suitability for X-ray experiments. Crystal data for all the compounds studied are presented in Table 1, while selected bond lengths and valence angles are summarized in Table 2. All bis-cyanoximes exhibited interesting H-bonding in the crystals studied and is tabulated in ESI† Table S10.| Data | 1,3-BCO·DMF | 1,3-BCO·H2O | 1,4-BCO | 1,4-BCO·DMSO |
|---|---|---|---|---|
| Empirical formula | C13H13N5O3 | C20H14N8O5 | C10H6N4O2 | C14H18N4O4S2 |
| Formula weight | 287.28 | 446.39 | 214.19 | 374.48 |
| Temperature, K | 150(2) | 120(2) | 153(2) | 129(2) |
| Crystal system | Orthorhombic | Orthorhombic | Orthorhombic | Triclinic |
| Space group | Pbcm | Fdd2 | Pbca |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| Unit cell constants | a = 7.9182(10) Å, α = 90° | a = 33.749(13) Å, α = 90° | a = 5.1519(9) Å, α = 90° | a = 6.2397(4) Å, α = 81.3740(10)° |
| b = 13.0263(16) Å, β = 90° | b = 34.175(13) Å, β = 90° | b = 11.244(2) Å, β = 90° | b = 7.8663 (5) Å, β = 80.6960(10)° | |
| c = 13.5001(16) Å, γ = 90° | c = 3.7634(15) Å, γ = 90° | c = 16.370(3) Å, γ = 90° | c = 9.6240(6) Å, γ = 72.0250(10)° | |
| Volume, Z | 1392.5(3) Å3, 4 | 4341(3) Å3, 8 | 948.2(3) Å3, 4 | 440.88(5) Å3, 1 |
| Reflections coll., indep | 21181, 1281 [Rint = 0.0491] |
11902, 2294 [Rint = 0.0602] |
21292, 1441 [Rint = 0.0677] |
5053, 1904 [Rint = 0.0152] |
| Final R indices [I > 2σ(I)] | R 1 = 0.0645, wR2 = 0.1688 | R 1 = 0.0600, wR2 = 0.1413 | R 1 = 0.0635, wR2 = 0.1809 | R 1 = 0.0269, wR2 = 0.0706 |
| Goodness of fit | 1.108 | 1.037 | 1.087 | 1.070 |
| Max/Min residual electron density, e Å−3 | 0.905/−0.7 | 0.297/−0.223 | 0.331/−0.360 | 0.329/−0.216 |
| F(000) | 600 e− | 1840 e− | 440 e− | 198 e− |
| Compound | Bonds, Å | Angles, ° |
|---|---|---|
| 1,3-BCO·H2O | C(1)–N(1) | 1.320(5) |
| C(1)–C(2) | 1.447(6) | |
| C(1)–C(3) | 1.474(5) | |
| C(2)–N(2) | 1.122(5) | |
| N(1)–O(1) | 1.334(4) | |
| N(3)–O(2) | 1.305(5) | |
| N(1)–C(1)–C(2) | 121.7(4) | |
| N(1)–C(1)–C(3) | 120.5(3) | |
| N(1)–C(1)–C(3) | 120.5(3) | |
| C(2)–C(1)–C(3) | 117.7(3) | |
| N(2)–C(2)–C(1) | 179.5(5) | |
| C(1)–N(1)–O(1) | 112.3(3) | |
| 1,3-BCO·DMF | C(1)–N(1) | 1.288(3) |
| C(1)–C(2) | 1.444(4) | |
| C(1)–C(3) | 1.482(4) | |
| C(2)–N(2) | 1.140(4) | |
| N(1)–O(1) | 1.377(3) | |
| N(1)–C(1)–C(2) | 122.0(2) | |
| N(1)–C(1)–C(3) | 119.4(2) | |
| C(2)–C(1)–C(3) | 118.6(2) | |
| N(2)–C(2)–C(1) | 178.4(3) | |
| C(1)–N(1)–O(1) | 112.4(2) | |
| 1,4-BCO | C(2)–N(2) | 1.145(2) |
| C(2)–C(1) | 1.448(3) | |
| C(1)–N(1) | 1.289(2) | |
| C(1)–C(3) | 1.480(3) | |
| N(1)–O(1) | 1.380(2) | |
| N(1)–C(1)–C(2) | 121.06(17) | |
| N(1)–C(1)–C(3) | 121.01(16) | |
| C(2)–C(1)–C(3) | 117.63(15) | |
| N(2)–C(2)–C(1) | 176.6(2) | |
| C(1)–N(1)–O(1) | 111.48(16) | |
| 1,4-BCO·DMSO | C(1)–N(1) | 1.2892(17) |
| C(1)–C(2) | 1.4526(18) | |
| C(1)–C(3) | 1.4732(17) | |
| C(2)–N(2) | 1.1437(18) | |
| O(1)–N(1) | 1.3787(13) | |
| O(1)–H(1) | 0.92(2) | |
| N(1)–O(1)–H(1) | 105.1(13) | |
| N(1)–C(1)–C(2) | 120.21(11) | |
| N(1)–C(1)–C(3) | 120.82(11) | |
| C(2)–C(1)–C(3) | 118.97(11) | |
| N(2)–C(2)–C(1) | 179.02(14) | |
Crystal structure of 1,3-BCO·H2O
Crystals of this cyanoxime demonstrated a strong propensity for twinning. The only crystal large enough for single-crystal X-ray diffraction experiments proved to be a merohedral twin with a 10.2(5)% second component contribution. The twin components are related by a 180.0° rotation about the [110] direction. In addition to twinning, there is also the presence of two geometrical isomers in the structure: 85% represent anti isomers while 15% are syn isomers. Both are statistically distributed throughout the crystal and appear as two-positional disorder in the oxime group8b (Fig. 1). The molecular structure and numbering scheme for 1,3-BCO·H2O are shown in Fig. 1, while a packing diagram and the organization of the crystal lattice are shown in Fig. 2 and ESI† Fig. S11. This compound crystallized in the non-centrosymmetric Fdd2 (#43) space group, but an absolute configuration of this light organic molecule was not possible to determine using the Mo Kα radiation of our diffractometer.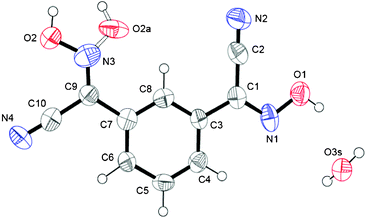 | ||
| Fig. 1 Numbering scheme for 1,3-BCO·H2O. An ORTEP drawing here and in other figures done at 50% thermal ellipsoids probability level. Bonding in the syn isomer in the oxime group is shown as a dotted line. | ||
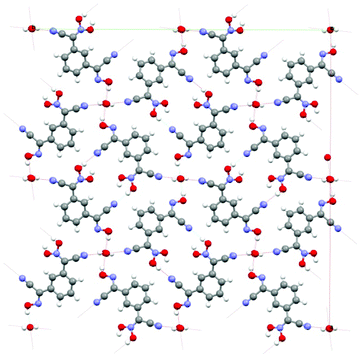 | ||
| Fig. 2 The organization of the crystal structure of 1,3-BCO·H2O: view along c-direction. The channels shown in the structure are occupied by water molecules. There is a system of H-bonds both between the cyanoxime molecules and trapped H2O in the structure. Coloring scheme here and later: red – O, blue – N, grey – C, white – H atoms. | ||
The compound adopts a non-planar structure in the solid state, with different configurations of both cyanoxime fragments, with one being trans- and another one being cis-oriented with respect to the phenyl group (Fig. 1; ESI† Fig. S1). Thus, in the crystal the bis-cyanoxime adopts trans–cis/anti–anti and trans–cis/syn–anti structures (2 and 5 respectively, ESI† Fig. S1). Both cyanoxime fragments have different degrees of deviation from the planar structure. The dihedral angle between the planes O1–N1–C1–C2–N2 and the phenyl group is 4.75°, while a similar angle between the O2N3–C9–C10 plane and the aromatic ring is 3.98° in the opposite direction.
The crystal packing in the structure can be described as the formation of 1D columns of molecules of the bis-cyanoxime running along c-directions that are held together by slipped π–π stacking interactions and extensive H-bonding. The latter is formed between both bis-cyanoxime molecules that assembled in dimeric units (ESI† Fig. S11), and a water molecule that is trapped inside the channel formed along the c-direction (ESI† Fig. S12). Water molecules occupy special positions in the lattice (ESI† Fig. S12) and are essential for the crystal lattice formation because of its crucial role in H-bonding between 1D columns of molecules that run in opposite directions (ESI† Fig. S12 and S13). Water molecules in the channel are aligned along the c-axis 3.749 Å apart, tilted relatively to each other at a 22.07° angle (ESI† Fig. S12), and occupy 2.51% of volume in the structure (109.11 Å3). The structure # in CCDC is 894545.
Crystal structure of 1,3-BCO·DMF
This structure of this bis-cyanoxime contains a solvent molecule that is disordered over two positions (Fig. 3; ESI† Fig. S14), which occupies alternating layers of voids in the structure that can be described as channels (ESI† Fig. S15). The shortest distance between loosely packed DMF molecules is 6.514 Å which are also aligned with a tilt angle of 11.50° relative to each other (ESI† Fig. S15). The solvent occupies 327.19 Å3 per unit cell (23.5%). The mirror plane passes through the C6, C5 and O1s atoms. The dihedral angle between the cyanoxime plane O1–N1–C1–C2–N2 and the phenyl ring is 3.42°. The bis-cyanoxime adopts a cis–cis/anti–anti structure 3, which is different from the monohydrate compound (ESI† Fig. S1). Crystal packing for the 1,3-BCO·DMF can be described as layered, practically planar 2D sheets. Van der Waals forces between the nitrogen atoms N2 of the cyano groups connect sheets into the 3D framework (Fig. 4). There is an intermolecular, interlayer interaction at a distance of 3.331 Å (Fig. 4).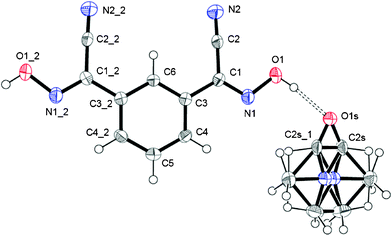 | ||
| Fig. 3 The molecular structure and numbering scheme in 1,3-BCO·DMF. The shown structure is in GROW mode with a disordered solvent molecule. Symmetry codes for #1: x, y, 1/2 − z; for #2: x, y, 3/2 − z. | ||
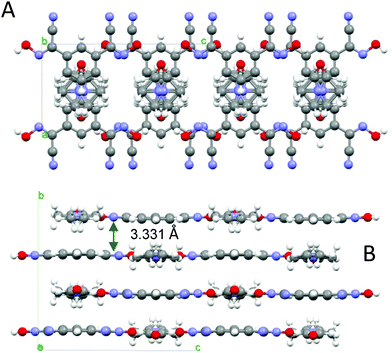 | ||
| Fig. 4 The organization of the crystal lattice in the structure of 1,3-BCO·DMF. A – view of two unit cells along the a-axis, showing the shortest interlayer distance and B – view along the b-axis, showing the direction of H-bonding and alternating voids filled with disordered solvent molecules (indicated as dotted circles). | ||
Not even the slightest overlap between traditionally π–π stacking phenyl groups was found in this structure. Another rather peculiar donor–acceptor interaction at a distance of 3.482 Å was determined between the centroid of the ring and the oxygen atom O1s of the solvent. Thus, both aforementioned contacts are responsible for the interlayer interactions. A strong symmetrical H-bonding between solvent molecules and cyanoxime fragments assembles bis-cyanoxime molecules into planar sheets that are formed along the c-axis (Fig. 4). The structure # in CCDC is 894546.
Crystal structure of 1,4-BCO
This bis-cyanoxime adopts a trans/anti–anti configuration (I, ESI† Fig. S2), but is not completely planar in the solid state (Fig. 5). The dihedral angle between the mean plane C2–C1–N1–O1 and the plane of the phenyl ring is 12.72°. Interestingly, the cyanoxime fragment is noticeably not planar itself: two planes N2–C2–C1–N1 and O1–N1–C1 have the dihedral angle between them equal to 5.85°, and the torsion angle C2–C1–N1–O1 = 4.23°. The packing of 1,4-BCO in the crystal can be best described as a layered H-bonded 2D “herring bone” framework (Fig. 6). Thus, weak π-stacking between the phenyl ring and N1 atoms of the cyanoxime fragment in neighboring molecules at a distance of 3.509 Å is responsible for the formation of 1D “slipped” columns running along the a-axis. These columns run in opposite directions and are interconnected via H-bonding along the b-axis forming the “herring bone” layers parallel to the b direction (Fig. 6; ESI† Fig. S17). The H-bonding in the structure is depicted in detail in ESI† Fig. S18 and shows the separation between two H-bonded parallel molecules (10.330 Å) in the layer running along a (ESI† Fig. S19 and S20). The structure # in CCDC is 894547.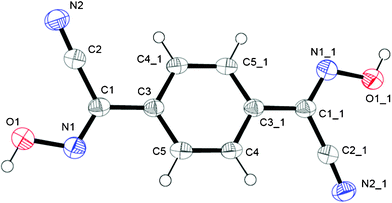 | ||
| Fig. 5 The molecular structure and numbering scheme for 1,4-BCO. Symmetry code for #1: 1 − x, −y, 1 − z. | ||
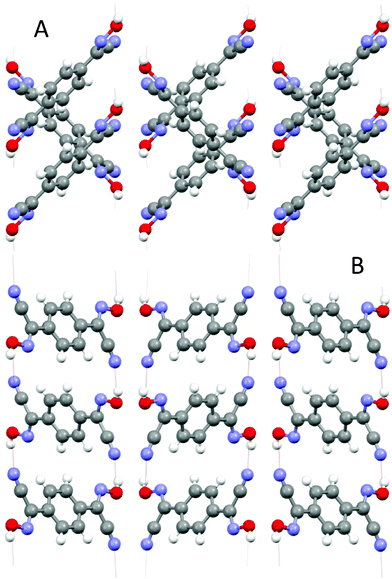 | ||
| Fig. 6 Two orthogonal views of the crystal lattice of 1,4-BCO: A – view along a, B – view along b. Both projections show H-bonding between layers of bis-cyanoxime molecules that organizes the structure into a 3D network. | ||
Crystal structure of 1,4-BCO·DMSO
This compound as clear, colorless blocks crystallized from a closed solution in pure DMSO after ∼3 weeks (ESI† Fig. S21), which is a noticeably rare case of crystallizing of a well soluble organic compound from that solvent. When removed from the solution onto filter paper, crystals of 1,4-BCO·DMSO within a day became opaque, and then white in color, presumably due to the partial or complete loss of the solvent trapped in the crystal (ESI† Fig. S21). The molecular structure and numbering scheme for this compound is shown in Fig. 7. The bis-cyanoxime adopts a trans/anti–anti structure and is not planar in the solid state (ESI† Fig. S2). There are two planar fragments, a cyanoxime group and a phenyl ring, with the dihedral angle between them 8.43°. The solvent molecules occupy voids between the layers of the 1,4-BCO molecules that form channels running along the a axis (ESI† Fig. S22) occupying 124.03 Å3 per unit cell (28.13%).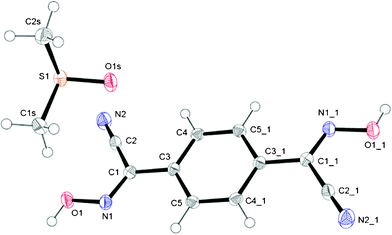 | ||
| Fig. 7 GROW fragment and numbering scheme in the structure of 1,4-BCO·DMSO; an ORTEP drawing at 50% thermal ellipsoids probability level. Symmetry codes for #1 position: 1 − x, −y, −z. | ||
Also, DMSO molecules play an important role in the structure arrangement by forming symmetrical H-bonds between the oxygen atom O1s and OH groups on both ends of the bis-cyanoxime molecule (Fig. 8A). The closest S⋯S distance in the structure is 4.006 Å (ESI† Fig. S23). The crystal structure of 1,4-BCO·DMSO can be described as 2D, layered, practically planar sheets with solvent molecules in between. The shortest distance between layers is 3.377 Å and represents an interaction between π-bonds in the cyanoxime groups of neighboring molecules (Fig. 8B; ESI† Fig. S24). The structure # in CCDC is 894548.
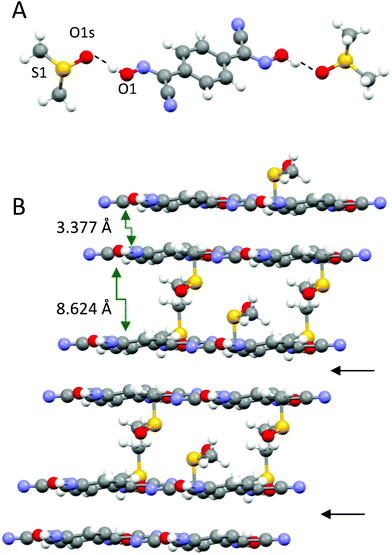 | ||
| Fig. 8 The solvent molecule that is H-bonded to the dioxime is the building block in the structure of 1,4-BCO·DMSO (A), and a prospective view of the 2D layered arrangement in the crystal showing only van der Waals forces between double H-bonded sheets (B) is indicated with black arrows. | ||
The two new bis-cyanoximes presented here readily form complexes with monovalent Ag and Tl, and react with 3d-metalloligands such as trans-MEn2X2 (M=Cu, Ni), and only cis-ML2X2 (L = Dipy or Phen; X = Cl, Br, NO3). As examples of ability of new bis-cyanoximes to form the metal–organic frameworks, views of the Tl2(1,3-BCO) and Tl2(1,4-BCO) elegant structures are shown in the ESI† Fig. S25 and S26, with selected crystal data presented in the ESI† Table S27. A detailed description and structural characterization of those main group complexes, and other transition metal complexes is the subject of a larger forthcoming publication.
Conclusions
Two isomeric bifunctional ligands containing ionizable oxime groups have been synthesized and characterized by conventional spectroscopic methods and X-ray analysis. Crystal structures demonstrated extensive H-bonding and inclusion of solvents of crystallization in the lattice in three out of four studied samples. Thus, both compounds have shown to have potential to form extended coordination polymers with MOF-like behavior. First and preliminary data evidenced the ability of both isomeric bis-cyanoximes to complex metal ions. Future work will include the synthesis and detailed characterization of coordination polymers followed with their thermal properties and gas sorption studies.Acknowledgements
MSU Graduate College and ACS PRF award to NG, for partial financial support, Dr. C. L. Barnes (University of Missouri–Columbia) for the data collection for the single-crystal specimen of 1,3-BCO·H2O, and to Dr. I. A. Guzei (University of Wisconsin–Madison) for help with the resolution of the complicated disorder of the solvent in the structure of 1,3-BCO·DMF.References
- O. T. Ilkun, S. J. Archibald, C. L. Barnes, N. Gerasimchuk, S. Silchenko, O. A. Gerasimchuk and V. N. Nemykin, Dalton Trans., 2008, 5715–5729 RSC.
- (a) Molecular Electronics, ed. J. Jortner and M. Ratner, Blackwell Science, Malden, MA, USA, 1997, pp. 215–239 Search PubMed; (b) Optoelectronic Properties of Inorganic Compounds, ed. D. M. Roundhill and J. P. Fackler, Plenum, New York, 1999 Search PubMed; (c) J. M. Williams, A. J. Schultz, A. E. Underhill and K. Carniero, in Extended Linear Chain Compounds, ed. J. S. Miller, Plenum Press, New York, 1982, vol. 1 Search PubMed; (d) G. Cuniberti, G. Fagas and K. Richter, Introducing Molecular Electronics, Springer, New York, 2005 Search PubMed; (e) W. Chen, F. Liu, D. Xu, K. Matsumoto, S. Kishi and M. Kato, Inorg. Chem., 2006, 45, 5552–5560 CrossRef CAS.
- (a) S. Wang, G. Garzon, C. King, J.-C. Wang and J. P. Fackler, Inorg. Chem., 1989, 28, 4623–4629 CrossRef CAS; (b) G. J. Ferraudi, Elements of Inorganic Photochemistry, John Wiley & Sons, New York, 1988 Search PubMed; (c) M. A. Omary, A. A. Mohamed, M. Rawashdeh-Omary and J. P. Fackler, Jr., Coord. Chem. Rev., 2005, 249, 1372–1381 CrossRef CAS; (d) S.-Y. Chang, J. Kavitha, S.-W. Li, C.-S. Hsu, Y. Chi, Y.-S. Yeh, P.-T. Chou, G.-H. Lee, A. J. Carty, Y.-T. Tao and C.-H. Chien, Inorg. Chem., 2006, 45, 137–146 CrossRef CAS; (e) V. J. Catalano, M. A. Malwitz and A. O. Etogo, Inorg. Chem., 2004, 43, 5714–5724 CrossRef CAS.
- (a) J. L. C. Roswell, A. R. Millward, K. S. Park and O. M. Yaghi, J. Am. Chem. Soc., 2004, 126, 5666–5667 CrossRef; (b) A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS; (c) K. Sanderson, Nature, 2007, 448, 746–748 CrossRef CAS; (d) D. Britt, D. Tranchemontagne and O. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11623–11627 CrossRef CAS; (e) R. Snurr, Nat. Chem., 2009, 1, 426–427 CrossRef CAS; (f) A. Vimont, P. H. Cortes, Y. K. Hwang, G. Ferey, M. Daturi, J. Chang, C. Serre and J. Yoon, US Pat., 0129684A1, 2012 Search PubMed.
- (a) O. Castillo, A. Luque, P. Roman, F. Lloret and M. Julve, Inorg. Chem., 2001, 40, 5526–5535 CrossRef CAS; (b) G. Ferguson, J. F. Gallagher and A. J. McAlees, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 454–458 CrossRef.
- (a) A. G. Wong-Foy, A. J. Matzger and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 3494–3495 CrossRef CAS; (b) S. Serin, Y. Gok, S. Karabocek and N. Gultekin, Analyst, 1994, 119, 1629–1631 RSC; (c) C. V. Banks, R. W. Vander Haar and R. P. Vander Wal, J. Am. Chem. Soc., 1955, 77, 324–325 CrossRef CAS.
- (a) S. J. Strickler and M. Kasha, J. Am. Chem. Soc., 1963, 85, 2899–2901 CrossRef CAS; (b) W. L. Jolly, The Synthesis and Characterization of Inorganic Compounds, Prentice Hall, Englewood Cliffs, NJ, 1970, pp. 320–322 Search PubMed; (c) D. Marcano, N. Gerasimchuk, V. Nemykin and S. Silchenko, Cryst. Growth Des., 2012, 12, 2877–2889 CrossRef CAS.
- (a) N. Gerasimchuk, L. Goeden, P. Durham, C. Barnes, J. F. Cannon, S. Silchenko and I. Hidalgo, Inorg. Chim. Acta, 2008, 361, 1983–2001 CrossRef CAS; (b) D. Robertson, J. F. Cannon and N. Gerasimchuk, Inorg. Chem., 2005, 44, 8326–8342 CrossRef CAS.
- N. Gerasimchuk and H. Charlier, Cyanoxime Inhibitors of Carbonyl Reductase and Methods of Using Said Inhibitors in Treatments Involving Antracyclines, US Pat., 7727967B2, 2010 Search PubMed.
- D. Eddings, C. Barnes, N. Gerasimchuk and K. V. Domasevitch, Inorg. Chem., 2004, 43, 3894–3909 CrossRef CAS.
- (a) A. Kolbe and H. Kohler, Z. Anorg. Allg. Chem., 1970, 373, 230 CrossRef CAS; (b) G. Glower, N. Gerasimchuk, R. Biagioni and K. V. Domasevitch, Inorg. Chem., 2009, 48, 2371–2382 CrossRef.
- (a) C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, Weinheim, Germany, 3rd edn, 2003, p. 629 and references therein Search PubMed; (b) Solvatochromism: Problems and Methods, ed. N. G. Bakhsiev, Izd-vo Leningradskogo Universiteta, Leningrad, USSR, 1989, Chem. Abstr., 1990, vol. 112, p. 186279g Search PubMed.
- (a) SAINT, Data Integration Program Bruker AXS, 1998; (b) Dr. Sergey Lindeman (private communication: provided with the table for full-sphere data collection at ∼95% coverage for P1 group using Mo Kα radiation at 5.00 cm distance to the detector).
- (a) R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33 CrossRef; (b) G. M. Sheldrick, SADABS Area-detector Absorption Correction, 2.03, University of Göttingen, Göttingen, Germany, 1999 Search PubMed.
- Software Package for Crystal Structure Solution, APEX 2, Bruker AXS, Madison, WI, 2009.
- (a) The Chemistry of the Nitro- and Nitroso-Groups, ed. H. Feuer, R. Krieger, Publishing Co., Huntington, New York, Part 1, 1981, p. 771 Search PubMed; (b) H. Kohler and G. Lux, Inorg. Nucl. Chem. Lett., 1968, 4, 133–136 CrossRef; (c) H. Kohler and B. Seifert, Z. Anorg. Allg. Chem., 1970, 379, 1–8 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Possible geometrical isomers and conformers for 1,3-BCO (Fig. S1); possible geometrical isomers and conformers for 1,4-BCO (Fig. S2); TLC, X-ray data and 13C NMR spectrum of the 1,3-BCO-monooxime product and its precursor (Fig. S3–S6); 1H and 13C NMR spectra of the 1,3-BCO (Fig. S7 and S8); solvatochromic series for both ligands as dianions (Fig. S9); H-bonding in crystals of studied compounds (Fig. S10); organization of 1,3-BCO structure and H-bonding inside two layers (Fig. S11); position of water molecules in the lattice of 1,3-BCO crystallohydrate (Fig. S12); H-bonding and geometry of trapped water molecule (Fig. S13); disordered DMF molecule in the structure of 1,3-BCO DMF and H-bonding pattern inside one layer (Fig. S14); arrangements of solvent molecules trapped inside channels (Fig. S15); organization of layered structure of 1,3-BCO DMF (Fig. S16); organization of crystal structure of 1,4-BCO (Fig. S17); H-bonding in “herring bone” motif of 1,4-BCO (Fig. S18); details of geometry of H-bonded columns in the structure of 1,4-BCO (Fig. S19 and S20); microscope photographs of single crystals of 1,4-BCO DMSO (Fig. S21); arrangements of the guest DMSO molecule in layers in voids in the structure (Fig. S22 and S23); details of layers of the bis-cyanoxime structure (Fig. S24); fragments of structures of Tl2(1,3-BCO), Tl2(1,4-BCO) and their selected crystal data (Fig. S25 and S26, Table S27). CCDC 894545–894548. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ce26395e |
| This journal is © The Royal Society of Chemistry 2013 |