A dramatic effect of the ionic liquid structure in esterification reactions in protic ionic media
Cinzia
Chiappe
*a,
Sunita
Rajamani
a and
Felicia
D'Andrea
b
aDipartimento di Chimica e Chimica Industriale, via Bonanno 33, 56126 Pisa, Italy. E-mail: cinziac@farm.unipi.it
bDipartimento di Scienze Farmaceutiche, via Bonanno 33, 56126 Pisa, Italy
First published on 9th October 2012
Abstract
Brønsted acidic ionic liquids (ILs) have been synthesized and investigated as catalysts in esterification and transesterification reactions: simple and non-corrosive salts characterized by aliphatic cations associated with an “acidic” anion (in particular, [HSO4]−) gave the higher yields. A quite good correlation between IL acidity (H0) and catalytic ability was found although also the hydrophilic nature of the ionic medium probably affects the process efficiency.
Introduction
The possibility to use Brønsted acidic ionic liquids (ILs) as catalysts and solvents in various reactions, instead of the corrosive acids, represents an attractive research area.1 In this context, the Fischer esterification of carboxylic acids with alcohols has been the object of extensive investigation.1,2 This equilibrium reaction, of industrial importance,3 generally requires an acid catalyst and an excess of one of the reagents or a continuous removing of the formed water to shift the equilibrium towards the ester; water scavengers and/or the use of an additional solvent have been largely employed in the past to carry over the water in the form of an azeotrope. However, such operations require large energy input to recycle the solvents and/or remove the excess of reactants. Furthermore, the loss of volatile organic solvents to the atmosphere results in increase in the cost of production and also damage to the environment.4Starting from 2002, Brønsted acidic ionic liquids have been proposed as catalysts and/or media to improve the Fischer esterification of organic substrates. Initially, Lewis acidic ILs (1-butylpyridinium chloride + aluminium(III) chloride) have been tested5 but rapidly the interest has moved towards the more manageable Brønsted acidic salts and both ILs bearing a protonated acidic group on the cation alkyl chain (such as –SO3H and –CO2H) or a proton on the quaternary nitrogen atom of the cation as well as ILs having an available proton on the anion (HSO4−, H2PO4−) have been screened.6 Despite the variety of cations and anions which can give Brønsted acidic ionic liquids, the majority of the ILs (not necessarily Brønsted acidic ILs) nowadays reported for esterification are imidazole and pyridine derivatives7 although simple triethylammonium salts ([(C2H5)3NH][HSO4], [(C2H5)3NH][H2PO4] and [(C2H5)3NH][BF4]) have been used with success in esterification of some carboxylic acids with primary alcohols.8
With the aim to develop new classes of “greener” ILs (i.e. salts prepared through more simple procedures and having a lower environmental impact9), we have prepared some Brønsted acidic salts simply by addition of an equimolar amount of a strong inorganic acid (HCl, H2SO4, HNO3, CF3COOH and H3PO4) to a commercial N-base, like morpholine, pyrrolidine, piperidine, betaine and imidazole.
Here, we report on the application of these salts (some of them have melting points higher than 100 °C and, consequently, cannot be defined as ILs) in esterification and transesterification reactions proving a simple, sustainable and effective method to prepare and isolate esters. In particular, they have been tested in esterification of acetic acid with butanol, octanol and methyl β-D-glucopyranoside. In all the investigated reactions, [HPyrr][HSO4] (followed by [HPip][HSO4] and [Hmim][HSO4]) was found to be the best catalytic environment suggesting that cation structure and counteranion acidity have an important role in the process. A quite good correlation between IL acidity (H0) and catalytic ability has been found although the ability of these media to bind water and give a biphasic system with the formed ester surely favours the shift of the esterification reaction towards products, contemporaneously favouring ester recovery. Finally, the same acidic salts were also tested in the transesterification of ethyl trans-cinnamate with methanol and octanol. Also in this case, the non-corrosive [HPyrr][HSO4] gave the best results among all.
Experimental
Materials and methods
Chemicals, including N-methylimidazole, N-methylmorpholine, N-methylpyrrolidine, N-methylpiperidine, DABCO and betaine, were purchased from Sigma-Aldrich with purities >98%. N-Methylimidazolium hydrogen sulfate [Hmim][HSO4], N-methylimidazolium sulfate [Hmim]2[SO4], N-methylimidazolium dihydrogen phosphate [Hmim][H2PO4], N-methylimidazolium trifluoroacetate [Hmim][CF3CO2], N-methylimidazolium nitrate [Hmim][NO3], N-methylimidazolium chloride [Hmim]Cl, N-methylmorpholinium hydrogen sulfate [HMor][HSO4], N-methylmorpholinium sulfate [HMor]2[SO4], N-methylmorpholinium chloride [HMor]Cl, N-methylpiperidinium hydrogen sulfate [HPip][HSO4], N-methylpiperidinium sulfate [HPip]2[SO4], N-methylpyrrolidinium hydrogen sulfate [HPyrr][HSO4], N-methylpyrrolidinium chloride [HPyrr]Cl, betaine chloride ([Bet]Cl), betaine nitrate ([Bet][NO3]), betaine hydrogen sulfate ([Bet][HSO4]) and betaine tetrafluoroborate ([Bet][BF4]) were prepared by addition of an equimolar amount of the proper acid dropwise to the selected base or zwitterion. The mixtures were stirred for 4 to 5 h at 60 °C to ensure that all of the bases are reacted. Then, water was removed at reduced pressure to obtain a colorless viscous liquid or a solid.N-Methylimidazolium hydrogen sulfate [Hmim][HSO4]; m.p. 39 °C: 1H NMR (250 MHz, D2O δ, ppm) 3.21 (s, 3H, CH3N–); 6.77 (s, 2H, aromatic); 7.97 (s, 1H, aromatic). 13C NMR (63 MHz, DMSO-d6, δ ppm) 34.27, 118.18, 121.83, 134.18.
N-Methylimidazolium sulfate [Hmim]2[SO4], hygroscopic white solid, m.p. 62 °C: 1H NMR (250 MHz, DMSO-d6, δ ppm) 3.77 (s, 3H, CH3N); 7.22 (s, 1H, aromatic); 7.39 (s, 1H, aromatic); 8.33 (s, 1H, aromatic); 7.20 (br, 1H, N+–H). 13C NMR (63 MHz, DMSO-d6, δ ppm) 34.02, 121.76, 123.67, 136.69.
N-Methylimidazolium dihydrogen phosphate [Hmim][H2PO4]; m.p. 137 °C: 1H NMR (250 MHz, CDCl3, δ ppm) 3.7 (s, 3H, NCH3); 6.88 (s, 1H, aromatic); 7.35 (s, 1H, aromatic); 8.92 (s, 1H, aromatic); 9.87 (N+–H).
N-Methylimidazolium trifluoroacetate [Hmim][CF3CO2], hygroscopic white solid, m.p. 55 °C: 1H NMR (250 MHz, CDCl3, δ, ppm) 3.93 (s, 3H, NCH3); 7.14 (s, 1H, aromatic); 7.35 (s, 1H, aromatic); 8.92 (s, 1H, aromatic); 9.87 (br N+–H). 13C NMR (63 MHz, CDCl3, δ ppm): 35.8, 121.3, 122.1, 136.3; 162.1.
N-Methylimidazolium nitrate [Hmim][NO3], hygroscopic white solid, m.p. 67 °C: 1H NMR (250 MHz, D2O δ, ppm) 3.75 (s, 3H, NCH3); 7.33 (s, 2H, aromatic); 8.46 (s, 1H, aromatic). 13C NMR (63 MHz, coaxial tube at 70 °C, δ ppm): 35.2, 118.3, 122.1, 134.3.
N-Methylimidazolium chloride [Hmim]Cl, hygroscopic white solid, m.p. 78 °C: 1H NMR (250 MHz, CDCl3, δ ppm) 3.5 (s, 3H, NCH3); 6.82 (s, 1H, aromatic); 6.87 (s, 1H, aromatic); 8.54 (s, 1H, aromatic); 10.7 (N+–H). 13C NMR (63 MHz, coaxial tube, δ ppm): 35.2, 118.3, 122.1, 134.3.
N-Methylmorpholinium hydrogen sulfate [HMor][HSO4], hygroscopic white solid, m.p. 32 °C: 1H NMR (250 MHz, D2O, δ ppm) 2.85 (s, 3H, N+CH3); 3.20 (m, 2H, HCN+); 3.46 (m, 2H, HCN+); 3.80 (m, 2H, HCO); 4.09 (m, 2H, HCO).13C NMR (63 MHz, D2O, δ ppm) δ: 42.66; 52.60; 63.31.
N-Methylmorpholinium sulfate [HMor]2[SO4], deliquescent white solid: 1H NMR (250 MHz, D2O, δ ppm) 2.90 (s, 3H, N+CH3); 3.20 (m, 2H, HCN+); 3.53 (m, 2H, HCN+); 3.80 (m, 2H, HCO); 4.09 (m, 2H, HCO). 13C NMR (63 MHz, D2O, δ, ppm) δ: 42.60; 52.70; 63.42.
N-Methylmorpholinium chloride [HMor]Cl, deliquescent crystals: 1H NMR (250 MHz, D2O, δ ppm) 2.93 (s, 3H, N+CH3); 3.20 (m, 2H, HCN+); 3.50 (m, 2H, HCN+); 3.80 (m, 2H, HCO); 4.09 (m, 2H, HCO).13C NMR (63 MHz, D2O, δ, ppm) δ: 42.66; 52.70; 63.38.
N-Methylpiperidinium hydrogen sulfate [HPip][HSO4], m.p. 32 °C: 1H NMR (250 MHz, D2O δ ppm): 1.2–1.7 (m, 6H); 2.74 (s, 3H, N+CH3); 2.85 (m, 2H); 3.38 (m, 2H).
N-Methylpiperidinium sulfate [HPip]2[SO4], hygroscopic white solid: 1H NMR (250 MHz, D2O δ ppm): 1.2–1.7 (m, 6H); 2.74 (s, 3H, N+CH3); 2.85 (m, 2H); 3.38 (m, 2H).
N-Methylpyrrolidinium hydrogen sulfate [HPyrr][HSO4], m.p. 34 °C: 1H NMR (250 MHz, D2O, ppm): 3.36 (t, 2H, NCH2), 2.76 (t, 2H, NCH2), 2.62 (s, 3H, NCH3), 1.85–1.73 (m, 4H, NCH2CH2).
N-Methylpyrrolidinium chloride [HPyrr]Cl, m.p. 158–160 °C: 1H NMR (250 MHz, D2O, ppm): 3.30 (t, 2H, NCH2), 2.75 (t, 2H, NCH2), 2.60 (s, 3H, NCH3), 1.85–1.73 (m, 4H, NCH2CH2).
Betaine chloride ([Bet]Cl), m.p. 240 °C: 1H NMR (250 MHz, D2O, δ ppm): 3.28 (s, 9H, (CH3)3N+); 4.26 (s, 1H, COOH); 4.85 (s, 2H, CH2). 13C NMR (63 MHz, D2O, δ ppm): 54.5, 64.0, 168.3.
Betaine nitrate ([Bet][NO3]), highly hygroscopic white solid, m.p. 128 °C: 1H NMR (250 MHz, D2O, δ ppm): 3.28 (s, 3H, (CH3)3N+); 4.25 (s, 1H, COOH); 4.85 (s, 2H, CH2). 13C NMR (63 MHz, D2O, δ ppm): 54.5, 64.2, 171.2.
Betaine hydrogen sulfate ([Bet][HSO4]); m.p. 148 °C: 1H NMR (250 MHz, D2O, δ ppm): 3.28 (s, 3H, (CH3)3N+); 4.25 (s, 1H, –COOH); 4.85 (s, 2H, CH2). 13C NMR (63 MHz, D2O, δ ppm): 54.5, 64.0, 171.3.
Betaine tetrafluoroborate ([Bet][BF4]), m.p. 200 °C: 1H NMR (250 MHz, DMSO-d6, δ ppm): 3.19 (s, 3H, (CH3)3N+); 4.29 (s, 2H, CH2). 13C NMR (63 MHz, D2O, δ ppm): 53.5, 63.0, 166.3.
Procedure for esterification of acetic acid with alcohol (butanol or octanol)
Generally, to 1 g of the selected Brønsted acidic IL, 2 ml of the selected alcohol (butanol or octanol) and 1 equivalent mole of acetic acid (Carlo Erba, 99.9%) were added. In the case of octanol, the reaction mixtures were heated for 4 h at 110 °C. Then, products were separated just by decanting, when two layers were formed, or by extraction with ethyl ether. The crude reaction mixtures were analyzed by NMR and GC-MS. With some selected ILs, reactions were carried out also modifying temperature and time. Results are shown in Tables 1 and 3.Procedure for esterification of acetic acid with methyl β-D-glucopyranoside
To 1.5 g of the selected Brønsted acidic ILs ([Hmim]Cl, [Hmim][NO3], [Hmim][HSO4] or [Hmim][H2PO4]) 0.5 g (0.00258 moles) of methyl β-D-glucopyranoside and 1.1 equivalent mole of acetic acid (Carlo Erba, 99.9%) were added. The reaction mixtures were heated for 4 h at 60 °C. Then, products were extracted with ethyl ether and, after solvent evaporation at reduced pressure, the crude reaction mixtures were analyzed by NMR. Results are reported in Table 4.Procedure for transesterification of ethyl trans-cinnamate
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and [Hmim][HSO4] (1:1.1)) were added. After stopping the reaction, products were extracted with ethyl ether and, after solvent evaporation at reduced pressure, the crude reaction mixtures were analyzed by NMR. Results are reported in Table 6.
:1) and [Hmim][HSO4] (1:1.1)) were added. After stopping the reaction, products were extracted with ethyl ether and, after solvent evaporation at reduced pressure, the crude reaction mixtures were analyzed by NMR. Results are reported in Table 6.
| H0 = pK(I)a + log ([I]/[IH+]) |
According to Lambert–Beer's Law, the values of [I] and [HI+] were calculated from the UV-vis spectra, on the basis of the absorbance values at 380 nm (absorption maximum).
Results and discussion
Several imidazolium, morpholinium, pyrrolidinium, piperidinium, dabco and betaine based Brønsted acidic ILs were used as solvents and catalysts in esterification and transesterification reactions (Scheme 1, not all of them were used in each reaction).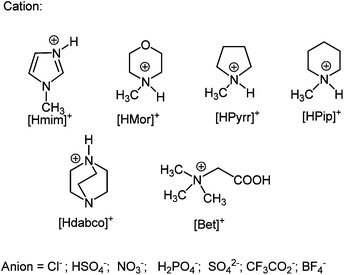 | ||
| Scheme 1 Protic cations used in combination with the reported anions to obtain Brønsted acidic ILs. | ||
Preliminary experiments were carried out at 110 °C, working with a 1:1 molar ratio of octanol and acetic acid (Scheme 2) and using ca. 1 g of the selected IL, the solvent-catalyst loading being around 60% w/w (based on the mass of octanol) corresponding to the IL/octanol molar ratios % reported in Table 1. To assess the catalytic ability of the selected ILs conversions were evaluated by NMR. Data are reported in Table 1. Experiments carried out at shorter reaction times showed that 4 h assured equilibrium conditions, at least in the case of the more efficient media.
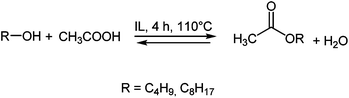 | ||
| Scheme 2 Fischer esterification of acetic acid. | ||
No by-product arising from side reactions has been detected in all the examined reactions. In the case of [Hmim][HSO4], experiments were also carried out at 75 °C and varying IL loading from 60% w/w to 30% w/w (Table 1, entries 1–3). A moderate effect of the temperature and an extremely low effect of IL loading were observed. Similar results were also obtained in the case of [HPyrr][HSO4] and [HPip][HSO4] showing that, at least with these highly active systems, lower amounts of IL can be sufficient.
| Entry | IL | IL/alcohol (%) | Ester:alcohola |
Recovered material (%) |
|---|---|---|---|---|
| a Determined by NMR. b Reaction carried out at 75 °C. | ||||
| 1 | [Hmim][HSO4]b | 46 | 76:24 |
83 |
| 2 | [Hmim][HSO4] | 46 | 82:18 |
85 |
| 3 | [Hmim][HSO4] | 25 | 80:20 |
82 |
| 4 | [HMor][HSO4] | 42 | 80:20 |
85 |
| 5 | [HPyrr][HSO4] | 45 | 92:8 |
95 |
| 6 | [HPip][HSO4] | 42 | 88:12 |
87 |
| 7 | [HPip]2[SO4] | 30 | 55:45 |
63 |
| 8 | [HMor]2[SO4] | 28 | 41:59 |
68 |
| 9 | [Hmim]2[SO4] | 32 | 20:80 |
63 |
| 10 | [Hmim][H2PO4] | 46 | 21:79 |
83 |
| 11 | [Hmim][NO3] | 48 | 62:38 |
65 |
| 12 | [HPyrr]Cl | 60 | 24:76 |
81 |
| 13 | [Hdabco]Cl | 55 | 20:80 |
75 |
| 14 | [HMor]Cl | 60 | 34:66 |
55 |
| 15 | [Hmim][CF3CO2] | 42 | 43:57 |
85 |
| 16 | [HBet]Cl | 54 | 85:15 |
85 |
It is noteworthy that for all ILs having HSO4−, SO42−, NO3− and H2PO4− as counteranions, after the stopping of the reactions, when the temperature was brought to room conditions, two phases were formed (Fig. 1); the lower phase was constituted by the IL and water and the upper layer by the formed ester and unreacted alcohol. This latter could be easily separated from the IL by simply decanting into another flask. A product recovery (ester and eventually unreacted alcohol) higher than 80% characterized ILs having HSO4− and H2PO4− as counteranions. Since this procedure gave lower recoveries in the case of SO42− and NO3− based ILs (ranging from 50–60%) for these latter catalytic systems product extraction with ethyl ether was also applied, a moderate increase in product recovery was observed. In all examined cases, the IL (lower layer) could be dried under reduced pressure to remove the water formed during the esterification process and eventually recycled.
![Situation at the end of the reaction. Left: Two different layers are formed. Top layer (ester + unreacted alcohol) lower layer (IL + water formed during the reaction). Right (reaction in [Hdabco]Cl): Just one solid layer was formed.](/image/article/2013/GC/c2gc35941c/c2gc35941c-f1.gif) | ||
| Fig. 1 Situation at the end of the reaction. Left: Two different layers are formed. Top layer (ester + unreacted alcohol) lower layer (IL + water formed during the reaction). Right (reaction in [Hdabco]Cl): Just one solid layer was formed. | ||
In the case of [Hdabco]Cl and [HPyrr]Cl, since at room temperature the reaction mixture was practically a unique solid phase enclosing the reaction product, ethyl ether was used to recover the product. On the other hand, another situation characterized the same process in [HMor]Cl; at room temperature the IL solidified and formed the lower layer whereas the ester constituted the upper liquid layer. Hence, the solvent-free separation could be applied also in this case.
On the basis of the data reported in Table 1 it is possible to establish that the catalytic activity of the investigated Brønsted acidic ILs follows the order [HPyrr][HSO4] > [HPip][HSO4] > [HBet]Cl > [Hmim][HSO4] > [HMor][HSO4] > [Hmim][NO3] > [HPip]2[SO4] > [Hmim][CF3COO] > [HMor]2[SO4] > [HMor]Cl > [HPyrr]Cl > [Hmim][H2PO4] > [Hmim]2[SO4] = [Hdabco]Cl. The Brønsted acidic ILs having hydrogen sulfate as the counteranion give the best results: [HPyrr][HSO4] is able to assure up to 92% of conversion in 4 h at 110 °C with a product recovery higher than 95%. On the other hand, Brønsted acidic ILs having the unprotonated sulfate as an anion give significantly lower conversions. This behavior suggests that the presence of a sufficiently acidic proton on the anionic component of the IL plays an important role in the activation of the reaction. About this, it is noteworthy that when [Hmim][H2PO4] was used as a catalyst and solvent only very low conversions were obtained (21%) suggesting a lower acidity of the [H2PO4]− protons with respect to the acidic proton of [HSO4]− also in the non-aqueous environment characterizing neat ILs.
On the other hand, only a moderate conversion was observed in [Hmim][NO3] and [Hmim][CF3CO2], 62% and 43% respectively, whereas the Brønsted acidic ILs having Cl− as a counteranion gave lower conversions; the ester percentages ranged from 20 to 34%. The stronger interactions between cations and anions in ILs bearing highly coordinating anions (such as chloride and trifluoroacetate)10 probably reduce the ability of the IL cation to act as a catalyst although the role of other factors (such as water binding ability) cannot be excluded.
An evaluation of the role of the IL acidity in this reaction was performed determining the Hammett acidity functions (H0) of seven ILs which give significantly different conversions under comparable conditions.
The H0 values, reported in Table 2, were measured spectrophotometrically determining the protonation of an uncharged indicator (4-nitroaniline) in water in terms of the measurable ratio of [I]/[IH+]. From the data reported in Table 2 the acidity decreases in the following order, [HBet]Cl > [HPip][HSO4] > [Hmim][HSO4] > [Hmim][NO3] > [Hmim][CF3CO2] > [Hmim]2[SO4] > [Hmim][H2PO4]. The strongest acids ([HBet]Cl, [HPip][HSO4], [Hmim][HSO4]) gave the highest yields, whereas considerably lower conversions were achieved using [Hmim]2[SO4] and [Hmim][H2PO4], which are the weakest acids.
As previously observed,11 the acidity of Brønsted acidic ILs in water, as well as in other polar solvents, depends on anion and cation nature: a fairly good correlation between the strength of the acid and the conversion (%) can be evidenced (Fig. 2).
| ILs | H 0 | Corrosivenessa (mg cm−2) |
|---|---|---|
| a The stainless steel sheets were polished, cleaned and dried to constant weight, then fully immersed in the same amount of IL (15 g). After 24 h at 85 °C, the stainless steel sheets were taken out of the IL, washed in water, dried and weighed. Being this IL a solid, having a high melting point, the experiment was carried out in the presence of 5% of water. b The absorbance change was too small to obtain a significant value. | ||
| [Hmim][HSO4] | 1.93 | 0.08 |
| [HPip][HSO4] | 1.58 | 0.05 |
| [Hmim]2[SO4] | 2.87 | 0.07b |
| [Hmim][CF3CO2] | 2.55 | 0.03 |
| [Hmim][NO3] | 2.22 | 0.06 |
| [Hmim][H2PO4] | ∼3b | 0.01 |
| [Bet]Cl | 1.47 | ND |
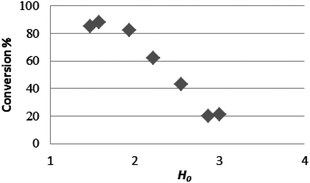 | ||
| Fig. 2 Relation between IL acidity (H0) and conversion of acetic acid esterification with octanol. | ||
However, the deviations from linear behavior characterizing the plot of Fig. 2 suggest that the acidity of these media is not the sole factor affecting the efficiency of the process. Probably, as previously suggested6h in a recent paper, when the IL is used as in this study in a relevant amount its ability to bind the formed water and contemporaneously to give a biphasic system with the formed ester can shift the equilibrium towards products, increasing the process efficiency.
Finally, it is noteworthy that when a task-specific ammonium-based salt, [HBet]Cl, bearing a carboxy group on the alkyl chain has been used, despite the presence of chloride as a counteranion, a conversion comparable to that found in [HSO4]− based ILs was obtained; the presence of the acidic group (–COOH) on the cation is able to assure the catalytic effect. Although in principle the carboxylic group on the IL could be esterified, probably the presence of the adjacent positive charge reduces this ability: no transformation on the IL has been detected by NMR analysis. It is also to note that since this IL is a high melting point solid and the product is not soluble in this medium it has been possible to recover the formed ester through a simple decanting procedure. [HBet]Cl may therefore represent a valid alternative to the other protic Brønsted acidic ILs.
On the other hand, since the best results were obtained using [HPyrr][HSO4], this IL has been selected for recycling experiments. After three recycles, conversion decreased from 92% to 85%, although the recovery of the product by simple decantation of the upper phase becomes less efficient (from 85% to 70%).
[HPyrr][HSO4] was also used as a test solvent and catalyst for the esterification of acetic acid with butanol. Considering the lower boiling point of butanol, the reaction was initially carried out at four different temperatures ranging from 85–100 °C by working with a 1:1 molar ratio of butanol–acetic acid and an IL loading of 60% w/w (based on the mass of butanol). To evaluate the catalytic ability of the different ILs, reactions were stopped after 2–4 h and the conversions were evaluated by NMR. On the basis of these data, we decided that a temperature between 80–90 °C and 2 h of reflux might represent the best conditions to evaluate the efficiency of acidic ILs in this reaction having practically reached the equilibrium.
Therefore, the same conditions were applied to test the Brønsted acidic ILs reported in Table 3, which have been found to give the higher conversions in octyl acetate synthesis. In these media, at the end of the reaction also in the case of butyl acetate synthesis two phases were obtained and the product was recovered simply by upper phase separation.
| Entry | IL | IL/alcohol (%) | Ester:alcoholb |
|---|---|---|---|
| a Yields of the recovered product (ester + unreacted alcohol) by decantation ranged around 85–90%. b Determined by NMR. | |||
| 1 | [HPyrr][HSO4] | 24 | 90:10 |
| 2 | [HPip][HSO4] | 23 | 75:25 |
| 3 | [Hmim][HSO4] | 25 | 88:12 |
| 4 | [HBet]Cl | 29 | 85:15 |
| 5 | [HBet][HSO4] | 21 | 79:21 |
| 6 | [HBet][NO3] | 25 | 67:33 |
| 7 | [HBet][BF4] | 22 | 65:35 |
Once again, [HPyrr][HSO4] assured the higher conversion although comparable values were also obtained in [Hmim][HSO4] and [HBet]Cl. Nevertheless, recycling experiments confirmed the possibility to carry out at least three consecutive processes in these ILs without significant reduction in conversion.
The selectivity of the esterification reaction was checked using a more complex alcohol, methyl β-D-glucopyranoside, characterized by a primary hydroxyl group and three secondary equatorial hydroxyl groups (Scheme 3). As a consequence of the low solubility of the substrate in most of the synthesized acidic ILs the reactions were conducted at a lower reagent concentration (around 1.5 M) only in the ILs reported in Table 4. The behavior of the reaction was checked by stopping the esterification at prefixed times (1, 2 and 4 h and eventually longer). The reaction mixtures were extracted with ethyl ether (extraction yield higher than 75%) and product distribution was analyzed by NMR (Fig. 3).
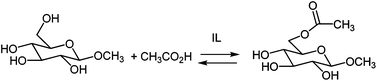 | ||
| Scheme 3 Esterification between methyl β-D-glucopyranoside with acetic acid. | ||
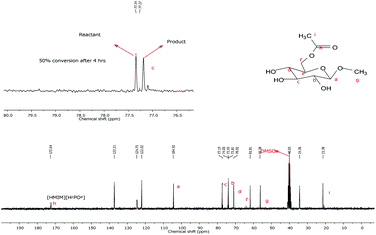 | ||
| Fig. 3 13C NMR of the crude reaction mixture arising from acetylation of methyl β-D-glucopyranoside. | ||
In all the investigated ILs, the reaction occurred with high regioselectivity: only the acetyl derivative arising from esterification of the primary hydroxyl group on C(6) was detected besides the unreacted product, conversions at different reaction times are reported in Table 4.
It is noteworthy that in the case of [Hmim][NO3] a decrease in conversion on increasing the reaction time from 2 to 4 h was observed. This peculiar behavior has to be attributed to the nature of [Hmim][NO3] which, probably less than [Hmim][HSO4], is able to “capture” the produced water. In this case the product is soluble in the ionic liquid and the presence of increasing amounts of free-water after a given conversion (the water coordination ability of the nitrate anion is not infinite!) may favor the hydrolysis process and shift the equilibrium position towards reagents. It is noteworthy that also in this reaction the higher conversion was obtained in the IL having hydrogen sulfate as a counteranion, [Hmim][HSO4]. Finally, attempts to increase the conversion prolonging the reaction beyond 4 h failed due to the formation of relevant amounts of polyacetylation products.
In this study also the possibility to use Brønsted acidic ILs in transesterification processes was checked investigating the reaction of ethyl trans-cinnamate with a small alkyl-chain alcohol (methanol) and with a longer alkyl-chain alcohol (octanol) (Scheme 4). Initially, the reaction with methanol was investigated in [HPip][HSO4] and temperature and reaction time were varied to find the best conditions.
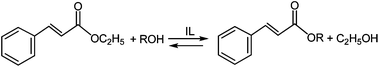 | ||
| Scheme 4 Transesterification of ethyl trans-cinnamate. | ||
Practically, no reaction occurred, even after 24 h, until the temperature was raised to 90 °C. Since at this temperature we could not exclude that the moderate conversion (50%) was at least partially due to the volatility of methanol the reaction was carried out also using an excess (3 equiv.) of this reagent: conversion increased at 73%. The efficiency of other acidic ILs was also tested at 90 °C. Results are reported in Table 5.
| Entry | IL | Conversion % | Recovered material % |
|---|---|---|---|
| a All the reactions were carried out at 90 °C for 28 h. b In this experiment 1.1 equiv. of acid with respect to the nitrogen base was used to prepare [HPyrr][HSO4]. | |||
| 1b | [HMor]Cl | NR | 0 |
| 2c | [HMor]Cl | NR | 0 |
| 3b | [HBet][HSO4] | NR | 0 |
| 4c | [HPyrr][HSO4] | 95 | 95 |
| 5b | [Hmim][HSO4] | 37 | 87 |
| 6b | [Hmim][HSO4]b | 75 | 92 |
| 7b | [HPip][HSO4] | 73 | 90 |
Whereas no reaction was observed in chloride based ILs, also working with an excess of alcohol, conversions ranging from 40 to 95% characterized reactions performed in hydrogen sulfate-based ILs. The best results were obtained, also for the transesterification process, in [HPyrr][HSO4]. It is also noteworthy that, in particular in [Hmim][HSO4], a significant increase in conversion was obtained working in the presence of a small excess of H2SO4, probably as a consequence of the superacidic behavior of species of the type A⋯H–A−, which should be present in the reaction mixture.
When the transesterification process of ethyl trans-cinnamate in [Hmim][HSO4] was carried out using octanol, and a 1:1 molar ratio ester: alcohol, lower conversions were obtained under comparable conditions. However, the presence of a small excess of inorganic acid (H2SO4) significantly increased the conversion also in this case (Table 6).
Finally, corrosive tests were carried out with six ILs using stainless steel plates and determining the weight loss after complete immersion in the selected ILs for 24 h at 85 °C. The results (Table 2) showed that all the investigated ILs are practically non-corrosive media, in agreement with recent studies performed on analogous ILs.6h,12
Conclusions
In conclusion, these data show that the water scavenging nature of many Brønsted acidic ILs and the insolubility of many esters in these media strongly affect the catalytic efficiency of these salts in esterification and transesterification reactions. In both cases, protic ILs bearing Brønsted acidic onium (pyrrolidinium) or imidazolium cations, associated with the “acidic” [HSO4]− anion, gave the highest yields. In the case of esterification of acetic acid with octanol and butanol a high conversion was also obtained using a task-specific ammonium-based IL, [HBet]Cl, bearing an acidic group (–COOH) on the alkyl chain. Nevertheless, acetylation of a relatively complex alcohol, such as methyl β-D-glucopyranoside, in Brønsted acidic ILs occurs with high regioselectivity: only the acetyl derivative arising from esterification of the primary hydroxyl group on C(6) was detected, besides the unreacted product (30%), when the reaction was performed in [Hmim][HSO4]. Finally, the improved catalytic ability of the 1:0.1 [Hmim][HSO4]–H2SO4 mixture in the transesterification reaction, probably as a consequence of the superacidic behavior of species of the type A⋯H–A−, has been evidenced.
Acknowledgements
We thank the University of Pisa and Fondazione CariPisa for financial support.Notes and references
- C. Chiappe and S. Rajamani, Eur. J. Org. Chem., 2011, 5517 CrossRef CAS.
- S. J. Zhang and X. M. Lv, Ionic Liquid-from Foundational Research to Industrial Application, Science Publication, Beijing, 2006 Search PubMed.
- R. C. Larock, Comprehensive Organic Transformations, VCH, New York, 1999 Search PubMed.
- P. A. Ganeshpure, G. George and J. Das, ARKIVOC, 2007, 273 CAS.
- Y. Q. Deng, F. Shi, J. J. Beng and K. Qiao, J. Mol. Catal. A: Chem, 2001, 165, 33 CrossRef CAS.
- (a) H.-P. Zhu, F. Yang, J. Tang and M. Y. He, Green Chem., 2003, 5, 38 RSC; (b) J. Fraga-Dubreuil, K. Bourahla, M. Rahmouni, J. P. Bazureau and J. Hamelin, Catal. Commun., 2002, 3, 185 CrossRef CAS; (c) T. Joseph, S. Sahoo and S. B. Halligudi, J. Mol. Catal. A: Chem., 2005, 234, 107 CrossRef CAS; (d) S. H. Chen, Q. Zhao and X. U. Xu, J. Chem. Sci., 2008, 120, 481 Search PubMed; (e) C. Yue, Q. Liu, T. Yi and Y. Chen, Monatsh. Chem., 2010, 141, 975 Search PubMed; (f) Z. Wei, F. Li, H. Xing, S. Deng and Q. Ren, Korean J. Chem. Eng., 2009, 26, 666 Search PubMed; (g) D. Fang, X.-L. Zhou, Z. W. Ye and Z. L. Liu, Ind. Eng. Chem. Res., 2006, 45, 7982 CrossRef CAS; (h) D.-J. Tao, Y. T. Wu, Z. Zhou, J. Geng, X. B. Hu and Z. B. Zhang, Ind. Eng. Chem. Res., 2011, 50, 1989 Search PubMed.
- A. C. Cole, J. L. Jensen, I. Ntai, K. L. T. Tran, K. J. Weaver, D. C. Forbes and J. H. Davis Jr., J. Am. Chem. Soc., 2002, 124, 5962 CrossRef CAS; J. Fraga-Dubreuil, K. Bourahla, M. Rahmouni, J. P. Bazureau and J. Hamelin, Catal. Commun., 2002, 3, 185 CrossRef CAS; H. P. Nguyen, S. Znifeche and M. Baboulene, Synth. Commun., 2004, 34, 2085 CrossRef CAS; A. Arfan and J. P. Bazureau, Org. Process Res. Dev., 2005, 9, 743 Search PubMed; H. Xing, T. Wang, Z. Zhou and Y. Dai, Ind. Eng. Chem. Res., 2005, 44, 4147 CrossRef CAS; S. A. Siddiqui, Synlett., 2006, 155 Search PubMed; Z. Zhang, W. Wu, B. Han, T. Jiang, B. Wang and Z. Liu, J. Phys. Chem. B, 2005, 109, 16176 CrossRef CAS; J. Gui, X. Cong, D. Liu, X. Zhang, Z. Hu and Z. Sun, Catal. Commun., 2004, 5, 473 CrossRef CAS.
- P. A. Ganeshpure, G. George and J. Das, J. Mol. Catal. A: Chem., 2008, 279, 182 CrossRef CAS.
- C. Pretti, C. Chiappe, I. Baldini, S. Brunini, G. Monni and L. Intorre, Ecotoxicol. Environ. Saf., 2009, 72, 1170 CrossRef; S. Bruzzone, C. Chiappe, S. E. Focardi, C. Pretti and M. Renzi, Chem. Eng. J., 2011, 175, 17 CrossRef CAS.
- R. Bini, O. Bortolini, C. Chiappe, D. Pieraccini and T. Siciliano, J. Phys. Chem. B, 2007, 111, 598 CrossRef CAS.
- R. Kore, T. J. D. Kumar and R. Srivastava, J. Mol. Catal. A: Chem, 2012, 360, 61 CrossRef CAS; X. Tong and Y. Li, ChemsusChem, 2010, 3, 350 CAS; N. Yan, Y. Yuan, R. Dykeman, Y. Kou and P. J. Dyson, Angew. Chem., Int. Ed., 2010, 49, 5549 CrossRef CAS; R. Kore and R. Srivastava, Tetrahedron Lett., 2012, 53, 3245 CrossRef CAS.
- B. Zhou, Y. Fang, H. Gu, S. Zhang, B. Huang and K. Zhang, Front. Chem. Eng. China, 2009, 3, 211 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |