 Open Access Article
Open Access ArticleStille couplings in water at room temperature†
Guo-ping
Lu
ab,
Chun
Cai
b and
Bruce H.
Lipshutz
*a
aDepartment of Chemistry & Biochemistry, University of California, Santa Barbara, California 93106, USA. E-mail: lipshutz@chem.ucsb.edu
bChemical Engineering College, Nanjing University of Science & Technology, Nanjing, Jiangsu 210094, P. R. China
First published on 1st October 2012
Abstract
A nonionic amphiphile, TPGS-750-M, enables efficient Stille couplings between a wide range of substrates to be conducted in water as the only medium, in most cases at room temperature.
Introduction
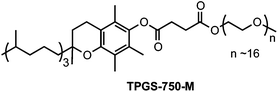
In a recent review on Nobel Prize-winning Suzuki, Heck, and Negishi couplings by Colacot, Snieckus and co-workers,1 data was presented indicative of the number of papers and patents associated with each of the commonly used cross-coupling reactions from the last decade. While Suzuki, Heck, and Sonogashira reactions ranked among the top three, fourth was Stille couplings with ca. 2000 citations. Thus, Pd-catalyzed Stille couplings of various organo- and pseudo-halides with organostannanes represent one of the most powerful tools for the formation of carbon–carbon bonds.2 Their popularity is usually attributed to the stability and functional group tolerance of stannanes, as well as their chemoselectivity and broad scope in terms of reaction partners.3 Traditionally, Stille couplings are carried out in organic solvents (e.g., THF, DMF, NMP), and oftentimes at temperatures above ambient.4 Although many attempts have been made to perform these reactions under thermally mild conditions,5 use of (potentially toxic) organic solvents is always the norm, which by definition fails to meet the 12 Principles of Green Chemistry.6Ideally, Stille couplings would be carried out at room temperature, in water as the only medium. Several nanometal catalysts have been designed to enable Stille couplings to be run under such conditions.7 However, opportunities for variation in substrate type are limited. More commonly, high temperatures are required in the presence of various Pd catalysts.8 Recently, we have reported that TPGS-750-M (polyethanol-α-tocopherylsuccinate; shown below) is an excellent commercially available “designer” surfactant that self-assembles in water to form nanomicelles within which several cross-couplings efficiently take place.9 Herein, we describe a new protocol for conducting Stille couplings in an aqueous solution containing TPGS-750-M, in most cases at room temperature.
Results and discussion
Scattered literature reports have mentioned that Pd(P(t-Bu)3)2 is an unusually reactive catalyst for Stille couplings of aryl bromides and chlorides.4a,5c,10 Thus, three aryl bromides were initially coupled with vinyl- or 2-furyltributyltin using Pd(P(t-Bu)3)2 (2 mol%). Using 2 wt.% aqueous TPGS-750-M at room temperature (0.25 M), excellent yields of products 1–3 were obtained in two to four hours (Scheme 1).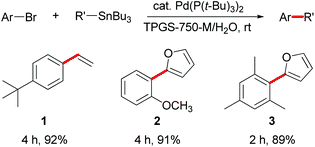 | ||
| Scheme 1 Stille couplings with aryl bromides in water at room temperature. | ||
Based on these initial results, both 3-chlorotoluene and 2-furyltributyltin were selected to investigate potential couplings between aryl chlorides, and to further optimize reaction conditions to be applied to couplings involving more challenging organotin partners (Table 1). Initially, several commonly used catalysts and additives were screened (entries 1–14). The combination of Pd(P(t-Bu)3)2 and DABCO4c,11 emerged as the best choice. Remarkably, the limited conversion initially observed could be increased dramatically by adding one equivalent of NaCl (entries 15, 16), but not NaF (entry 17), presumably due to the enlarged, reorganized micelles which offer increased surface area and thus, increased binding constants for substrates and catalysts.9 Alternatively, the positive effects of chloride on the palladium center may be operative.12 A good yield of product 4 (85%) could be achieved by slight heating to 50 °C. The corresponding coupling run “on water” (i.e., in the absence of TPGS-750-M), led to lower conversion (entry 16 vs. entry 18).13 Screening various surfactants indicated that both CTAB, an ionic surfactant (entry 22), and the neutral amphiphile TPGS-750-M were equally effective in this model reaction.
|
|
|||
|---|---|---|---|
| Entry | Surfactant | Additive | Conversionb (%) |
| a Conditions: aryl halide (0.250 mmol), organotin reagent (0.275 mmol), Pd(P(t-Bu)3)2 (0.005 mmol for entries 1–22, 0.010 mmol for entries 23–28), solution of aqueous surfactant (2 wt.%, 1 mL), additive (0.750 mmol) rt, 24 h. b Conversion determined by GC. c The catalyst is Pd(P(t-Bu)2OH)2Cl2 (2 mol%). d The catalyst is Pd(OAc)2/2Xphos (2 mol%). e The catalyst isPd2(dba)3/4P(o-Tol)3 (1 mol%). f 20 mol%. g 1.0 equiv. h At 50 °C. i Isolated yield. j At 40 °C. | |||
| 1c | TPGS-750-M | / | NR |
| 2d | TPGS-750-M | / | 20 |
| 3e | TPGS-750-M | / | NR |
| 4 | TPGS-750-M | / | 27 |
| 5 | TPGS-750-M | CuIf | NR |
| 6 | TPGS-750-M | LiCl | 14 |
| 7 | TPGS-750-M | CsF | 4 |
| 8 | TPGS-750-M | Bu4NF | 17 |
| 9 | TPGS-750-M | Net3 | 30 |
| 10 | TPGS-750-M | NaOH | 7 |
| 11 | TPGS-750-M | K2CO3 | 34 |
| 12 | TPGS-750-M | K2CO3 + CuClf | 17 |
| 13 | TPGS-750-M | K2CO3 + ZnBr2f | 12 |
| 14 | TPGS-750-M | DABCO | 37 |
| 15 | TPGS-750-M | NaClg | 32 |
| 16 | TPGS-750-M | DABCO + NaCl g | 50, 85 h , i |
| 17 | TPGS-750-M | DABCO + NaFg | 35 |
| 18 | None | DABCO + NaClg | 39 |
| 19 | SDS | DABCO + NaClg | 50 |
| 20 | Trition X-100 | DABCO + NaClg | 40 |
| 21 | Brij 30 | DABCO + NaClg | 39 |
| 22 | CTAB | DABCO + NaClg | 55 |
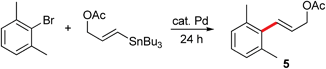
|
|||
| 23 | TPGS-750-M | / | 12 |
| 24 | TPGS-750-M | K2CO3 | 14 |
| 25 | TPGS-750-M | DABCO | 44 |
| 26 | TPGS-750-M | DABCO + NaCl g | 76 i , 85 i , j |
| 27 | CTAB | DABCO + NaClg | 68i |
| 28 | None | DABCO + NaClg | 12 |
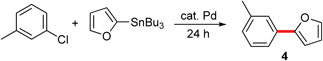
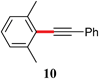
On the other hand, in the reaction between 2-bromo-1,3-dimethylbenzene and (E)-3-(tributylstannyl)allyl acetate, TPGS-750-M proved to be more effective than CTAB, run under otherwise identical conditions (entry 26 vs. 27). Very low conversion to the desired product 5 was observed when the reaction was conducted “on water” (entry 28), indicative of the vagaries associated with this type of approach to cross-couplings, as observed previously on many occasions.13
With optimized conditions in hand, several combinations of aryl halides and organotin reagents were investigated to ascertain the scope of the protocol (Table 2). In general, aryl bromides coupled smoothly at room temperature. Tributylphenyltin appeared to be a less active coupling partner than other organotin reagents. Although in the case of p-bromoanisole (entry 5), P(P(t-Bu)3)2 led to full conversion at 50 °C, a poor yield of desired product was obtained due to homocoupling of the bromide. Switching to Pd2(dba)3/P(o-Tol)3 with increased catalyst loading (4 mol%) was found to enhance the yield at the expense of homocoupling.
|
|
|||||
|---|---|---|---|---|---|
| Entry | X | Time (h) | T (°C) | Product | Yieldb (%) |
a Conditions: aryl halide (0.250 mmol), organotin reagent (0.275 mmol), Pd(P(t-Bu)3)2 (0.005 mmol), aqueous TPGS-750-M solution (2 wt.%, 1 mL), DABCO (0.750 mmol), NaCl (0.250 mmol).
b Isolated yield.
c 2 mol% Pd2(dba)3 and 8 mol% P(o-Tol)3 are used as catalyst.
d 4 mol% Pd(P(t-Bu)3)2 is used.
e A mixture of 15 and 4-butyl-2-nitrotoluene (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) determined by 1H NMR.
f Trimethylphenyltin (1.1 equiv) used. :1) determined by 1H NMR.
f Trimethylphenyltin (1.1 equiv) used.
|
|||||
| 1 | Br | 24 | rt |
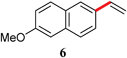
|
93 |
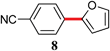
| 2 | Br | 4 | rt |
|
88 |
| 3 | Br | 4 | rt |
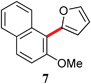
|
94 |
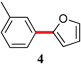
| 4 | Cl | 24 | rt |
|
94 |
| 5 | Br | 24 | 50 |
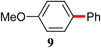
|
39, 80c |
| 6 | Br | 4 | rt |
|
91 |
| 7 | Br | 5 | rt |
|
92 |
| 8 | Br | 1 | rt |
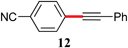
|
95 |
| 9 | Cl | 24 | 50 | 75d | |
| 10 | Br | 4 | rt |
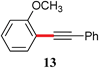
|
97 |
| 11 | Cl | 24 | 50 | NRd | |
| 12 | Br | 5 | rt |
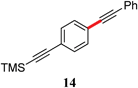
|
97 |
| 13 | Cl | 24 | 60 |
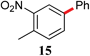
|
78d,e |
| 14 | Cl | 24 | 60 | 92d,f | |
| 15 | Cl | 24 | 50 |
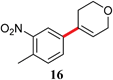
|
45d |
| 16 | Cl | 24 | 60 | 73d | |

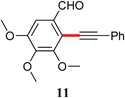
The analogous reactions of aryl chlorides, not surprisingly, were more sluggish. However, in some cases, e.g., 2-furyltributyltin and p-chlorobenzonitrile, the coupling was successful even at room temperature (entry 4). Generally, the couplings took place under the influence of mild heat, along with an increase in catalyst loading to 4 mol% (entries 9, 13–16). No product was formed, however, in the reaction of an electron-rich aryl chloride (e.g., o-chloroanisole) with tributyl(phenylethynyl)-stannane (entry 11). It should be noted that the byproduct 4-butyl-2-nitrotoluene was formed when 4-chloro-2-nitrotoluene was coupled with either of two tributylstannanes (entries 13, 15). The by-product in the case of Bu3SnPh could be avoided by switching to the trimethylstannyl analog (entry 14).
To compare and contrast these couplings, guided by the principles of green chemistry,5 with traditional Stille coupling conditions, a direct comparison was performed between 3-bromobenzothiophene and 2-furyltributyltin (Scheme 2). Rather than an organic solvent (dioxane), additives (excess cesium fluoride), and heat (80 °C),14 the “green” protocol afforded a considerably higher yield, done in water at room temperature, and in a shorter time frame.
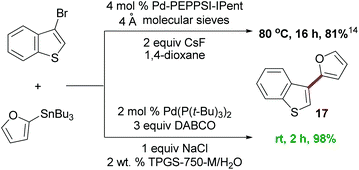 | ||
| Scheme 2 The reaction of 3-bromobenzothiophene with 2-furyltributyltin. | ||
Likewise, Stille couplings with alkenyl halides could also be effected under otherwise identical conditions (Table 3). Both alkenyl iodides and bromides readily participate at room temperature to afford products 18–21 (entries 1–4). In the case of (Z)-β-bromostyrene, significant undesired Z-to-E isomerization,15 as well as homocoupling, took place (entry 5). However, switching catalysts from Pd(P(t-Bu)3)2 to Pd2(dba)3/P(o-Tol)3 led to retention of stereochemistry giving Z-22 in high yield, while avoiding homocoupling (entry 6).
|
|
|||||
|---|---|---|---|---|---|
| Entry | X | Time (h) | Product | Yieldb (%) | |
| a Conditions: alkenyl halide (0.250 mmol), organotin reagent (0.275 mmol), Pd(P(t-Bu)3)2 (0.005 mmol), aqueous TPGS-750-M solution (2 wt.%, 1.0 mL), DABCO (0.750 mmol), NaCl (0.250 mmol), rt. b Isolated yield. c The Z/E ratio of β-bromostyrene is 96/4. d Z/E ratio of product is determined by GC on the crude products. e 1 mol% Pd2(dba)3 and 4 mol% P(o-Tol)3 are used as catalyst. | |||||
| 1 | I | 6 |

|
18 | 88 |
| 2 | I | 4 |

|
19 | 91 |
| 3 | Br | 24 |

|
20 | 89 |
| 4 | I | 2 |
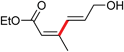
|
21 | 94 (Z/E = 99/1) |
| 5c | Br | 24 |
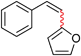
|
22 | 50 (Z/E = 90/10)d |
| 6c | Br | 24 | 90 (Z/E = 96/4)d,e | ||
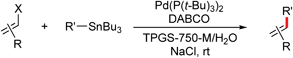
Interestingly, while the literature illustrates a traditional coupling between Z-alkenyl triflate 23 and tributyl-4-methoxyphenylstannane in NMP leading to isomerized E-product 24,16 the desired Z-24 is formed under micellar conditions (Scheme 3). To further demonstrate the potential of this methodology, dienoate 25, a crucial intermediate en route to mammalian V-ATPase inhibitor 26,17 was smoothly generated by reaction between (E)-ethyl 3-iodoacrylate and (E)-3-(tributylstannyl)-prop-2-en-1-ol, yielding the unprotected alcohol 25 in water at room temperature (Scheme 4).
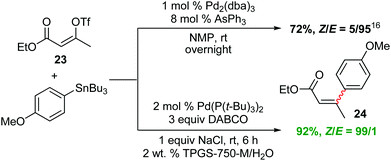 | ||
| Scheme 3 Stille couplings with a Z-alkenyl triflate. | ||
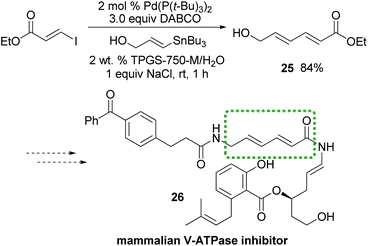 | ||
| Scheme 4 A potential application of Stille couplings to total synthesis. | ||
Finally, the prospects for recycling aqueous solutions of TPGS-750-M used in Stille couplings were studied (Table 4). After the successful coupling of 2-bromo-1,3-dimethylbenzene and tributyl(phenylethynyl)stannane under optimized conditions (entry 1), in-flask extractions with minimal amounts of hexane led to facile product isolation. The extraction also removed much of the Pd catalyst, accounting for the reduced yield (22%; entry 2). The addition of fresh catalyst to each recycle increased the level of conversion and the resulting yields returned to that seen in the initial experiment (entries 3–5).
|
|
|||
|---|---|---|---|
| Entry | Run | Time (h) | Yieldb (%) |
| a Conditions: 2,6-dimethylbromobenzene (0.250 mmol), tributyl phenylethynyltin (0.275 mmol), Pd(P(t-Bu)3)2 (0.005 mmol), aqueous TPGS-750-M solution (2 wt.%, 1.0 mL), DABCO (0.750 mmol), NaCl (0.250 mmol), rt. b Isolated yield. c Additional 2 mol% Pd(P(t-Bu)3)2 is used. d Yield after 4 h with additional 2 mol% Pd(P(t-Bu)3)2. e No additional Pd(P(t-Bu)3)2 added; conversion determined by GC. | |||
| 1 | 1 | 4 | 91 |
| 2c | 2 | 4 | 80d (22)e |
| 3c | 3 | 6 | 92 |
| 4c | 4 | 6 | 89 |
| 5c | 5 | 6 | 88 |
Conclusions
In summary, Stille couplings can be performed in water typically at room temperature by employing nanoreactors formed from the nonionic designer surfactant TPGS-750-M. The catalyst system Pd(P(t-Bu)3)2/DABCO leads, in many cases, to a wide range of couplings between various organostannanes and both aryl and alkenyl halides. The newly developed procedures are environmentally friendly in that no organic solvent is required in these couplings, limited amounts of water are invested, and workup entails only an in-flask extraction with a minimal amount of a single, recoverable organic solvent. These reactions take place in high yields and stereoselectivity, thereby offering considerable potential for applications to complex targets in organic synthesis.Experimental
General procedure for Stille couplings in water
The palladium catalyst (0.005 mmol), organohalides (0.250 mmol), DABCO (0.750 mmol) and NaCl (0.250 mmol) were weighed into a microwave vial at room temperature, and the organotin reagent (0.275 mmol) and 2 wt.% aqueous TPGS-750-M solution (1.0 mL) were then added by syringe. (The liquid organohaildes were also added by syringe.) The resulting solution was allowed to stir at room temperature (slight heating was required in some cases) and monitored by GC or TLC. Upon completion, the reaction mixture was then diluted with NEt3 (0.3 mL) and EtOAc (4.0 mL), filtered through a bed of silica gel layered over Celite. The volatiles were removed in vacuo to afford the crude product. The extent of conversion and Z/E ratios were determined by GC. Further column chromatography on silica gel afforded the pure desired product.Procedure for recycling aqueous solutions of TPGS-750-M
After completion of the reaction, the mixture was extracted by hexane (4 × 1 mL). The organic layer was separated from the aqueous layer by syringe. The remaining hexane in the micellar solution was removed in vacuo. For the second run, fresh starting materials and Pd(P(t-Bu)3)2 (2 mol%) were added to the aqueous solution, and the reaction was conducted as described for the initial run.Characterization data of new compounds
Notes and references
- C. C. Carin, J. Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef CAS.
- (a) M. Kosugi, K. Sasazawa, Y. Shimizu and T. Migita, Chem. Lett., 1977, 301 CAS; (b) D. Milstein and J. K. Stille, J. Am. Chem. Soc., 1978, 100, 3636 CrossRef; (c) M. Kosugi and K. Fugami, J. Organomet. Chem., 2002, 653, 50 CrossRef CAS . For an updated review, see: Y. Peng and W.-D. Z. Li, Eur. J. Org. Chem., 2010, 6703 Search PubMed , and references therein.
- (a) M. D. Shair, T. Y. Yoon, K. K. Mosny, T. C. Chou and S. J. Danishefsky, J. Am. Chem. Soc., 1996, 118, 9509 CrossRef CAS; (b) P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704 CAS; (c) H. Fuwa, N. Kainuma, K. Tachibana and M. Sasaki, J. Am. Chem. Soc., 2002, 124, 14983 CrossRef CAS; (d) J. H. Chang, H.-U. Kang, I.-H. Jung and C.-G. Cho, Org. Lett., 2010, 12, 2016 CrossRef CAS.
- (a) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 1999, 38, 2411 CrossRef CAS; (b) S. P. H. Mee, V. Lee and J. E. Baldwin, Chem.–Eur. J., 2005, 11, 3294 CrossRef CAS; (c) J.-H. Li, Y. Liang, D.-P. Wang, W.-J. Liu, Y.-X. Xie and D.-L. Yin, J. Org. Chem., 2005, 70, 2832 CrossRef CAS.
- (a) J. K. Stille and B. L. Groh, J. Am. Chem. Soc., 1987, 109, 813 CrossRef CAS; (b) J. K. Stille and J. H. Simpson, J. Am. Chem. Soc., 1987, 109, 2138 CrossRef CAS; (c) A. F. Littke, L. Schwarz and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 6343 CrossRef CAS; (d) D.-H. Lee, A. Taher, W.-S. Ahn and M.-J. Jin, Chem. Commun., 2010, 46, 478 RSC; (e) W. Su, S. Urgaonkar, P. A. McLaughlin and J. G. Verkade, J. Am. Chem. Soc., 2004, 126, 16433 CrossRef CAS.
- (a) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, UK, 1998 Search PubMed; (b) P. T. Anastas and T. Williamson, Green Chemistry, Frontiers in Benign Chemical Synthesis and Process, Oxford University of Press, Oxford, UK, 1998 Search PubMed; (c) C. Capello, U. Fischer and K. Hungerbuhler, Green Chem., 2007, 9, 927 RSC.
- (a) J. C. Garcia-Martinez, R. Lezutekong and R. M. Crooks, J. Am. Chem. Soc., 2005, 127, 5097 CrossRef CAS; (b) M. A. Bernechea, M. E. de Jesús, C. López-Mardomingo and P. Terreros, Inorg. Chem., 2009, 48, 4491 CrossRef CAS; (c) L. Wu, X. Zhang and Z. Tao, Catal. Sci. Technol., 2012, 2, 707 RSC.
- C. Wolf and R. Lerebours, J. Org. Chem., 2003, 68, 7551 CrossRef CAS.
- (a) B. H. Lipshutz, S. Ghorai, A. R. Abela, R. Moser, T. Nishikata, C. Duplais, A. Krasovskiy, R. D. Gaston and R. C. Gadwood, J. Org. Chem., 2011, 76, 4379 CrossRef CAS; (b) B. H. Lipshutz, B. R. Taft and A. R. Abela, Platin. Met. Rev., 2012, 56, 62 Search PubMed; (c) B. H. Lipshutz and S. Ghorai, Aldrichimica Acta, 2008, 41, 59 CAS; (d) B. H. Lipshutz and S. Ghorai, Aldrichimica Acta, 2012, 45, 3 Search PubMed.
- C. G. Fu, Acc. Chem. Res., 2008, 41, 1555 CrossRef CAS.
- J.-H. Li, X.-C. Hu, Y. Liang and Y.-X. Xie, Tetrahedron, 2006, 62, 31 CrossRef CAS.
- (a) B. H. Lipshutz, S. Ghorai, W. W. Y. Leong, B. R. Taft and D. V. Krogstad, J. Org. Chem., 2011, 76, 5061 CrossRef CAS; (b) T. Kobayashi, M. Nakashima, T. Hakogi, K. Tanaka and S. Katsumura, Org. Lett., 2006, 8, 3809 CrossRef CAS.
- (a) J. E. Klijn and J. B. Engberts, Nature, 2005, 435, 746 CrossRef CAS; (b) S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275 CrossRef CAS; (c) B. H. Lipshutz and B. R. Taft, Org. Lett., 2008, 10, 1329 CrossRef CAS; (d) B. H. Lipshutz, D. W. Chung and B. Rich, Org. Lett., 2008, 10, 3793 CrossRef; (e) T. Nishikata and B. H. Lipshutz, Org. Lett., 2009, 11, 2377 CrossRef CAS.
- M. Dowlut, D. Mallik and M. G. Organ, Chem.–Eur. J., 2010, 16, 4279 CrossRef CAS.
- (a) A. Krasovskiy and B. H. Lipshutz, Org. Lett., 2011, 13, 3818 CrossRef CAS; (b) G. P. Lu, K. R. Voigtritter, C. Cai and B. H. Lipshutz, J. Org. Chem., 2012, 77, 3700 CrossRef CAS.
- V. Farina and G. P. Roth, Tetrahedron Lett., 1991, 32, 4243 CrossRef CAS.
- R. Shen, T. Inoue, M. Forgac and J. A. Porco, J. Org. Chem., 2005, 70, 3686 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: More experimental details and characterization data of all products. See DOI: 10.1039/c2gc36042j |
| This journal is © The Royal Society of Chemistry 2013 |