Catalytic fixation of CO2 to cyclic carbonates by phosphonium chlorides immobilized on fluorous polymer†
Qing-Wen
Song
a,
Liang-Nian
He
*ab,
Jin-Quan
Wang
a,
Hiroyuki
Yasuda
b and
Toshiyasu
Sakakura
*b
aState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin, 300071, P. R. China. E-mail: heln@nankai.edu.cn; Tel: +86 22 2350 3878
bNational Institute of Advanced Industrial Science and Technology (AIST), Tsukuba Central 5, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan. E-mail: t-sakakura@aist.go.jp; Tel: +81 29 861 4559
First published on 9th October 2012
Abstract
Phosphonium chloride covalently bound to the fluorous polymer is proved to be an efficient and recyclable homogeneous CO2-soluble catalyst for organic solvent-free synthesis of cyclic carbonates from epoxides and CO2 under supercritical CO2 conditions. The catalyst can be easily recovered by simple filtration after reaction and reused with retention of high activity and selectivity. In addition, the effects of various reaction variables on the catalytic performance are also discussed in detail. The process represents a simpler access to preparing cyclic carbonates with the ease of homogeneous catalyst recycling.
Introduction
Recently, CO2 fixation has received much attention because CO2 is the most inexpensive and renewable carbon resource from the viewpoint of green chemistry and atom economy.1 Development of a truly environmentally friendly process utilizing CO2, which is the largest single source of greenhouse gas and can also be regarded as a typical renewable natural resource, has drawn current interest in synthetic chemistry from the viewpoint of environmental protection and resource utilization. In this regard, chemical fixation of CO2 onto industrial useful materials is one of the most promising methods because there are many possibilities for CO2 to be used as a safe and cheap C1 building block in organic synthesis.2 On the other hand, supercritical CO2 (scCO2) is considered to be a green solvent, and thus substantial effort has been devoted to developing chemical processes in which scCO2 is used to replace hazardous organic solvent.3Organic cyclic carbonates such as ethylene carbonate and propylene carbonate (PC) have been widely used for various purposes, for instance, electrolytic elements of lithium secondary batteries, polar aprotic solvents, monomers for synthesizing polycarbonates, and chemical ingredients for preparing medicines or agricultural chemicals.4
For the cyclic carbonate synthesis from CO2 and epoxides, numerous effective catalytic systems have been developed including homogeneous5 and heterogeneous catalysis.6 We have reported that a fluorous phosphonium catalyst5k can be used in scCO2 as a homogeneous catalyst, and this process offers the advantage of allowing the synthesis of e.g. PC to take place under supercritical conditions with the direct and spontaneous separation of the carbonate. Recently, attachment of fluorinated chains to chelating agents, surfactants, and catalyst ligands could generally enhance the solubility of such compounds in scCO2, and thus can be used to design CO2-soluble catalysts.7 In this context, a phosphonium chloride covalently bound to the fluorous polymer (Scheme 1) was employed to design such a supported catalyst that can homogeneously dissolve during the reaction and precipitate quantitatively in the separation stage.
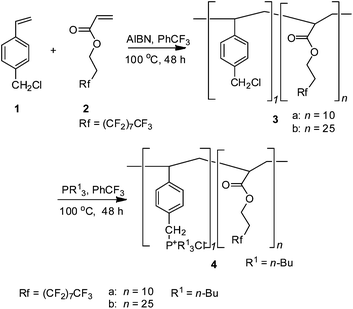 | ||
| Scheme 1 Synthesis of fluorous polymers supported phosphonium chloride. | ||
The fluorous polymers 4a and 4b were identified by 1H NMR, 13C NMR, 19F NMR, FT-IR elementary analysis7d and GPC measurement. The reaction of propylene oxide (PO) and CO2 performed well with 98% of PC yield and excellent selectivity, as shown in Scheme 2. Notably, incorporation of the fluorinated chain in the polymer effectively enhance the solubility of phosphonium salts which are usually insoluble in scCO2, thus providing a simple way for performing homogeneous catalysis under scCO2 conditions.
 | ||
| Scheme 2 Synthesis of propylene carbonate from propylene oxide and CO2. | ||
Results and discussion
The coupling of PO and CO2 was conducted in a batch-wise operation in the presence of 1 mol% of the catalyst with respect to the initial amount of PO under identical conditions (150 °C, 8 MPa, 8 h). The results are summarized in Table 1. It is worthwhile mentioning that the fluorous polymer (3a) itself can hardly catalyze the cycloaddition reaction under the reaction conditions (entry 3, Table 1). Interestingly, phosphonium chloride covalently bound to fluorous polymers (4a and 4b) exhibited high activity, higher than Bu4PCl (entries 1, 2 vs. 4), the corresponding lower molecular weight catalyst; even higher than KI and Bu4NBr (entries 1, 2 vs. 5, 6), traditional homogeneous catalysts for PC synthesis in industrial application. It is most possible that the incorporation of the fluorinated side chain in the polymer effectively enhances the solubility of phosphonium salts that are usually insoluble in scCO2. Therefore, the catalyst (4a) was selected as the model catalyst for further investigation.| Entry | Cat. | Temperature (°C) | Yieldb (%) | Selectivityb (%) |
|---|---|---|---|---|
| a Reaction conditions: propylene oxide (28.6 mmol); 1 mol% of the catalyst based on propylene oxide; CO2 8 MPa; 8 h. b Determined by GC using biphenyl as an internal standard. c 0.5 mol% of the catalyst. d 2 mol% of the catalyst. | ||||
| 1 | 4a | 150 | 98 | 99 |
| 2 | 4b | 150 | 97 | 97 |
| 3 | 3a | 150 | 5 | – |
| 4 | Bu4PCl | 150 | 90 | 99 |
| 5 | Bu4NBr | 150 | 95 | 98 |
| 6 | KI | 150 | 89 | 96 |
| 7 | 4a | 90 | 21 | 99 |
| 8 | 4a | 120 | 58 | >99 |
| 9 | 4a | 180 | 81 | 90 |
| 10c | 4a | 150 | 82 | 99 |
| 11d | 4a | 150 | 98 | 99 |
The influence of reaction temperature on PC yield was further studied. It was found that the yield of PC was strongly affected by the reaction temperature. The yield and selectivity of PC increased with the reaction temperature up to 150 °C (entries 1 and 7–9, Table 1), whereas further increase in the temperature caused a decrease in the selectivity, possibly due to facile formation of side-products at the higher temperature, i.e. the isomerisation of PO to acetone and the ring opening by water to propylene glycol. From a practical viewpoint, apparently, 150 °C could be the optimal and desirable temperature.2f Moreover, 1 mol% of the catalyst is a proper amount for the reaction (entries 1, 10, 11).
Fig. 1 shows the influence of reaction time on PC yield. The supported phosphonium chloride-catalyzed PC synthesis from PO and CO2 proceeded rapidly, and more than 80% PC yield was obtained within the first 2 h at 150 °C. The PC yield experienced a continuing growth within 6 h, while the selectivity of PC remained almost constant during the whole course of the reaction. Consequently, a reaction time of 6 h was required for almost complete PO conversion.
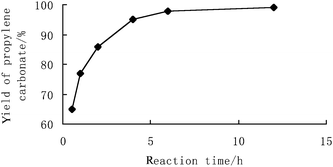 | ||
| Fig. 1 Reaction time – the PC yield profile. Reaction conditions: propylene oxide (28.6 mmol); catalyst 4a (1 mol%); 150 °C; 8 MPa. | ||
It is well-known that the properties of supercritical fluids are sensitive to pressure, and thus pressure may drastically influence the reaction outcome when a reaction takes place in supercritical conditions. The effect of CO2 pressure on the yield and selectivity of PC is depicted in Fig. 2. The selectivity slightly changed in the range of 3–14 MPa. As easily seen, the best PC yield could be achieved under scCO2 conditions, demonstrating the preferential effect of the supercritical conditions, while the yield was remarkably influenced by the pressure below 8 MPa. Possibly, the isomerisation to acetone increased or the CO2 insertion step was limited at low CO2 pressure.
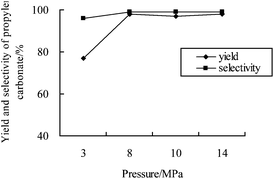 | ||
| Fig. 2 Reaction pressure – the PC yield profile. Reaction conditions: propylene oxide (28.6 mmol); catalyst 4a (1 mol%); 150 °C; 8 h. | ||
The scope of the substrates was further explored as shown in Scheme 3 and Table 2. Several terminal epoxides were chosen to be tested under the optimized reaction conditions. The catalyst (4a) was found to be applicable to a variety of terminal epoxides, yielding the corresponding cyclic carbonates in excellent yields with more than 99% selectivity (entries 1–5, Table 2). This catalyst can also be utilized in the coupling reaction of CO2 and optical pure epoxide, (R)-styrene oxide to conveniently obtain (R)-1-phenyl-1,2-ethanediol carbonate (RR-6c) in 99% ee with retention of stereochemistry (entry 6).8
 | ||
| Scheme 3 Synthesis of various carbonates catalyzed by fluorous polymers-R3P+X−. | ||
| Entry | Substrate | Product | Yieldb (%) | Selectivity (%) |
|---|---|---|---|---|
| a Reaction conditions: epoxide (28.6 mmol); catalyst 4a (1 mol%); temperature 150 °C; CO2 8 MPa; 8 h. b Determined by GC using biphenyl as an internal standard; value in parentheses refers to isolated yield column chromatography with ethyl acetate–petroleum as the eluent. | ||||
| 1 |
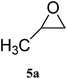
|
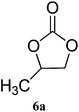
|
98 (93) | 99 |
| 2 |
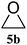
|
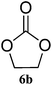
|
97 (95) | >99 |
| 3 |
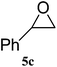
|
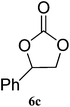
|
95 (91) | 99 |
| 4 |
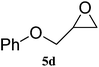
|
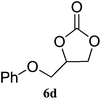
|
99 (94) | 99 |
| 5 |
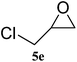
|
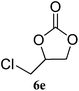
|
91 (88) | 98 |
| 6 |
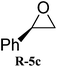
|
|
97 | 99 |
The facility of recycling the catalysts is one of the key advantages in this catalytic system. As shown in Fig. 3, a single homogeneous phase was formed under reaction conditions (100 °C, 14 MPa5k or 150 °C, 8 MPa). Indeed, the phase behaviour of the reaction visually inspected through a sapphire window attached to the autoclave6q revealed that the reaction mixture initially formed a uniform phase, while propylene carbonate formed a new phase upon the reaction. After the reaction, the catalyst was precipitated as a solid upon venting CO2. As a result, the catalyst was easily recovered by simple filtration. A series of repeated reactions run continuously to investigate the constancy of the catalyst activity. In each cycle, the catalyst was separated by filtration and then used for the next run directly. The results show that the catalyst can be reused at least 7 times without significant loss of catalytic activity, while the excellent selectivity of propylene carbonate is nearly unchanged. Furthermore, the purity of the product separated directly by filtration from the reaction mixture, reached 99% without further purification process.
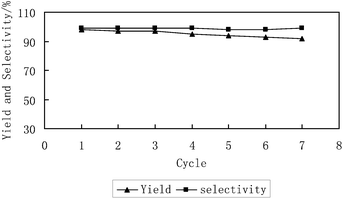 | ||
| Fig. 3 Catalytic activity of the reused CO2-soluble catalyst (4a). Reaction conditions: propylene oxide (28.6 mmol); catalyst 4a (1 mol%); 150 °C; 8 MPa; 8 h. | ||
A possible mechanism for the fluorous polymer supported phosphonium chloride-catalyzed coupling of epoxides with CO2 in this study would be similar to that of ammonium/phosphonium salt catalysis.5d,s The proposed mechanism involves the activation of propylene oxide by the phosphonium cation, the ring-opening of the epoxide via nucleophilic attack of chloride at the least-hindered carbon, and the insertion of CO2 into the N–O bond. Subsequent cyclization via an intramolecular nucleophilic attack leads to the cyclic carbonate and the regeneration of the catalyst.
Experimental
Chemical reagents
All chemicals, except CO2, were handled under a nitrogen atmosphere. The catalysts were synthesized by a modified method described in the literature.7d–l
Compound
3a3a3a
: 1H NMR (perfluoro-compound FC 77 with drops CDCl3) δ 7.01–7.35 (br m, 1H), 4.27 (s, 5H), 2.46 (s, 5H), 1.40–1.90 (br m, 15H); 13C NMR (100.6 MHz, FC 77 with an external chloroform-d lock) δ 175.52 (s, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 105.73–126.03 (m, Rf), 58.01 (s, OCH2), 31.78 (s, CH2Rf).
O), 105.73–126.03 (m, Rf), 58.01 (s, OCH2), 31.78 (s, CH2Rf).
Compound 3b3b : 1H NMR (FC 77) 7.14–7.70 (br m, 1H), 4.52 (s, 12H), 1.41–2.63 (br s, 30H).
Compound
4a4a4a
: colorless solid. 1H NMR (FC 77) 7.10–7.90 (br m, 2H), 4.46 (s, 25H), 1.40–2.57 (br s, 75H); 13C NMR (FC 77) 175.22 (s, CO), 102.98–125.57 (m, Rf), 57.73 (s, OCH2), 42.14 (s, CH2Ph), 31.39 (t, CH2CF2, JFC = 21.49 Hz), 25.00 (s); 31P NMR (FC 77) 32.97. IR (ν, cm−1), 2973 (C–H), 1737 (CO), 1249 (C–F), 1103, and 1012, and 711 (phosphonium salt), 653; Anal. Calcd for (4a): C, 32.76; H, 1.93; F, 58.3; P, 0.56. Found: C, 32.43; H, 1.90; F, 57.4; P, 0.44. Mw: 5536.
Compound
4b4b4b
: 1H NMR (FC 77 with an external chloroform-d lock) 7.08–7.64 (br m, 1H), 4.28 (br m, 5H), 3.49 (s, 5H), 2.50 (m, 5H), 1.57–1.89 (br m, 20H); 13C NMR (100.6 MHz, FC 77 with an external chloroform-d lock) 175.50 (s, CO), 106.61–125.67 (m, Rf), 57.76 (s, OCH2), 42.40 (s, CH2Ph), 31.48 (t, CH2Rf, JFC = 20.63 Hz), 25.33 (s), 14.47 (s); 31P NMR (161.9 MHz, FC 77 with an external chloroform-d lock) 34.64. IR (ν, cm−1) 2965 (C–H), 1740 (CO), 1255 (C–F); Anal. Calcd for (4b): C, 31.49; H, 1.66; F, 60.3; P, 0.29. Found: C, 31.14; H, 1.57; F, 60.2; P, 0.24. Mw: 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 718.
718.
Representative procedure for the cycloaddition reaction
In a typical reaction, the cycloaddition reaction of CO2 to propylene oxide was carried out in a stainless steel autoclave (25 cm3 inner volumes). Prior to the reaction, the catalyst was evacuated at 150 °C for 3 h. CO2 (gas, 4.0 MPa) was introduced to a mixture of propylene oxide (28.6 mmol), catalyst (4a, 1 mol%), and biphenyl (80 mg, an internal standard for GC analysis) at room temperature. The gauge was used to monitor the pressure of the system. The initial pressure was adjusted to 8 MPa at 150 °C and the autoclave was heated at that temperature for 8 h. After cooling, the products were analyzed on a gas chromatograph (Agilent 6890) equipped with a capillary column (HP-5 30 m × 0.25 μm) using a flame ionization detector. The structure and the purity of the products were further identified using GC-MS (HP G1800A) by comparing retention times and fragmentation patterns with those of authentic samples. The solvent was removed, and the residue was subjected to column chromatography with ethyl acetate–petroleum as the eluent to obtain the desired product.The products were also characterized by NMR. 1H NMR and 13C NMR spectra were recorded on a Varian Mercury-plus 400 spectrometer. Chemical shifts were given as δ values referenced in parts per million (ppm) from tetramethylsilane (TMS) as an internal standard. Spectral characteristics of the products (cyclic carbonates 2a–e) in Table 2 were provided as follows:
Conclusions
In summary, phosphonium chloride covalently bound to fluorous polymers is proved to be an efficient and recyclable homogeneous CO2-soluble catalyst for solvent-free synthesis of cyclic carbonate from propylene oxide and carbon dioxide under supercritical conditions. Incorporation of the fluorinated chain in the polymer effectively enhance the solubility of phosphonium salts which are usually insoluble in scCO2, thus providing a simple way for performing homogeneous catalysis under scCO2 conditions. Furthermore, the catalyst can be easily recovered upon releasing CO2 and is reusable for up to 7 cycles without significant loss of activity, while the selectivity of propylene carbonate remained >99%.Acknowledgements
This work was partially supported by the National Science Foundation of China (Grant 21172125).Notes and references
- For recent reviews on fixation of carbon dioxide, see: (a) D. J. Darensbourg, R. M. Mackiewicz, A. L. Phelps and D. R. Billodeaux, Acc. Chem. Res., 2004, 37, 836 CrossRef CAS; (b) P. Braunstein, D. Matt and D. Nobel, Chem. Rev., 1988, 88, 747 CrossRef CAS; (c) K. C. Nicolaou, Z. Yang, J. J. Liu, H. Ueno, P. G. Nantermet, R. K. Guy, C. F. Claiborne, J. Renaud, E. A. Couladouros, K. Paulvannanand and E. J. Sorensen, Nature, 1994, 367, 630 CrossRef CAS; (d) D. H. Gibson, Chem. Rev., 1996, 96, 2063 CrossRef CAS; (e) W. Leitner, Coord. Chem. Rev., 1996, 153, 257 CrossRef CAS.
- For recent reviews on transformation of carbon dioxide, see: (a) X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462 RSC; (b) I. Omae, Coord. Chem. Rev., 2012, 256, 1384 CrossRef CAS; (c) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510 CrossRef CAS; (d) T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312 RSC; (e) K. M. K. Yu, I. Curcic, J. Gabriel and S. C. E. Tsang, ChemSusChem, 2008, 1, 893 CrossRef; (f) T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS; (g) D. B. Dell'Amico, F. Calderazzo, L. Labella, F. Marchetti and G. Pampaloni, Chem. Rev., 2003, 103, 3857 CrossRef; (h) I. Omae, Catal. Today, 2006, 115, 33 CrossRef CAS; (i) G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618 CrossRef CAS; (j) D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 2003, 103, 3857 Search PubMed; (k) X. L. Yin and J. R. Moss, Coord. Chem. Rev., 1999, 181, 27 CrossRef CAS.
- For recent reviews on chemical reaction in CO2, see: (a) Chemical Synthesis Using Supercritical Fluids, ed. P. D. Jessop and W. Leitner, Willy-VCH, Weinheim, 1999 Search PubMed; (b) P. D. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1999, 99, 475 CrossRef CAS; (c) A. Baiker, Chem. Rev., 1999, 99, 453 CrossRef CAS; (d) W. Leitner, Top. Curr. Chem., 1999, 206, 107; (e) P. Licence, J. Ke, M. Sokolova, S. K. Ross and M. Poliakoff, Green Chem., 2003, 5, 99 RSC; (f) W. Keim, Green Chem., 2003, 5, 105 RSC; (g) R. A. Sheldon, Green Chem., 2005, 7, 267 RSC; (h) Green Chemistry Using Liquid and Supercritical Carbon Dioxide, ed. J. M. DeSimone and W. Tumas, Oxford University Press, 2003 Search PubMed; (i) E. J. Beckman, J. Supercrit. Fluids, 2004, 28, 121 CrossRef CAS; (j) P. G. Jessop, J. Supercrit. Fluids, 2006, 38, 211 CrossRef CAS; (k) H. R. Hobbs and N. R. Thomas, Chem. Rev., 2007, 107, 2786 CrossRef CAS; (l) P. Jessop and B. Subramaniam, Chem. Rev., 2007, 107, 2666 CrossRef CAS.
- (a) A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951 CrossRef CAS; (b) M. Yoshida and M. Ihara, Chem.–Eur. J., 2004, 10, 2886 CrossRef CAS; (c) B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554 CrossRef CAS.
- For recent typical references on coupling of CO2 and epoxides catalyzed by homogeneous catalytic system, see: (a) Z.-Z. Yang, Y.-N. Zhao, L.-N. He, J. Gao and Z.-S. Yin, Green Chem., 2012, 14, 519 RSC; (b) Z.-Z. Yang, L.-N. He, Y.-N. Zhao, B. Li and B. Yu, Energy Environ. Sci., 2011, 4, 3971 RSC; (c) Z.-Z. Yang, L.-N. He, C.-X. Miao and S. Chanfreau, Adv. Synth. Catal., 2010, 352, 2233 CrossRef CAS; (d) C.-X. Miao, J.-Q. Wang, Y. Wu, Y. Du and L.-N. He, ChemSusChem, 2008, 1, 236 CrossRef CAS; (e) A. Ghosh, P. Ramidi, S. Pulla, S. Z. Sullivan, S. L. Collom, Y. Gartia, P. Munshi, A. S. Biris, B. C. Noll and B. C. Berry, Catal. Lett., 2010, 137, 1 CrossRef CAS; (f) J. Sun, L.-J. Han, W.-G. Cheng, J.-Q. Wang, X.-P. Zhang and S.-J. Zhang, ChemSusChem, 2011, 4, 502 CrossRef CAS; (g) F. Wu, X.-Y. Dou, L.-N. He and C.-X. Miao, Lett. Org. Chem., 2010, 7, 73 Search PubMed; (h) X.-Y. Dou, J.-Q. Wang, Y. Du, E. Wang and L.-N. He, Synlett, 2007, 3058 CAS; (i) L.-N. He, H. Yasuda and T. Sakakura, Green Chem., 2003, 5, 92 RSC; (j) H. Yasuda, L.-N. He and T. Sakakura, J. Catal., 2005, 233, 119 CrossRef CAS; (k) R. L. Paddock and S. T. Nguyen, J. Am. Chem. Soc., 2001, 123, 11498 CrossRef CAS; (l) X.-B. Lu, B. Liang, Y.-J. Zhang, Y.-Z. Tian, Y.-M. Wang, C.-X. Bai, H. Wang and R. Zhang, J. Am. Chem. Soc., 2004, 126, 3732 CrossRef CAS; (m) T. Aida and S. Inoue, J. Am. Chem. Soc., 1983, 105, 1304 CrossRef CAS; (n) H. S. Kim, J. J. Kim, B. G. Lee, O. S. Jung, H. G. Jang and S. O. Kang, Angew. Chem., Int. Ed., 2000, 39, 4096 CrossRef CAS; (o) J.-S. Tian, C.-X. Miao, J.-Q. Wang, F. Cai, Y. Du, Y. Zhao and L.-N. He, Green Chem., 2007, 9, 566 RSC; (p) R. L. Paddock and S. T. Nguyen, Chem. Commun., 2004, 1622 RSC; (q) V. Caló, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561 CrossRef CAS; (r) H. Kawanami, A. Sasaki, K. Matsui and Y. Ikushima, Chem. Commun., 2003, 896 RSC; (s) Y. Du, J.-Q. Wang, J.-Y. Chen, F. Cai, J.-S. Tian, D.-L. Kong and L.-N. He, Tetrahedron Lett., 2006, 47, 1271 CrossRef CAS.
- For recent typical references on coupling of CO2 and epoxides catalyzed by heterogeneous catalytic system, see: (a) J. Gao, Q.-W. Song, L.-N. He, C. Liu, Z.-Z. Yang, X. Han, X.-D. Li and Q.-C. Song, Tetrahedron, 2012, 68, 3835 Search PubMed; (b) J.-N. Lu and P. H. Toy, Synlett, 2011, 659 Search PubMed; (c) H. C. Cho, H. S. Lee, J. Chun, S. M. Lee, H. J. Kim and S. U. Son, Chem. Commun., 2011, 47, 917 RSC; (d) I. Shibata, I. Mitani, A. Imakuni and A. Baba, Tetrahedron Lett., 2011, 52, 721 Search PubMed; (e) A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212 RSC; (f) W.-L. Dai, L. Chen, S.-F. Yin, S.-L. Luo and C.-T. Au, Catal. Lett., 2010, 135, 295 Search PubMed; (g) L.-P. Guo, C.-M. Wang, X.-Y. Luo, G.-K. Cui and H.-R. Li, Chem. Commun., 2010, 46, 5960 RSC; (h) Y. Xie, K.-L. Ding, Z.-M. Liu, J.-J. Li, G.-M. An, R.-T. Tao, Z.-Y. Sun and Z.-Z. Yang, Chem.–Eur. J., 2010, 16, 6687 CAS; (i) Y. Tsutsumi, K. Yamakawa, M. Yoshida, T. Ema and T. Sakai, Org. Lett., 2010, 12, 5728 CrossRef CAS; (j) Y.-B. Xiong, H. Wang, R.-M. Wang, Y.-F. Yan, B. Zheng and Y.-P. Wang, Chem. Commun., 2010, 46, 3399 RSC; (k) A. Decortes, M. M. Belmonte, J. B. Buchholz and A. W. Kleij, Chem. Commun., 2010, 46, 4580 RSC; (l) C.-R. Qi, J.-W. Ye, W. Zeng and H.-F. Jiang, Adv. Synth. Catal., 2010, 352, 1925 CrossRef CAS; (m) X.-X. Zheng, S.-Z. Luo, L. Zhang and J.-P. Cheng, Green Chem., 2009, 11, 455 RSC; (n) J.-L. Song, Z.-F. Zhang, S.-Q. Hu, T.-B. Wu, T. Jiang and B.-X. Han, Green Chem., 2009, 11, 1031 RSC; (o) Y. Kayaki, M. Yamamoto and T. Ikariya, Angew. Chem., Int. Ed., 2009, 48, 4194 CrossRef CAS; (p) J.-Q. Wang, X.-D. Yue, F. Cai and L.-N. He, Catal. Commun., 2007, 8, 167; (q) Y. Du, F. Cai, D.-L. Kong and L.-N. He, Green Chem., 2005, 7, 518 RSC; (r) J.-L. He, T.-B. Wu, Z.-F. Zhang, K.-L. Ding, B.-X. Han, Y. Xie, T. Jiang and Z.-M. Liu, Chem.–Eur. J., 2007, 13, 6992 CrossRef CAS; (s) K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526 CrossRef CAS; (t) A.-L. Zhu, T. Jiang, B.-X. Han, J.-C. Zhang, Y. Xie and X.-M. Ma, Green Chem., 2007, 9, 169 RSC; (u) F. Shi, Q. Zhang, Y. Ma, Y. He and Y. Deng, J. Am. Chem. Soc., 2005, 127, 4182 CrossRef CAS.
- For the reactions based on tethering fluorinated chains to catalysts in scCO2, see: (a) T. Sarbu, T. Styranec and E. J. Beckman, Nature, 2000, 405, 165 CrossRef CAS; (b) W. Leitner, Nature, 2000, 405, 129 CrossRef CAS; (c) E. J. Beckman, Chem. Commun., 2004, 1885 RSC; (d) D. E. Bergbreiter, J. G. Franchina and B. L. Case, Org. Lett., 2000, 2, 393 CrossRef CAS; (e) K. Burgemeister, G. Francio, H. Hugl and W. Leitner, Chem. Commun., 2005, 6026 RSC; (f) D. J. Adams, J. A. Bennett, D. J. Cole-Hamilton, E. G. Hope, J. Hopewell, J. Kight, P. Pogorzelec and A. M. Stuart, Dalton Trans., 2005, 3862 RSC; (g) Y. Hu, W. Chen, A. M. B. Osuna, J. A. Iggo and J. Xiao, Chem. Commun., 2002, 788 RSC; (h) W. Chen, Y. Hu and J. Xiao, Chem. Commun., 2000, 839 RSC; (i) I. Kani, M. A. Omary, M. A. Rawashdeh-Omary, Z. K. Lopez-Castillo, R. Flores, A. Akgerman and J. P. Fackler, Jr., Tetrahedron, 2002, 58, 3923 CrossRef CAS; (j) I. Kani, R. Flores, J. P. Facler, Jr. and A. Akgerman, J. Supercrit. Fluids, 2004, 31, 287 Search PubMed; (k) R. Flores, Z. K. Lopez-Castillo, I. Kani, J. P. Facler, Jr. and A. Akgerman, Ind. Eng. Chem. Res., 2003, 42, 6720 Search PubMed; (l) S. Kainz, Z. Y. Luo, D. P. Curran and W. Leitner, Synthesis, 1998, 1425 CrossRef CAS.
- For the reaction of enantiomerically pure epoxides with CO2 yielding enantiomerically pure cyclic carbonates with complete retention of the original stereochemistry, see: (a) X.-B. Lu, Y.-J. Zhang, K. Jun, L.-M. Luo and H. Wang, J. Catal., 2004, 227, 537 CrossRef CAS; (b) R. L. Paddock, Y. Hiyama, J. M. McKay and S. B. T. Nguyen, Tetrahedron Lett., 2004, 45, 2023 CrossRef CAS.
Footnote |
| † Safety warning: Experiments using compressed CO2 gas are potentially hazardous and must only be carried out by using the appropriate equipment and under rigorous safety precautions. |
| This journal is © The Royal Society of Chemistry 2013 |