Promoting effect of SnOx on selective conversion of cellulose to polyols over bimetallic Pt–SnOx/Al2O3 catalysts†
Tianyin
Deng
and
Haichao
Liu
*
Beijing National Laboratory for Molecular Sciences, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: hcliu@pku.edu.cn; Fax: +86-10-6275-4031
First published on 1st October 2012
Abstract
Cellulose is the most abundant source of biomass in nature, and its selective conversion into polyols provides a viable route towards the sustainable synthesis of fuels and chemicals. Here, we report the marked change in the distribution of polyols in the cellulose reaction with the Sn/Pt atomic ratios in a wide range of 0.1–3.8 on the SnOx-modified Pt/Al2O3 catalysts. Such a change was found to be closely related to the effects of the Sn/Pt ratios on the activity for the hydrogenation of glucose and other C6 sugar intermediates involved in the cellulose reaction as well as to the notable activity of the segregated SnOx species for the selective degradation of the sugar intermediates on the Pt–SnOx/Al2O3 catalysts. At lower Sn/Pt ratios of 0.1–1.0, there existed electron transfer from the SnOx species to the Pt sites and strong interaction between the catalysts, as characterized by temperature-programmed reduction in H2 and infrared spectroscopy for CO adsorption, which led to their superior hydrogenation activity (per exposed Pt atom), and in-parallel higher selectivity to hexitols (e.g. sorbitol) in the cellulose reaction, as compared to Pt/Al2O3. The hexitol selectivity reached the greatest value of 82.7% at the Sn/Pt ratio of 0.5, nearly two times that of Pt/Al2O3 at similar cellulose conversions (∼20%). As the Sn/Pt ratios exceeded 1.5, the Pt–SnOx/Al2O3 catalysts exhibited inferior hydrogenation activity (per exposed Pt atom), due to the formation of the crystalline Pt–Sn alloy, which led to the preferential conversion of cellulose to C2 and especially C3 products (e.g. acetol) over hexitols, most likely involving the isomerization of glucose to fructose and retro-aldol condensation of these sugars on the segregated SnOx species, apparently in the form of Sn(OH)2. These findings clearly demonstrate the feasibility for rational control of the cellulose conversion into the target polyols (e.g. acetol or propylene glycol), for example, by the design of efficient catalysts based on the catalytic functions of the SnOx species with tunable hydrogenation activity.
1 Introduction
Catalytic conversion of cellulose to polyols has attracted great attention as one of the most viable primary routes for cellulose utilization because of the versatile uses of polyols in the synthesis of fuels and value-added chemicals.1–7 This conversion route traditionally involves the use of mineral acids for cellulose hydrolysis to sugars, which are subsequently hydrogenated to polyols on metal-based catalysts.6,8,9 Since the pioneering work on Pt/Al2O3 in water and Ru nanoclusters in an ionic liquid,10,11 recent studies on this cellulose reaction have been dedicated to the development of greener approaches to avoid the use of mineral acids and to improve polyol productivity.Fukuoka and Dhepe reported the direct conversion of cellulose into hexitols (i.e. sorbitol and mannitol) in a 31% yield at 463 K on Pt/Al2O3 in the absence of mineral acids.10 By taking advantage of the unique properties of water under near-critical conditions, Luo et al. obtained a ∼40% yield of hexitols on Ru/C in combination with the hydrolysis role of H+ ions reversibly formed from hot water at 513 K.12 Further studies on different hydrogenation catalysts led to improved yields.13–15 For example, Pang et al. achieved a 59.8% yield of hexitols using a mesoporous carbon supported Ni–Rh catalyst;13 Sels and co-workers reported a >90% yield of hexitols at a 100% conversion of ball-milled cellulose on bifunctional Ru/H-USY catalyst in the presence of a trace amount of HCl, and even the quantitative conversion of ball-milled cellulose into hexitols on Ru/C together with heteropolyacids.14b,c More noticeably, Zhang and co-workers found that cellulose can even be converted directly to ethylene glycol on Ni–W2C/C.16 These results demonstrate that the polyol selectivities are strongly dependent on the properties of the hydrogenation catalysts, which accordingly influence the relative rates between the competitive hydrogenation of the sugar intermediates and their degradation. Therefore, it is necessary to further understand the underlying reasons for such a dependence in order to design more efficient catalysts towards the controllable conversion of cellulose to polyols.
Compared to monometallic catalysts, bimetallic catalysts frequently exhibit superior activity and selectivity in many reactions. Concerning the aforementioned Pt/Al2O3 catalyst, its modification with SnOx as an example leads to wide applications in industry.17–19 Extensive studies on the SnOx-modified Pt/Al2O3 (Pt–SnOx/Al2O3) catalysts have revealed the drastically electronic and geometric effects of SnOx on the catalytic properties of the Pt sites.20,21 Moreover, the presence of SnOx as a Lewis acid in the Pt catalysts can largely improve the hydrogenation of unsaturated aldehydes (e.g. citral, crotonaldehyde and carvone) via its close interaction with the carbonyl groups, to form desirable products of unsaturated alcohols.19,22–24 Therefore, we herein study the Pt–SnOx/Al2O3 catalysts with varying Sn/Pt atomic ratios in the range of 0–3.8 in the cellulose reaction and for comparison in the reactions of glucose and fructose. Their structures are characterized by X-ray powder diffraction (XRD), X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), temperature-programmed reduction in H2 (H2-TPR), Fourier transform infrared (FT-IR) spectroscopy using a CO probe molecule and H2 chemisorption. These detailed studies lead to our understanding of the SnOx modification effects on the structures of the Pt–SnOx/Al2O3 catalysts and their hydrogenation activities, and consequently on the polyol distribution in the cellulose reaction.
2 Experimental
Catalyst preparation
Pt/Al2O3 and SnOx-modified Pt/Al2O3 (denoted as Pt–SnOx/Al2O3) catalysts with seven different nominal Sn/Pt molar ratios in the range of 0.1–3.8, were prepared by the incipient wetness impregnation of γ-Al2O3 (Alfa Aesar, 208 m2 g−1) with aqueous solutions of H2PtCl6·6H2O (AR, Beijing Chemical), H2PtCl6·6H2O (AR, Beijing Chemical) and SnCl2·2H2O (AR, Shantou Xilong Chemical), respectively. The impregnated samples were dried at room temperature for about 12 h and then at 383 K overnight. Afterwards, they were calcined in flowing air at 673 K for 4 h, and then reduced in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 H2/N2 flow at 673 K for 4 h. The compositions of these samples were measured by profile spec inductively coupled plasma-atomic emission spectrometry (ICP-AES), showing that they contained 1.88–2.07 wt% Pt and that the Sn/Pt atomic ratios for the Pt–SnOx/Al2O3 samples were 0, 0.1, 0.3, 0.5, 1.0, 1.5, 2.1, and 3.8 (Table 1). The Pt–SnOx/Al2O3 samples were denoted as Pt–SnOx(m)/Al2O3, where the m in parentheses represents their measured Sn/Pt atomic ratio. For example, Pt–SnOx(0.1)/Al2O3 represents the sample with a Sn/Pt atomic ratio of 0.1.
:4 H2/N2 flow at 673 K for 4 h. The compositions of these samples were measured by profile spec inductively coupled plasma-atomic emission spectrometry (ICP-AES), showing that they contained 1.88–2.07 wt% Pt and that the Sn/Pt atomic ratios for the Pt–SnOx/Al2O3 samples were 0, 0.1, 0.3, 0.5, 1.0, 1.5, 2.1, and 3.8 (Table 1). The Pt–SnOx/Al2O3 samples were denoted as Pt–SnOx(m)/Al2O3, where the m in parentheses represents their measured Sn/Pt atomic ratio. For example, Pt–SnOx(0.1)/Al2O3 represents the sample with a Sn/Pt atomic ratio of 0.1.
| Catalyst | Pt loadinga/wt% | d b/nm | D c/% |
|---|---|---|---|
| a Measured by ICP. b Measured by TEM. c Measured by H2 chemisorption. | |||
| Pt/Al2O3 | 2.07 | 0.8 ± 0.2 | 100.0 |
| Pt–SnOx(0.1)/Al2O3 | 2.07 | 1.1 ± 0.2 | 83.6 |
| Pt–SnOx(0.3)/Al2O3 | 2.04 | 0.9 ± 0.2 | 94.4 |
| Pt–SnOx(0.5)/Al2O3 | 1.92 | — | 76.1 |
| Pt–SnOx(1.0)/Al2O3 | 1.88 | 1.0 ± 0.3 | 55.9 |
| Pt–SnOx(1.5)/Al2O3 | 1.98 | — | 44.9 |
| Pt–SnOx(2.1)/Al2O3 | 1.88 | 0.9 ± 0.2 | 38.5 |
| Pt–SnOx(3.8)/Al2O3 | 1.90 | 1.2 ± 0.4 | 31.8 |
ReOx, SbOx and GaOx-modified Pt/Al2O3 catalysts were prepared in the same way using HReO4 (75–80% aqueous solution, Alfa Aesar), Ga(NO3)3·xH2O (99.9%, Alfa Aesar) and SbCl3 (AR, Sinopharm Chemical), respectively. They contained 1.98–1.99 wt% Pt, and their Re/Pt, Sb/Pt or Ga/Pt atomic ratios were each 0.3.
In a similar way, SnOx/Al2O3 (0.36 wt% Sn) was prepared by the incipient wetness impregnation of Al2O3 with ethanol solutions of SnCl2·2H2O. SnOx (4.87 wt% Sn)/Al2O3 was prepared using tri-n-butyltin acetate (AR, Alfa Aesar). Sn(OH)2 and Sn(OH)4 were prepared by hydrolysis of aqueous SnCl2·2H2O and SnCl4·5H2O (AR, Shantou Xilong Chemical) with stoichiometric NH3·2H2O (AR, 25%–28%, Beijing Chemical), respectively, followed by washing the precipitates with deionized water until the filtrate was neutral. The precipitates were dried at room temperature under vacuum.
Catalyst characterization
The contents of Pt, Sn, Re, Sb, and Ga in the catalysts were measured by ICP-AES after they were dissolved in the aqua regia solutions.XRD patterns were recorded on a Rigaku D/MAX-2400 diffractometer using Cu Kα1 radiation (λ = 1.5406 Å) operated at 40 kV and 100 mA. The 2θ angle was scanned in the range of 25–50° at a rate of 4° min−1.
XPS studies were conducted on an Axis Ultra spectrometer (Kratos Analytical Ltd.) using monochromatic Al Kα radiation at a source power of 15 kV. The binding energies were referenced to the Al 2p peak of the Al2O3 support at 74.5 eV.
TEM images were taken on a FEI Tecnai G2 T20 instrument operated at 300 kV. Samples were prepared by uniformly dispersing in ethanol and then placing onto carbon-coated copper grids. The average sizes of metal particles and their size distributions were determined by measuring more than 300 particles randomly distributed in the images. Energy dispersive X-ray (EDX) analysis was carried out on a Philips Tecnai F30 FEI TEM equipped with an EDX spectrometer operated at 300 kV.
Temperature programmed reduction (TPR) experiments were performed on the TP-5000 flow unit (Tianjin Xianquan) by ramping the temperature from 298 to 1073 K at 10 K min−1 in a 5.02% H2/N2 flow (30 mL min−1). Prior to the measurements, the samples were calcined at 673 K in an air flow for 4 h. The amounts of consumed H2 for the samples were measured by a TCD detector and then estimated by referring to the amount obtained from the CuO reduction.
Hydrogen chemisorption measurements were taken on a Quantachrome instrument (Autosorb-1). Calcined samples were reduced in situ at 673 K under a 5.02% H2/N2 flow for 4 h, then out-gassed at 673 K for 6 h and finally cooled to room temperature. The total H2 chemisorption isotherm was performed at 303 K between 4 and 43 kPa H2.
FT-IR spectra for CO adsorption were taken in the wavenumber range 4800–600 cm−1 at 298 K on a Bruker Tensor 27 spectrometer equipped with a MCT detector with a resolution of 4 cm−1 by averaging 100 scans per spectrum. The samples were pressed into self-supported thin wafers and placed into a quartz cell with CaF2 windows. Prior to each measurement, the samples were treated in the cell in a 5.02% H2/N2 flow (40 mL min−1) at 673 K for 30 min, followed by evacuation at 673 K for 30 min. Upon cooling to ambient temperature in the vacuum, the background spectra of the samples were recorded. Afterwards, the samples were exposed to ca. 4 kPa CO for 10 min, and then evacuated for 30 min at 298 K for taking their spectra. All spectra were obtained as difference spectra by subtracting the background spectra of the samples from the spectra after exposure to CO.
Cellulose (microcrystalline, Alfa Aesar) reactions were carried out in a stainless steel autoclave (100 mL) typically at 473 K and 6 MPa H2 for 30 min with vigorous stirring at a speed of 800 rpm. In a typical run, 1 g cellulose and 0.4 g catalyst were introduced to the autoclave containing 50 mL H2O. Afterwards, the reactor was fully purged with H2 (>99.999%, Beijing Longhui Jingcheng), pressurized with H2 to 6.0 MPa and then heated to 473 K which was kept constant during the reaction. After cooling to room temperature in water, the reaction mixture was filtrated and the solids were washed several times with deionized water. The filtrate was diluted with deionized water to 100 mL. The solids including the catalyst and remaining cellulose were washed with acetone three times and then fully dried in an oven at 333 K for 24 h. Cellulose conversions were determined by the change in the weight of cellulose loaded before and after the reactions. The products in the liquid phase (e.g. polyols) were analyzed by high-performance liquid chromatography (Shimadzu LC-20A) using Bio-Rad Aminex HPX-87H and HPX-87C columns with an RID detector. The products in the gas phase (e.g. methane) were analyzed by gas chromatography (Shimadzu 2010) using a HJ-OV-101 column connected to a FID detector. The product selectivities were reported on a carbon basis.
3 Results and discussion
Fig. 1 shows the XPS spectra of the Pt 4d core level for the monometallic Pt/Al2O3 and bimetallic Pt–SnOx/Al2O3 catalysts with different Sn/Pt atomic ratios between 0.1–3.8. The Pt 4d lines were analyzed instead of the most intense Pt 4f lines because of their superimposition with the strong Al 2p peak. Only two peaks were observed at binding energies of around 332.5 and 315.0 eV, the characteristic peaks of Pt 4d3/2 and Pt 4d5/2 for metallic Pt, respectively, which indicates that the surface Pt species were present as zero-valent Pt in these catalysts.25,26 In the Sn 3d region, as shown in Fig. 2, the bimetallic Pt–SnOx/Al2O3 samples exhibited two symmetrical signals at binding energies of 486.3–487.1 and 494.8–495.6 eV, which can be respectively assigned to Sn3d5/2 and Sn 3d3/2 for oxidized tin, Sn2+ and Sn4+, although XPS cannot distinguish between the two oxidation states.25–27 These results indicate that the SnOx species, i.e. SnO and/or SnO2, were the main Sn species at the surfaces of the bimetallic samples. The SnOx species did not reveal XRD peaks (Fig. 3), suggesting that they were highly dispersed. But a diffraction peak at 2θ = 41.7° appeared when the Sn/Pt molar ratios exceeded 1.5, and the intensity increased with increasing the Sn/Pt ratios (Fig. 3). This peak is tentatively assigned to the Pt–Sn (Sn/Pt atomic ratio = 1:1) alloy (JCPDS: 25-0614 and JCPDS: 65-0959), which is confirmed by the representative TEM image for the Sn-rich Pt–SnOx(3.8)/Al2O3 catalyst with a Sn/Pt ratio of 3.8, showing a parallel variation of the relative Pt and Sn compositions against the line-scan positions in the energy-dispersive X-ray spectrum (Fig. S1 in the ESI†).21,28,29 No detection of zero-valent Sn in the XPS spectra (Fig. 2) may be due to the facile surface oxidation of Sn after exposure to air induced by the known strong affinity of tin for oxygen.30
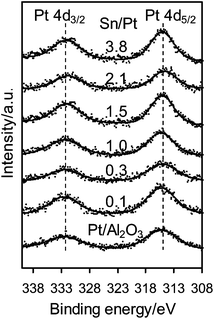 | ||
| Fig. 1 XPS spectra of Pt 4d for Pt/Al2O3 and Pt–SnOx/Al2O3 (∼2 wt% Pt) with Sn/Pt atomic ratios in the range of 0.1–3.8. | ||
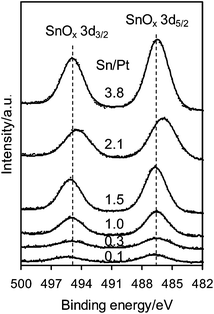 | ||
| Fig. 2 XPS spectra of Sn 3d for Pt–SnOx/Al2O3 (∼2 wt% Pt) with Sn/Pt atomic ratios in the range of 0.1–3.8. | ||
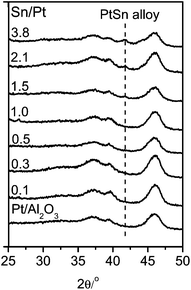 | ||
| Fig. 3 XRD patterns of Pt/Al2O3 and Pt–SnOx/Al2O3 (∼2 wt% Pt) with different Sn/Pt atomic ratios in the range of 0.1–3.8. | ||
The Pt particle sizes for the Pt/Al2O3 and Pt–SnOx/Al2O3 samples were characterized by TEM. As shown in Fig. 4 and Fig. S2,† these samples possessed similar Pt particle sizes (0.8–1.2 nm), irrespective of the Sn/Pt ratios. However, their Pt dispersions measured by H2 chemisorption at 303 K changed with the Sn/Pt ratios (Table 1). An almost 100% Pt dispersion was obtained on Pt/Al2O3. But the dispersion decreased to ca. 83–94% on the bimetallic samples with the Sn/Pt ratios of 0.1 and 0.3, which then sharply decreased to 31.8% with increasing the Sn/Pt ratio up to 3.8. Such a decrease in the Pt dispersion is obviously not due to the change in the Pt particle sizes, which may arise from the partial blockage of the Pt sites by the SnOx species and the formation of the large PtSn alloy crystallites at the higher Sn/Pt ratios.19,20
 | ||
| Fig. 4 TEM images and histograms of Pt particle size distributions of Pt/Al2O3 (a) and Pt–SnOx/Al2O3 (∼2 wt% Pt) with different Sn/Pt atomic ratios of (b) 0.3, and (c) 3.8. | ||
These catalysts were examined in the cellulose reaction. As shown in Table 2, they gave similar cellulose conversions (16.3–22.5%) after 30 min at 473 K and 6 MPa H2, reflecting the fact that the cellulose conversions are controlled by the cellulose hydrolysis step that is catalyzed mainly by the H+ ions generated from hot water under such reaction conditions.12 However, the selectivities to polyols on these catalysts changed significantly with their Sn/Pt ratios (Table 2). The hexitol selectivity on Pt/Al2O3 was 43.3% (including 34.5% sorbitol, 8.0% mannitol and trace amount of iditol, see Table S1 in ESI†), which is similar to the result (43.0%) reported by Fukuoka and co-workers.31 The other identified products mainly included pentitols (7.7%), tetritols (3.4%), glycerol (7.2%), 1,2-propanediol (6.3%), and ethylene glycol (3.5%). Glucose (2.9%), cellobiose alcohol (0.6%) and methane (3.2%) were also detected, which are listed in Table S1 in the ESI† for clarity. The total selectivity of the identified products was 78.1%, not including the unidentified compounds most likely derived from the condensation or degradation of the sugar intermediates. Upon addition of a small amount of SnOx to Pt/Al2O3 (Sn/Pt = 0.1), the hexitol selectivity sharply increased from 43.3 to 78.0% while the selectivity to the cracking products, e.g. C2 and C3 polyols, decreased concurrently from 17.0 to 3.3%. The total selectivity to the identified products reached 96.0%. With increasing the Sn/Pt ratios to 0.3 and 0.5, the hexitol selectivity increased to 81.3 and 82.7%, respectively, nearly twice the value of Pt/Al2O3. However, further increasing the Sn/Pt ratios led to a concurrent decline in the hexitol selectivity and an increase in the C2–C5 polyols. Specifically, as the Sn/Pt ratio increased to 2.1, the hexitol selectivity dramatically decreased to as low as 3.2%, whereas the selectivity to the C2 and C3 polyols increased to as high as 46.1%. It is noted that acetol, an important chemical intermediate,32,33 was formed with a selectivity of 19.6%, which further increased to 25.2% by increasing the Sn/Pt ratio from 2.1 to 3.8.
| Catalyst | Conversion% | Selectivity% | ||||||
|---|---|---|---|---|---|---|---|---|
| Hexitolsb | Pentitolsc | Tetritolsd | Glycerol | 1,2-Propanediol | Acetol | Ethylene glycol | ||
| C6 | C5 | C4 | C3 | C3 | C3 | C2 | ||
| a 1 g cellulose, 0.4 g catalyst, 473 K, 50 mL H2O, 6 MPa H2, 30 min. b Sorbitol, mannitol and iditol. c Xylitol and arabitol. d Erythritol and threitol. e Physical mixture of Pt–SnOx(0.3)/Al2O3 and Pt–SnOx(3.8)/Al2O3. f Physical mixture of Pt/Al2O3 and SnOx/Al2O3, Sn = 0.36 wt%. | ||||||||
| Pt/Al2O3 | 17.6 | 43.3 | 7.7 | 3.4 | 7.2 | 6.3 | 0.0 | 3.5 |
| Pt–SnOx(0.1)/Al2O3 | 16.3 | 78.0 | 6.9 | 1.7 | 2.2 | 0.5 | 0.0 | 0.6 |
| Pt–SnOx(0.3)/Al2O3 | 18.9 | 81.6 | 5.5 | 1.3 | 1.4 | 0.3 | 0.0 | 0.3 |
| Pt–SnOx(0.5)/Al2O3 | 17.1 | 82.8 | 4.1 | 1.1 | 1.5 | 0.3 | 0.0 | 0.4 |
| Pt–SnOx(1.0)/Al2O3 | 17.8 | 73.4 | 4.3 | 3.8 | 3.2 | 1.4 | 0.0 | 2.9 |
| Pt–SnOx(1.5)/Al2O3 | 19.6 | 43.2 | 2.6 | 5.2 | 4.8 | 5.8 | 0.0 | 8.7 |
| Pt–SnOx(2.1)/Al2O3 | 18.3 | 3.2 | 1.6 | 4.6 | 3.9 | 5.6 | 19.6 | 17.0 |
| Pt–SnOx(3.8)/Al2O3 | 22.5 | 0.8 | 1.6 | 1.1 | 2.0 | 1.4 | 25.2 | 7.4 |
| Pt–SnOx(0.3)/Al2O3 + Pt–SnOx(3.8)/Al2O3e |
23.8 | 3.9 | 1.5 | 2.4 | 2.4 | 13.5 | 12.3 | 22.0 |
| Pt/Al2O3 + SnOx/Al2O3f |
19.2 | 5.7 | 0.9 | 2.5 | 3.2 | 16.3 | 14.2 | 14.2 |
| Pt–ReOx(0.3)/Al2O3 | 18.0 | 63.7 | 14.1 | 3.2 | 2.8 | 1.0 | 0.0 | 0.7 |
| Pt–SbOx(0.3)/Al2O3 | 20.1 | 51.6 | 1.6 | 2.4 | 7.3 | 5.6 | 0.0 | 2.7 |
| Pt–GaOx(0.3)/Al2O3 | 17.7 | 37.1 | 4.5 | 2.6 | 7.5 | 7.3 | 0.0 | 3.3 |
Such a change in the polyol selectivities reflects the effect of SnOx on the hydrogenation property of Pt on Al2O3, as reported previously in many other reactions such as the hydrogenation of crotonaldehyde or citral.19 This effect is further examined here in the hydrogenation of glucose, the key primary intermediate in the cellulose conversion into polyols. As shown in Table 3, the glucose conversion was 35.2% on Pt/Al2O3 after 10 min at 393 K and 6 MPa H2, which increased to 63.3% on Pt–SnOx(0.5)/Al2O3 with a Sn/Pt ratio of 0.5, corresponding to an increase in their turnover frequency (TOF) from 4.6 to 11.4 min−1 (normalized per exposed surface Pt atom). Afterwards, a further increase in the Sn/Pt ratio up to 3.8 led to a monotonic decline in the glucose conversion and TOF to only 3.8% and 0.3 min−1, respectively. The Sn/Pt ratios also influenced the product selectivities in the glucose hydrogenation. Notably, the catalyst with the highest Sn/Pt ratio of 3.8 exhibited essentially no hydrogenation activity, and it catalyzed mainly the glucose isomerization to fructose with a selectivity of 82.3% (at a low conversion of only 3.8%). Clearly, the Pt–SnOx/Al2O3 samples with the Sn/Pt ratios of 0.1–1.0 afforded the superior hydrogenation activities, and thus favored the efficient synthesis of hexitols from the hydrogenation of glucose and other C6 sugar intermediates involved in the cellulose hydrolysis step, which would otherwise undergo degradation or condensation reactions, as observed at the Sn/Pt ratios higher than 1.0 (Table 2). To understand the underlying reason of such SnOx effects on the catalytic properties of the Pt–SnOx/Al2O3 catalysts, they were characterized by TPR in H2 (H2-TPR) and FT-IR of CO adsorption (CO-IR).
| Sn/Pt | Conversion% | TOFb/min−1 | Selectivity% | |
|---|---|---|---|---|
| Hexitols | Fructose | |||
| a 1 g glucose, 0.4 g catalyst, 393 K, 50 mL H2O, 6 MPa H2, 10 min. b TOF was calculated from the amount of glucose converted to polyols normalized per exposed Pt atom. | ||||
| Pt/Al2O3 | 35.2 | 4.6 | 94.0 | 1.4 |
| Pt–SnOx(0.1)/Al2O3 | 43.2 | 7.1 | 96.5 | 0.0 |
| Pt–SnOx(0.3)/Al2O3 | 50.9 | 7.4 | 94.8 | 0.0 |
| Pt–SnOx(0.5)/Al2O3 | 63.3 | 11.4 | 96.7 | 0.0 |
| Pt–SnOx(1.0)/Al2O3 | 38.9 | 9.6 | 98.3 | 0.0 |
| Pt–SnOx(1.5)/Al2O3 | 25.7 | 7.6 | 93.3 | 4.3 |
| Pt–SnOx(2.1)/Al2O3 | 6.1 | 2.2 | 63.0 | 30.1 |
| Pt–SnOx(3.8)/Al2O3 | 3.8 | 0.3 | 0.0 | 82.3 |
Fig. 5 shows the H2-TPR profiles of the Pt–SnOx/Al2O3 samples. For comparison, the profiles of Pt/Al2O3 and SnOx/Al2O3 are also included. SnOx/Al2O3 presents a broad SnOx reduction feature around 450 °C, as reported previously.26 For Pt/Al2O3, two reduction peaks were observed at 227 and 351 °C.34,35 The first peak is characteristic of the reduction of the PtOx species interacting weakly with the Al2O3 support, while the second one can be assigned to the PtOx or oxy-chlorided Pt species interacting strongly with Al2O3.34,35 The total consumption of H2 corresponds to the stoichiometric reduction of Pt4+ to Pt0. This TPR feature confirms the presence of merely metallic Pt on Pt/Al2O3 after the general treatment with H2 at 400 °C employed in this work, which is consistent with the XPS result (Fig. 1). Compared to Pt/Al2O3, an additional peak appeared around 400 °C on the Pt–SnOx/Al2O3 catalysts due to the reduction of their SnOx species. The PtOx reduction peak shifted to lower temperatures around 200 °C with increasing the Sn/Pt ratios to 0.3 and 0.5. It then shifted to the higher temperature around 225 °C, approaching that for Pt/Al2O3, as the Sn/Pt ratios exceeded 1.0. The higher reducibility of the PtOx species observed at the lower Sn/Pt ratios of 0.1–0.5 is related to the strong interaction between PtOx and SnOx, as reported previously, via the electron transfer from SnOx to PtOx on Pt–SnOx/Al2O3.36,37 The observed weak effect of SnOx on the PtOx reduction at the higher Sn/Pt ratios may be due to the partial coverage of the PtOx species by SnOx, as inferred from the decreased Pt dispersion (Table 1), and the formation of larger PtSn alloy particles with low ability of hydrogen activation.38 These results are in agreement with those obtained from the in situ FT-IR characterization of CO adsorption, as shown below in Fig. 6.
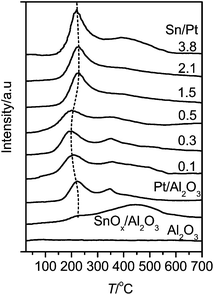 | ||
| Fig. 5 TPR profiles of Pt–SnOx/Al2O3 with different Sn/Pt atomic ratios in the range of 0.1–3.8, and for comparison Pt/Al2O3, SnOx/Al2O3 (0.36 wt% Sn) and Al2O3. | ||
 | ||
| Fig. 6 FT-IR spectra of CO adsorption on Pt/Al2O3 and Pt–SnOx/Al2O3 with Sn/Pt atomic ratios in the range of 0.1–3.8. | ||
It is known that the SnOx surfaces do not significantly adsorb CO.29,34 Therefore, the CO adsorption bands observed on the bimetallic Pt–SnOx/Al2O3 catalysts (Fig. 6) arose from the CO adsorption on the Pt sites. For Pt/Al2O3, an intense band was detected at 2081 cm−1, which corresponds to the linear adsorption of CO on the metallic Pt sites.34 This band shifted to ca. 2073 cm−1 on the Pt–SnOx/Al2O3 samples with Sn/Pt ratios of 0.1–1.0. Such a red-shift on the Pt–SnOx catalysts can be explained by the geometric effect of the SnOx species that block or dilute the Pt sites, leading to the weakening of dipolar coupling between the adsorbed CO molecules, or by the electronic effect of the SnOx species that transfer electrons to the Pt sites, leading to increasing electron back-donation from the d orbitals of Pt to the 2π* states of CO.29,39 However, a further increase in the Sn/Pt ratios to over 1.5 led to the appearance of the CO adsorption band at ca. 2081 cm−1, as observed on Pt/Al2O3, which excludes the geometric effect of the SnOx species. Therefore, the red-shift of the CO adsorption band on the samples with Sn/Pt ratios of 0.1–1.0 is attributed to the electron transfer from SnOx to Pt.37
It is known that the electron transfer from SnOx to Pt can increase the electron density of the Pt sites, and consequently promote H2 dissociation, required for the hydrogenation of glucose and other C6 sugar intermediates involved in the cellulose conversion. Moreover, the electron transfer can make the SnOx surfaces more positively charged and enhance their Lewis acidity, which, as reported by Gallezot et al., can favor the polarization of the carbonyl group of glucose via withdrawing electrons from the oxygen atom in its carbonyl group.40 Such enhanced polarization then facilitates the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O hydrogenation by H2 dissociatively adsorbed on the Pt sites that are in intimate contact with the SnOx species, as proposed in Scheme 1. Taken together, these electronic effects of the SnOx species led to the superior hydrogenation activities and efficiency for the synthesis of hexitols in the cellulose reaction on the Pt–SnOx/Al2O3 catalysts with the Sn/Pt ratios of 0.1–1.0, relative to Pt/Al2O3 (Tables 2 and 3).
O hydrogenation by H2 dissociatively adsorbed on the Pt sites that are in intimate contact with the SnOx species, as proposed in Scheme 1. Taken together, these electronic effects of the SnOx species led to the superior hydrogenation activities and efficiency for the synthesis of hexitols in the cellulose reaction on the Pt–SnOx/Al2O3 catalysts with the Sn/Pt ratios of 0.1–1.0, relative to Pt/Al2O3 (Tables 2 and 3).
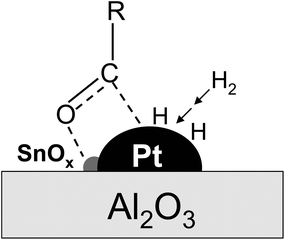 | ||
| Scheme 1 Schematic description of the promoting role of SnOx in glucose hydrogenation on Pt–SnOx/Al2O3 catalysts with atomic ratios of 0.1–1.5. | ||
Such electronic effects on Pt hydrogenation activity were further confirmed by the Pt/Al2O3 catalysts modified with ReOx, SbOx and GaOx in the cellulose reaction. These catalysts (∼2 wt% Pt) possessed similar Pt dispersions (∼72–75%) with low atomic ratios of Re, Sb or Ga to Pt of 0.3. Their CO-IR spectra show that the CO adsorption band shifted from 2081 cm−1 on Pt/Al2O3 to 2076 and 2074 cm−1 on Pt–ReOx/Al2O3 and Pt–SbOx/Al2O3, respectively, while it remained at 2081 cm−1 on Pt–GaOx/Al2O3 (Fig. 7). Such different effects on the CO adsorption reflect the electron transfer to Pt from ReOx and SbOx, but not from GaOx, which is consistent with their effects on the Pt hydrogenation activity probed by the cellulose reaction. As shown in Table 2, the selectivity to hexitols increased from 43.3% on Pt/Al2O3 to 63.7 and 51.6% on Pt–ReOx/Al2O3 and Pt–SbOx/Al2O3, respectively, but it was as low as 37.1% on Pt–GaOx/Al2O3 at the similar cellulose conversions (17.6–20%). These results reveal that ReOx and SbOx, similar to SnOx, can donate electrons to the Pt sites on Al2O3, and accordingly improve their hydrogenation activity.
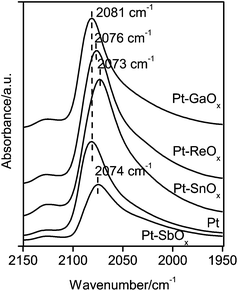 | ||
| Fig. 7 FT-IR spectra of CO adsorption on ReOx, SbOx, and GaOx-modified Pt/Al2O3 catalysts with atomic ratios of Sb, Re, or Ga to Pt of 0.3, and for comparison, on Pt/Al2O3 and Pt–SnOx(0.3)/Al2O3 with a Sn/Pt atomic ratio of 0.3. | ||
Next, we examine the underlying basis for the preferential conversion of cellulose to the C2 and C3 polyols at the expense of hexitols on the Pt–SnOx/Al2O3 catalysts with Sn/Pt ratios above 1.5. These catalysts exhibited inferior hydrogenation activity, which appears to lead to the degradation of glucose or other C6 sugar intermediates to form the C2 and C3 products, rather than the sugar hydrogenation to hexitols. However, our control experiments show that the low hydrogenation activity cannot fully account for the formation of the degradation products. For example, as aforementioned, the total selectivity of the C2 and C3 products was ∼36% on the Pt–SnOx(3.8)/Al2O3 catalyst with the Sn/Pt ratio of 3.8 (Table 2). After physically mixing it with the active hydrogenation catalyst of Pt–SnOx(0.3)/Al2O3, the selectivity to hexitols only increased slightly from 0.6 to 3.9%, but the C2 and C3 polyols were still the dominant products (50.2% selectivity) at similar cellulose conversions (Table 2). This result suggests that the SnOx species not in contact with the Pt sites on Pt–SnOx(3.8)/Al2O3 contributes to the cleavage of the C–C bonds in glucose or other sugar intermediates in the cellulose reaction, most likely following the retro-aldol condensation mechanism, to form the C2 and C3 products, which apparently occurs more readily on the SnOx species than the sugar hydrogenation to hexitols on the Pt sites. This proposition was indeed confirmed by the catalytic performance of another physical mixture containing Pt/Al2O3 and SnOx/Al2O3 (0.36 wt% Sn). As shown in Table 2, the addition of SnOx/Al2O3 to Pt/Al2O3 even at a low Sn/Pt ratio of 0.3, led to a sharp increase in the selectivity to the C2 and C3 polyols from 9.8 to 48.8% and a concurrent decline in the selectivity to hexitols from 43.3 to 5.7% at similar cellulose conversions. Such catalytic properties of the SnOx species were more directly evidenced from the cellulose reaction catalyzed only by SnOx, as discussed below.
To understand the nature of the active SnOx species responsible for breaking the C–C bonds, different tin samples including bulk SnO, SnO2, Sn(OH)2 and Sn(OH)4 were examined for the cellulose reaction in the absence of the Pt sites (Table 4). On these samples, three C3 products: glycerol, acetol and lactic acid, were detected. Other products included glucose and trace amounts of C2 compounds (e.g. glycolic acid). SnO, SnO2 and Sn(OH)4 exhibited low selectivities to glycerol, acetol and lactic acid in the range of 10–15%, which increased significantly to 34.4% on Sn(OH)2 at similar cellulose conversions under identical conditions. These results indicate that Sn(OH)2 is the active SnOx species for catalyzing the retro-aldol condensation of glucose or other C6 sugars to form the C2 and C3 products in the cellulose reaction. To further understand the catalytic properties of the SnOx species on the Pt–SnOx/Al2O3 catalysts, SnOx/Al2O3 (4.87 wt% Sn) was examined. Similar to Sn(OH)2, it catalyzed cellulose conversion to the C3 products at a selectivity of 28.8%, which remained essentially unaltered (i.e. 30.4%) when the SnOx/Al2O3 sample was recycled. These results imply that Sn(OH)2 species were formed on SnOx/Al2O3 and remained stable during the cellulose reaction.
| Catalyst | Conversion% | Selectivity% | |||
|---|---|---|---|---|---|
| Glucose | Glycerol | Acetol | Lactic acid | ||
| a 1 g cellulose, 0.4 g catalyst, 473 K, 50 mL H2O, 6 MPa H2, 30 min. b Sn = 4.87 wt%. | |||||
| SnO | 17.1 | 5.3 | 2.6 | 8.4 | 2.2 |
| SnO2 | 19.9 | 10.2 | 2.1 | 6.5 | 5.7 |
| Sn(OH)4 | 12.0 | 0.6 | 1.8 | 4.7 | 4.3 |
| Sn(OH)2 | 15.5 | 1.7 | 5.1 | 21.1 | 8.2 |
| SnOx/Al2O3b |
8.2 | 12.5 | 3.6 | 12.2 | 13.0 |
| SnOx/Al2O3-recycledb |
6.8 | 11.8 | 3.2 | 9.9 | 17.3 |
Furthermore, it is noted that the C3 products were dominant over the C2 products on the SnOx and Pt–SnOx/Al2O3 (with the higher Sn/Pt ratios) catalysts, as shown in Tables 2 and 4. Following the retro-aldol condensation mechanism, glucose tends to convert to the C2 products while the C3 products dominate in the fructose reaction. Therefore, we suggest that the formation of the C3 products in the cellulose reaction involves the isomerization of glucose to fructose prior to its retro-aldol condensation on the SnOx species, which is consistent with the observed conversion of glucose to fructose, as shown in Table 3, on the Pt–SnOx/Al2O3 catalysts with high Sn/Pt ratios of 2.1 and 3.8 at 393 K. This suggestion can account for the formation of the C3 products. The retro-aldol condensation of fructose is known to form the glyceraldehyde and dihydroxyacetone intermediates which undergo hydrogenation to glycerol (or further to 1,2-propanediol), or dehydration to pyruvaldehyde. The pyruvaldehyde intermediate then converts to acetol and lactic acid by hydrogenation and benzilic acid rearrangement, respectively.41 This suggestion is further confirmed by the reactions of glucose and fructose on Pt–SnOx(3.8)/Al2O3, predominantly forming acetol and lactic acid. For example, the yields to acetol and lactic acid were ∼40.0 and ∼55.0% from glucose and fructose, respectively, at 473 K and 6 MPa H2 (at a 100% conversion), in which the yields to acetol were 32.9 and 38.2%. Such high acetol yields, although obtained under non-optimized conditions, show the potential reactivity of the SnOx species for the selective cleavage of the C–C bonds in glucose and fructose to produce acetol, an important bio-platform chemical, which have not been reported before directly from the cellulose reaction.
Moreover, the Pt–SnOx/Al2O3 catalysts were stable and recyclable under the reaction conditions in this work. Figs. S3 and S4† show the representative results with Pt–SnOx(3.8)/Al2O3 in the glucose reaction at 473 K and 6 MPa H2. No significant decline in the yields of the C2 and C3 products was observed after five successive cycles (Fig. S3†), which is consistent with the essentially identical XRD patterns for this catalyst (Fig. S4†) before and after the five cycles. This result reveals that the hydrothermal stability of Al2O3 can be enhanced by loading SnOx on its surface, as is also found with the WOx/Al2O3 catalysts.5c In contrast, pure Al2O3 support readily transformed to AlO(OH) in water at 473 K (Fig. S4†).
4 Conclusions
The Sn/Pt atomic ratios of SnOx-modified Pt/Al2O3 catalysts significantly influence their hydrogenation activities and selectivities for cellulose conversion to polyols at 473 K and 6 MPa H2. At lower Sn/Pt ratios of 0.1–1.0, the SnOx species strongly interact with and transfer electrons to the Pt sites, as partly evidenced from the red-shift of the CO adsorption IR bands, leading to the superior hydrogenation activities of the Pt–SnOx/Al2O3 catalysts, and thus their higher selectivity to hexitols. The selectivity reaches a maximum of 82.7% at the Sn/Pt ratio of 0.5, which is nearly two times that of Pt/Al2O3 at similar cellulose conversions (∼20%). At higher Sn/Pt ratios than 1.5, the Pt–SnOx/Al2O3 catalysts favor the formation of C2 and especially C3 products (e.g. acetol), as a result of their low hydrogenation activities, and the activity of the segregated SnOx species from the Pt sites, apparently in the form of Sn(OH)2, for cleavage of the C–C bonds of glucose and other C6 sugar intermediates in the cellulose reaction, which most likely involves the isomerization of glucose to fructose and its subsequent retro-aldol condensation on SnOx. Taken together, these results demonstrate the bifunctional role of the SnOx species in tuning the reaction rate of the hydrogenation of glucose and other C6 sugar intermediates to hexitols and of their selective degradation to C2 and C3 products, which is crucial for the rational synthesis of target polyols in the cellulose reaction.Acknowledgements
This work was supported by the National Natural Science Foundation of China (20825310, 21173008 and 51121091) and the National Basic Research Project of China (2011CB201400 and 2011CB808700).References
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- (a) S. Van de Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 Search PubMed; (b) J. A. Geboers, S. Van de Vyver, R. Ooms, B. Op de Beeck, P. A. Jacobs and B. F. Sels, Catal. Sci. Technol., 2011, 1, 714–726 RSC.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2602 CrossRef CAS.
- (a) J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 RSC; (b) J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS.
- (a) Y. Shen, S. Wang, C. Luo and H. Liu, Prog. Chem., 2007, 19, 431–436 Search PubMed; (b) T. Deng, J. Sun and H. Liu, Sci. China: Chem., 2010, 53, 1476–1480 Search PubMed; (c) Y. Liu, C. Luo and H. Liu, Angew. Chem., Int. Ed., 2012, 51, 3249–3253 CrossRef CAS.
- J. M. Robinson, C. E. Burgess, M. A. Bently, C. D. Brasher, B. O. Horne, D. M. Lillard, J. M. Macias, H. D. Mandal, S. C. Mills, K. D. O'Hara, J. T. Pon, A. F. Raigoza, E. H. Sanchez and J. S. Villarreal, Biomass Bioenergy, 2004, 26, 473–483 CrossRef CAS.
- H. Kobayashi, H. Ohta and A. Fukuoka, Catal. Sci. Technol., 2012, 2, 869–883 RSC.
- V. I. Sharkov, Angew. Chem., Int. Ed. Engl., 1963, 2, 405–409 CrossRef.
- R. Palkovits, K. Tajvidi, A. M. Ruppert and J. Procelewska, Chem. Commun., 2011, 47, 576–578 RSC.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS.
- N. Yan, C. Zhao, C. Luo, P. J. Dyson, H. Liu and Y. Kou, J. Am. Chem. Soc., 2006, 128, 8714–8715 CrossRef CAS.
- C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636–7639 CrossRef CAS.
- J. Pang, A. Wang, M. Zheng, Y. Zhang, Y. Huang, X. Chen and T. Zhang, Green Chem., 2012, 14, 614–617 RSC.
- (a) S. Van de Vyver, J. Geboers, W. Schutyser, M. Dusselier, P. Eloy, E. Dornez, J. W. Seo, C. M. Courtin, E. M. Gaigneaux, P. A. Jacobs and B. F. Sels, ChemSusChem, 2012, 5, 1549–1558 Search PubMed; (b) J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Chem. Commun., 2011, 47, 5590–5592 RSC; (c) J. Geboers, S. Van de Vyver, K. Carpentier, K. de Blochouse, P. Jacobs and B. Sels, Chem. Commun., 2010, 46, 3577–3579 RSC.
- W. Deng, X. Tan, W. Fang, Q. Zhang and Y. Wang, Catal. Lett., 2009, 133, 167–174 CrossRef CAS.
- (a) N. Ji, T. Zhang, M. Zheng, A. Wang, H. Wang, X. Wang and J. G. Chen, Angew. Chem., Int. Ed., 2008, 47, 8510–8513 CrossRef CAS; (b) Y. Zhang, A. Wang and T. Zhang, Chem. Commun., 2010, 46, 862–864 RSC.
- C. L. Pieck, C. R. Vera, C. A. Querini and J. M. Parera, Appl. Catal., A, 2005, 278, 173–180 CrossRef CAS.
- L. Huang, B. Xu, L. Yang and Y. Fan, Catal. Commun., 2008, 9, 2593–2597 CrossRef CAS.
- I. M. J. Vilella, S. R. de Miguel and O. A. Scelza, J. Mol. Catal. A: Chem., 2008, 284, 161–171 CrossRef CAS.
- P. D. Zgolicz, V. I. Rodríguez, I. M. J. Vilella, S. R. de Miguel and O. A. Scelza, Appl. Catal., A, 2011, 392, 208–217 CrossRef CAS.
- C. Kappenstein, M. Guerin, K. Lazar, K. Matusek and Z. Paal, J. Chem. Soc., Faraday Trans., 1998, 94, 2463–2473 RSC.
- K. Liberková-Šebková, L. Červený and R. Touroude, Res. Chem. Intermed., 2003, 29, 609–617 Search PubMed.
- A. B. Merlo, V. Vetere, J. F. Ruggera and M. L. Casella, Catal. Commun., 2009, 10, 1665–1669 CrossRef CAS.
- G. C. Torres, S. D. Ledesma, E. L. Jablonski, S. R. de Miguel and O. A. Scelza, Catal. Today, 1999, 48, 65–72 CrossRef CAS.
- C. Guzmán, G. Del Angel, J. Fierro and V. Bertin, Top. Catal., 2010, 53, 1142–1144 Search PubMed.
- D. L. Hoang, S. A. F. Farrage, J. Radnik, M. M. Pohl, M. Schneider, H. Lieske and A. Martin, Appl. Catal., A, 2007, 333, 67–77 Search PubMed.
- Y. Zhou and S. M. Davis, Catal. Lett., 1992, 15, 51–55 Search PubMed.
- N. Kamiuchi, K. Taguchi, T. Matsui, R. Kikuchi and K. Eguchi, Appl. Catal., B, 2009, 89, 65–72 CrossRef CAS.
- G. J. Arteaga, J. A. Anderson, S. M. Becker and C. H. Rochester, J. Mol. Catal. A: Chem., 1999, 145, 183–201 CrossRef CAS.
- G. Neri, C. Milone, S. Galvagno, A. P. J. Pijpers and J. Schwank, Appl. Catal., A, 2002, 227, 105–115 CrossRef CAS.
- H. Kobayashi, Y. Ito, T. Komanoya, Y. Hosaka, P. L. Dhepe, K. Kasai, K. Hara and A. Fukuoka, Green Chem., 2011, 13, 326–333 RSC.
- S. Niu, Y. Zhu, H. Zheng, W. Zhang and Y. Li, Chin. J. Catal., 2011, 32, 345–351 Search PubMed.
- C. Liu, X. Dou and Y. Lu, Org. Lett., 2011, 13, 5248–5251 CrossRef CAS.
- V. A. Mazzieri, J. M. Grau, J. C. Yori, C. R. Vera and C. L. Pieck, Appl. Catal., A, 2009, 354, 161–168 Search PubMed.
- I. Contreras-Andrade, A. Vázquez-Zavala and T. Viveros, Energy Fuels, 2009, 23, 3835–3841 Search PubMed.
- M. Kang, W. Kim, J. Joo, N. Kim, H. Yun and J. Yi, Catal. Lett., 2009, 132, 417–421 CrossRef CAS.
- F. Somodi, I. Borbáh, M. Hegedus, K. Lázár, E. Sajó István, O. Geszti, S. Rojas, J. L. G. Fierro and J. L. Margitfalvi, Appl. Surf. Sci., 2009, 256, 726–736 Search PubMed.
- R. A. van Santen and M. Neurock, Molecular Heterogeneous Catalysis, Wiley, 2006 Search PubMed.
- J. Ruiz-Martíez, F. Coloma, A. Sepúveda-Escribano, J. A. Anderson and F. Rodríuez-Reinoso, Catal. Today, 2008, 133–135, 35–41 Search PubMed.
- P. Gallezot, P. J. Cerino, B. Blanc, G. Fleche and P. Fuertes, J. Catal., 1994, 146, 93–102 CrossRef CAS.
- E. P. Maris and R. J. Davis, J. Catal., 2007, 249, 328–337 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2gc36088h |
| This journal is © The Royal Society of Chemistry 2013 |