Nanostructured carbon–metal oxide composite electrodes for supercapacitors: a review
Mingjia Zhi, Chengcheng Xiang, Jiangtian Li, Ming Li and Nianqiang Wu*
Department of Mechanical and Aerospace Engineering, West Virginia University, Morgantown, WV 26505-6106, USA. E-mail: nick.wu@mail.wvu.edu; Fax: +1-304-293-6689; Tel: +1-304-293-3326
First published on 23rd October 2012
Abstract
This paper presents a review of the research progress in the carbon–metal oxide composites for supercapacitor electrodes. In the past decade, various carbon–metal oxide composite electrodes have been developed by integrating metal oxides into different carbon nanostructures including zero-dimensional carbon nanoparticles, one-dimensional nanostructures (carbon nanotubes and carbon nanofibers), two-dimensional nanosheets (graphene and reduced graphene oxides) as well as three-dimensional porous carbon nano-architectures. This paper has described the constituent, the structure and the properties of the carbon–metal oxide composites. An emphasis is placed on the synergistic effects of the composite on the performance of supercapacitors in terms of specific capacitance, energy density, power density, rate capability and cyclic stability. This paper has also discussed the physico-chemical processes such as charge transport, ion diffusion and redox reactions involved in supercapacitors.
 Mingjia Zhi | Mingjia Zhi received his B.S. and M.S. degrees from Department of Materials Science and Engineering at Zhejiang University in 2004 and 2006, respectively. He received his Ph.D. degree in Mechanical Engineering at West Virginia University in USA. He currently works as a Postdoctoral Fellow, co-supervised by Dr Nianqiang (Nick) Wu at West Virginia University and Dr A. Manivannan at National Energy Technology Laboratory, DOE. His research interests include nanomaterials, electrochemical energy conversion and storage devices. |
 Chengcheng Xiang | Chengcheng Xiang received her MS degrees in Material Physics at Central South University in China in 2007 and in Industrial Engineering at West Virginia University in 2011. Currently she is a Ph.D. student in Industrial and Management System Engineering at West Virginia University in USA. Her research interest is focused on experimental design and statistical analysis, nanomaterials and their application in supercapacitors. |
 Jiangtian Li | Jiangtian Li received his Ph.D. degree in Materials Physics and Chemistry at Shanghai Institute of Ceramics, Chinese Academy of Science in 2008. He then worked as an Assistant Professor with Prof. Fuqiang Huang. Afterwards, he worked as a Postdoctoral Fellow at University of Cologne, Germany. In 2010, he joined Prof. Nianqiang (Nick) Wu's group at West Virginia University. His current research interest is focused on nanostructured materials, solar energy conversion and storage. |
 Ming Li | Ming Li received his B.Sc. degree in Fine Chemicals from Dalian University of Technology in China in 2005 and his M.S. degree from Department of Materials Science and Engineering at Zhejiang University in China. He then joined Prof. Nianqiang (Nick) Wu's group as a Ph.D. student. He received his Ph.D. degree from Department of Mechanical and Aerospace Engineering at West Virginia University in USA in 2012. His research interest lies in functional nanomaterials, biosensors as well as energy conversion and storage devices. |
 Nianqiang Wu | Nianqiang Wu received his Ph.D. degree from Zhejiang University, China. After two years of postdoctoral training in University of Pittsburgh, he joined Keck Interdisciplinary Surface Science Center at Northwestern University. In 2005, he took a faculty position in West Virginia University. He is currently Associate Professor in Department of Mechanical and Aerospace Engineering at West Virginia University in USA. His research areas include (i) nanomaterials and nanolithography, (ii) electrochemical devices for energy conversion and storage, (iii) chemical sensors and biosensors, and (iv) photocatalysts and photoelectrochemical cells. |
1 Introduction
Supercapacitors, also called ultracapacitors, are finding increasing applications in hybrid electric vehicles, large industrial equipment, memory back-up devices and renewable energy power plants.1–5 Supercapacitors provide high power density, fast charge–discharge, and long service life as compared to batteries.6–10 The specific capacitance (C) of a supercapacitor is defined as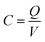 | (1) |
Supercapacitors store the energy via two operating mechanisms:
(i) Electrochemical double-layer capacitance (EDLC),11,12 which results from the electrical double-layer surrounding the surface of electrode. That is, the depletion of the oppositely charged species stores the energy at the interface of the electrode and the electrolyte, respectively. The accumulation of electrons at the electrode is a non-Faradaic process. The specific capacitance of an EDLC is measured as
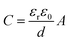 | (2) |
(ii) Pseudo-capacitance, which is originated from the redox reaction of the electrode material with the electrolyte.13–15 The accumulation of electrons at the electrode is a Faradaic process where the electrons produced by the redox reaction are transferred across the electrolyte–electrode interface. The theoretical pseudo-capacitance of metal oxide can be calculated as
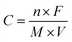 | (3) |
The energy density (E) of a supercapacitor is expressed as
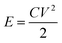 | (4) |
The maximum power density of a supercapacitor is determined by
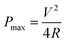 | (5) |
| Type of supercapacitor | Electrode material | Charge storage mechanism | Merits/shortcomings | |
|---|---|---|---|---|
| Electrochemical double layer capacitor (EDLC) | Carbon | Electrochemical double layer (EDL), non-Faradaic process | Good cycling stability, good rate capability, low specific capacitance, low energy density | |
| Pseudocapacitor | Redox metal oxide or redox polymer | Redox reaction, Faradaic process | High specific capacitance, relatively high energy density, relatively high power density, relatively low rate capability | |
| Hybrid capacitor | Asymmetric hybrid | Anode: pseudocapacitance materials, cathode: carbon | Anode: redox reaction, cathode: EDL | High energy density, high power density and good cyclability |
| Symmetric composite hybrid | Redox metal oxide/carbon or redox polymer/carbon | Redox reaction plus EDL | High energy density, moderate cost and moderate stability | |
| Battery-like hybrid | Anode: Li-insertion material, cathode: carbon | Anode: Lithiation/delithiation cathode: EDL | High energy density, high cost and requires electrode material capacity match | |
The key performance parameters of supercapacitors include specific capacitance (normalized by electrode mass, volume, or area), energy density, power density, rate capability (retained capacitance at a high current loading) and cycling stability.16 To increase the energy density and power density of a supercapacitor, it is desirable to increase the specific capacitance (C) and the operating voltage window (V) as well as reduce the equivalent series resistance (R). For EDLC supercapacitors, the maximum operating voltage window (Vm) is mainly dependent on the electrolyte used, which is limited by the stability of the electrolyte. Normally Vm is 1 V for the supercapacitors based on the aqueous electrolyte. One of the current research trends in supercapacitors is to develop non-aqueous electrolytes with high Vm. For example, an ionic liquid-based electrolyte is able to be operated at Vm as high as 3.5 V.17 For a review on the electrolytes refer to the previous paper.18 This paper is focused on the electrode materials. Ideal electrode materials are required to possess:
• high specific surface area, which governs the specific capacitance,
• controlled porosity, which affects the specific capacitance and the rate capability,
• high electronic conductivity, which is crucial to the rate capability and the power density,
• desirable electroactive sites, which enable pseudo-capacitance,
• high thermal stability and chemical stability, which affect the cyclic stability, and
• low costs of raw materials and manufacturing.
Currently the electrodes of most commercial supercapacitors are made of carbon that is inexpensive and has good resistance to corrosion. Carbon-based supercapacitors are operated under the EDLC. The carbon-based EDLC supercapacitors have excellent cyclic stability and long service lifetime since the electrode undergoes no chemical change during the charge/discharge processes. However, their maximum capacitance is restricted by the active electrode surface area and the pore size distribution (typically 0.15–0.4 F m−2, or ∼150 F g−1 for carbon).19,20 Overall, the energy density of currently commercial carbon-based EDLC supercapacitors is typically 3–5 W h kg−1, which is much lower than that of an electrochemical battery (30–40 W h kg−1 for a lead acid battery and 10–250 W h kg−1 for a lithium-ion battery).21 Such low energy-density cannot fulfill the need of energy storage devices for vehicles, wind-farms and solar power plants.
To improve the specific capacitance and the energy density, transition metal oxides are being investigated as the alternative materials for supercapacitor electrodes. The investigated metal oxides include RuO2, MnO2, NiO, Co3O4, SnO2, ZnO, TiO2, V2O5, CuO, Fe2O3, WO3, etc.22–33 In particular, some metal oxides show excellent pseudo-capacitance. Unlike a lithium battery in which the ions are deeply intercalated in the materials lattice, the pseudo-capacitance is originated from the weakly attached surface ions. The surface functional groups, the defects and the grain boundaries can serve as the excellent redox centers for the charge storage reactions. Table 2 lists the theoretical capacitance of typical metal oxides as well as the charge storage reactions. It can be seen that the transition metal oxide electrodes have one order higher specific capacitance than the carbon electrodes. Several reports have shown that bare metal oxide electrodes can deliver large specific capacitance and high energy density at a slow scan rate or at a low current density. For example, the electrodeposited NiO thin film electrode showed a specific capacitance of 1776 F g−1 in the 1 M KOH aqueous electrolyte at a scan rate of 1 mV s−1. However, only 23% of capacitance was retained when the scan rate increased from 1 mV s−1 to 100 mV s−1.44 Therefore metal oxides may not be employed alone as the supercapacitor electrodes for practical purpose due to the following drawbacks.
| Oxide | Electrolyte | Charge storage reaction | Theoretical capacitance (F g−1) | Conductivity (S cm−1) |
|---|---|---|---|---|
| MnO2 | Na2SO4 | MnO2 + M+ + e− = MMnO2 (M could be H+, Li+, Na+, K+) | 1380 (0.9 V)34 | 10−5 to 10−6(ref. 35) |
| NiO | KOH, NaOH | NiO + OH− = NiOOH + e− | 2584 (0.5 V)36 | 0.01–0.32 (ref. 37) |
| Co3O4 | KOH, NaOH | Co3O4 + OH− + H2O = 3CoOOH + e− CoOOH + OH− = CoO2 + H2O + e− | 3560 (0.45 V)38,39 | 10−4 to 10−2(ref. 40) |
| V2O5 | NaCl, Na2SO4 | V2O5 + 4M+ + 4e− = M2V2O5 (M could be H+, Li+, Na+, K+) | 2120 (1 V) | 10−4 to 10−2(ref. 41) |
| RuO2, xH2O | H2SO4, Na2SO4 | RuO2 + xH+ + xe− = RuO2−x, (OH)x (0 < x < 2) | 1200–2200 (1.23 V)42 | 103 for polycrystalline, ∼1 for amorphous43 |
• The conductivity of most metal oxides except for RuO2 is very low. The high resistivity of metal oxides increases both the sheet resistance and the charge transfer resistance of the electrode and especially causes a large IR loss at a high current density. Thus the power density and the rate capability are poor, which hinders the practical application.
• The strain developed in the pure metal oxide during the charge–discharge processes causes the cracking of the electrode, leading to poor long-term stability.
• The surface area, the pore distribution as well as the porosity are difficult to tailor in metal oxides.
It is logical to develop a composite electrode containing both carbon and transition metal oxide as the supercapacitor electrode, which combines the merits and mitigate the shortcomings of both the components. In such carbon nanostructure–metal oxide composite electrodes, the carbon nanostructures not only serve as the physical support of metal oxides but also provide the channels for charge transport. The high electronic conductivity of carbon nanostructures benefits to the rate capability and power density at a large charge/discharge current. The metal oxides are the main sources that store the charge and the energy. The electro-activities of metal oxides contribute to high specific capacitance and high energy density of the carbon nanostructure–metal oxide composite electrodes. A synergistic effect could be expected and the materials cost can be reduced. The compositional constituent, microstructure and physical properties of metal oxide–carbon composites govern the performance of the supercapacitor electrodes. The electrode porosity, electronic conductivity, pore size distribution, specific surface area affect the cell performance. Table 3 gives a comparison of carbon, metal oxide and metal oxide–carbon composite electrodes.
| Electrode materials | Surface area | Pore size distribution | Specific capacitance | Conductivity | Rate capability | Stability | Cost |
|---|---|---|---|---|---|---|---|
| Carbon | High | Various ways to tailor | Low | High | High | Good | Low |
| Metal oxide | Normally low | Difficult to tailor | High | Low | Low | Poor | High |
| Metal oxide–carbon composite | Normally controlled by a carbon support | Various ways to tailor depending on the carbon support | High | Tunable, depending on the carbon support | Good | Good | Moderate |
The aim of research on the carbon–metal oxide composite electrodes is to develop electrodes that simultaneously possess high power density, high energy density as well as good rate capability and cyclic stability. In the past decade, carbon materials were engineered at different dimensions as the building blocks to couple with metal oxides to form composites for supercapacitor electrodes. Therefore, this review article will be organized according to the roles of carbon nanostructures with different dimensions45,46 in the composite electrodes.
1. Zero-dimensional (0-D) carbon nanoparticles: 0-D carbon particles refer to round-shaped particles with an aspect ratio close to 1. 0-D carbon particles mainly include ultrafine activated carbon (AC), carbon nanospheres, and mesoporous carbon. They have high specific area (∼3000 m2 g−1 for AC47,48). More importantly, the pore content and size distribution can be tailored, which makes them suitable for supporting metal oxides for the supercapacitor electrodes.
2. One-dimensional (1-D) carbon nanostructures: 1-D nanostructures are defined as the fiber-shaped materials with a high aspect ratio. Carbon nanotubes (CNT), carbon nanofibers (CNF) and carbon nanocoils fall into the category of 1-D carbon nanostructures. They have high aspect ratio and good electronic transport properties, which is expected to facilitate the kinetics of the electrochemical reactions.
3. Two-dimensional (2-D) nanosheets: 2-D nanostructures are defined as the sheet-shaped materials with a high aspect ratio. Graphene, graphene oxide (GO) or reduced graphene oxide (rGO) are the representatives of the 2-D carbon nanosheets. Graphene and GO exhibit great mechanical strength, excellent electronic conductivity as well as high specific surface area, which are promising candidates for supercapacitor electrodes. For example, the theoretical surface area of single layer graphene is 2756 m2 g−1 with a charge mobility of 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1.49
000 cm2 V−1 s−1.49
4. Three-dimensional (3-D) porous architectures: 3-D architectures are made of low dimensional building blocks. 3-D porous carbon nano-architectures used in the supercapacitor electrodes are mainly carbon nanofoams or sponges. When used in supercapacitor electrodes, such a foam has high specific surface area, large area of electrolyte–electrode interface and continuous electron transport path.
2 Zero-dimensional carbon-supported metal oxide electrodes
Activated carbon (AC): AC is the most widely used carbon support material in supercapacitors. AC features moderate electronic conductivity and high specific surface area, which is suitable for enlarging the effective surface area of the metal oxide while providing electronic conductance.50 An example is the NiO–AC composite electrode.51 The surface areas for NiO and AC alone were 150 m2 g−1 and 776 m2 g−1, respectively. After a NiO–AC composite was formed, the specific surface area was maintained from 575 m2 g−1 to 700 m2 g−1 and the NiO agglomeration was prevented. As a result, a specific capacitance of ∼100 F g−1 was obtained for the NiO component at 7 wt% of NiO loading in the 6 M KOH electrolyte, which was measured by cyclic voltammetry (CV) at a scan rate of 5 mV s−1, which was 5 times higher than that of pristine NiO (∼20 F g−1). The reason was that the dispersed NiO had larger active surface area as compared with the sintered pristine NiO. Another example is the carbon–RuO2 composite obtained by dispersing 10 nm sized Ru metallic nanoparticles on the carbon aero-gel.52 Such a carbon–RuO2 composite had a specific surface area of 520 m2 g−1 and showed a specific capacitance of 256 F g−1 at a current density of 1 A g−1 in 30 wt% H2SO4 electrolyte. The high capacitance was estimated based on the total electrode mass, which corresponded to 50% utilization of the theoretical capacitance of RuO2. It was also reported that a hybrid supercapacitor electrode was constructed by mixing MnO2 nanopowder with active carbon black. This resulted in a specific capacitance of 60 F g−1 in 1 M LiOH electrolyte at a current density of 0.1 A g−1 and a working window of 1.5 V.53Mesoporous carbon: Besides high specific surface area, 0-D carbon can bring another advantage to the composite electrode which is its tunable pore size and fraction. The pore size of carbon is one of the critical factors that govern the specific capacitance and the rate capability. Desirable carbon electrodes have a hierarchical pore structure that contains macropores as the ion-buffering reservoirs, mesopores (2–50 nm) for promoting the ion transport, and nanopores (<2 nm) for enhancing the charge storage.54
The accessibility of redox molecules and ions into the nanopores (<2 nm) is dependent on both the solvated molecular size and the pore diameter.55 When the pores are smaller than the solvated ions, the ions will have difficulty in accessing to the pore inwall.56 Only when the pore diameter is comparable to the ion size (that is, typically 0.5–1 nm for the aqueous electrolyte), the nanopores will make maximum contribution to the EDLC since the strong interaction of ions with the pore inwall will occur, leading to high electro-adsorption. Partial desolvation of hydrated ions has been found to occur in the nanopores (<1 nm), which contributes to overall capacitance.57 However, the nanopores have negligible contribution to the ion transport in the electrolyte. In contrast, the mesopores in a diameter of 2–50 nm favor the rapid mass transport of ions to the electrode surface, which facilitates the charging/discharging of the electric double-layer.58 Especially, the ion transport time constant can be expressed as τ = L2/D, where L is the ion transport length and D is the ion diffusion constant. Thus suitable mesopores with large pore size and short ion diffusion length can reduce the ion transport barrier. Another effect that needs to be taken into account is the regularity of the pores. A narrow pore size distribution can reduce the ion scattering, which improves the electrode kinetics.54 It has been found that the differential capacitance in an aqueous solution increases linearly with the surface area of the mesopores.56 The existence of interconnected mesopores will allow the efficient propagation of solvated ions to the nanopores throughout the electrode, which is the key to high rate capability. In short, the optimal performance (high specific capacitance and good rate capability) is expected for the porous carbon electrodes with a mixture of a large fraction of nanopores with the narrow size distribution and a suitable amount of mesopores.54
An ideal pore structure for a supercapacitor should fulfill the following criteria: large pore volume for storage of energy, suitable ion transport channel for fast delivery of ions into the electrode and active pore walls for increasing the specific capacitance. Similar laws are also applicable to the metal oxide–carbon composite electrodes since the pseudo-capacitance of metal oxide is dependent on the fast surface redox reaction. Hence the access and transport of the ions in the composite electrode play a critical role. The carbon support offers a tunable platform for tailoring the fine pore microstructure and the specific surface area in the metal oxide–carbon electrodes. It is easier to adjust the property of carbon than the metal oxide. The unique features of the carbon support are desirable to be preserved in the composite.54 For example, mesocarbon microbeads (MCMBs) were activated in the KOH solution at high temperature to yield an active carbon with a high mesopore content of 64.5% and a specific surface area of ∼3000 m2 g−1. The physical mixture of the activated MCMBs and MnO2 nanosphere was determined to be suitable for the organic electrolyte (LiPF6) due to the optimized pore structure in the activated MCMBs in contrast to the commercial carbon–MnO2 composite electrode. The activated MCMB–MnO2 composite electrode was operated at a window of 3 V, and exhibited an energy density of 128 W h kg−1.59
Moreover, carbon can be tuned to have ordered mesoporous structure, which is also an attractive feature for supercapacitors. Since microporous (>10 μm) ACs have a long diffusion distance (>5 μm) and a high ion-transport resistance, a large IR drop and limited ion-accessible surface area are consequently expected at a high current density. In contrast, ordered mesoporous carbons exhibit a particle size of 1–2 μm, which results in a short diffusion distance of 0.5–1 μm. Ordered mesoporous carbons also have mesoporous channels (4–6 nm), which lead to a low ion-transport resistance.60 A recent review paper has summarized the progress of mesoporous/metal oxide as a supercapacitor electrode.14 For instance, MnO2 was deposited on the wall of ordered mesoporous carbon (denoted as CMK-3).61 The microstructure of the materials can be found in Fig. 1. It has been found that the CMK-3–MnO2 composite retained its high specific surface area (700–900 m2 g−1), which was comparable to bare CMK-3 carbon (968 m2 g−1). The pore size was also kept constant (3.6 nm) after different MnO2 loadings. The specific capacitance of such a nanostructured electrode reached 605 F g−1 for MnO2 at a scan rate of 5 mV s−1 in the aqueous Na2SO4 electrolyte. Mesoporous carbon was also used as the template for preparing nanostructured MnO2. As compared with MnO2 prepared with a CNT template, the mesoporous carbon-derived MnO2 not only showed higher specific surface area but also narrower pore size distribution (3–7 nm vs. 20–80 nm). Such a unique microstructure enhanced the capacitive behavior of MnO2. It exhibited a specific capacitance of 221 F g−1 at 5 mA cm−2 and a good rate capability (87.4% capacitance was retained when the current density increased from 2.5 to 10 mA cm−2). In contrast, 98 F g−1 of specific capacitance and 71.7% of capacitance remained at the same condition for the CNT-derived MnO2.62 Recently peapod-like Ni nanoparticles have been deposited on the mesoporous carbon with a specific surface area of 178 m2 g−1 and an average pore size of 3.6 nm. Such an electrode showed a specific capacitance of 972 F g−1 for Ni at a current density of 0.5 A g−1, which was higher than that of the NiO nanospheres and the NiO nanotubes.63
 | ||
| Fig. 1 TEM image of 26 wt% MnO2 loaded mesoporous carbon, inset is the high resolution TEM image, and the arrows indicate the embedded MnO2 (reprinted with permission from ref. 61. Copyright (2006) American Chemical Society). | ||
Carbon nanospheres: Another type of 0-D carbon nanomaterial is carbon nanospheres. Carbon nanospheres are prepared using the SiO2 sphere as the template and ferrocene as the carbon source.64 The as-grown hollow carbon spheres are highly graphitic, which ensures good electronic conductivity. MnO2 nanofibers were then grown by reduction of KMnO4 solution and a mild reflux method. Such carbon sphere–MnO2 nanofiber electrode showed a specific capacitance of 190 F g−1 at a current density of 0.1 A g−1 with 67 wt% MnO2. The electrode demonstrated good rate capability (55% capacitance retained at 10 A g−1). In addition, an asymmetric hybrid supercapacitor was fabricated using the carbon spheres and carbon sphere–MnO2 as the electrode and operated at a voltage window of 0–2 V, which increased the energy density to 22.1 W h kg−1. The MnO2 nanoribbons were grown on the onion-like carbon nanostructures to form the MnO2–C nanospheres composite, such a structure ensured the active materials to accept electrons and ions in all the directions, which yielded good rate capability (177.5 F g−1 at 2 A g−1, 56.3% of capacitance was retained when the current increased from 0.2 to 2 A g−1).65 Carbon-coated Li4Ti5O12 nanoparticles were used as the anode for a metal oxide–C/AC hybrid supercapacitor. The specific capacity of Li4Ti5O12 was as high as 271 F g−1 at a current loading of 980 mA g−1. The full cell can deliver a power density of 440 W kg−1 at an energy density of 16 W h kg−1.66
3 One-dimensional carbon–metal oxide electrodes
A collection of round-shaped 0-D nanoparticles are widely used for electrodes and current collectors, in which electrons are transported either via hopping between trap states on the neighboring nanoparticles or via diffusion within the extended states slowed down by the trapping–detrapping processes.67 In contrast, in the 1-D nanostructure-based electrodes, the long 1-D nanostructures form a continuous network for charge transport. Owing to the reduced contact resistance between neighboring nanoparticles, 1-D nanostructures show better charge transport ability than their 0-D nanoparticle counterparts.68–70 In particular, single-crystalline 1-D nanostructures can act as a “highway” for charge transport along the longitude direction.71,72 It has been proved that the conductivity of the electrode material is crucial to a high power density supercapacitor. Therefore, 1-D nanostructures are the promising candidates for supercapacitor electrodes due to their super charge transport property, which will greatly improve the rate capability of supercapacitors. In addition, 1-D nanostructures can form a percolating network to avoid high mass loading as compared with 0-D nanoparticles.69 Also, 1-D nanostructures, which have relatively high surface area, can form a porous network that acts as the scaffold/support of metal oxides.68,69 The porous network can facilitate the mass transport of the solvated ions in the electrolyte to the electrode material surface. Furthermore, the 1-D carbon nanostructure–metal oxide composites have better mechanical toughness than pure metal oxides, which will benefit to the long-term cyclic stability. Many approaches including infiltration, electrophoretic deposition (EPD), hydrothermal, co-precipitation and vapor growth have been successfully developed to deposit metal oxides to form the 1-D carbon nanostructure–metal oxide composites.73–83Carbon nanotubes (CNTs): CNTs are the mostly investigated material among the 1-D carbon nanostructures for supercapacitor electrode applications. CNTs have a much higher electronic conductivity than amorphous AC. It has been reported that pure CNT electrodes are characteristic of the EDLC and the pseudocapacitance. Broad Faradaic peaks are observed in the MWCNT (multi-walled carbon nanotube) electrode during CV scan in various aqueous electrolytes, which is due to the fact that the surface functional groups and/or the impurities act as the redox centers. The highest specific capacitance of pure CNT electrode is found to be ∼100 F g−1.73
CNTs have been coupled with NiO,84 V2O5,85 MnO286,87 and CuO88 to form the CNT–metal oxide composite electrodes. The NiO–CNT composite was prepared by in situ growing the NiO nanoparticles in the CNT suspension during a hydrothermal process.87 A more rectangular shape of CV was observed as compared with the pristine NiO powder electrode, indicating a more ideal capacitor character. The IR loss of the NiO–CNT composite electrode was reduced as the CNT content increased, which improved the rate capability of the electrode. After forming the composite, the specific capacitance of the active NiO component increased from 122 F g−1 to ∼1600 F g−1 at a scan rate of 2 mV s−1 in the alkaline electrolyte. The increase in the specific capacitance was also observed when the V2O5 nanowires were integrated with MWCNTs.85 The V2O5 component in the inter-weaved V2O5 nanowire–MWCNT composite delivered as high as 450 F g−1 of specific capacitance at a current density of 1 A g−1, which far exceeded that (100 F g−1) of the pure V2O5 nanowire electrode. The CV of the V2O5–MWCNT composite showed much better defined redox peaks associated with the redox reaction of V2O5, which suggested that charge transfer properties were improved when the MWCNTs acted as the electron transport channel. Thus a high power density of 3.75 kW kg−1 was achieved at an energy density of 5.5 W h kg−1. The advantages of the composite were further demonstrated by high performance of the MnO2–CNT electrode.86 To prepare this composite electrode, the CNTs were immersed in the KMnO4 solution to in situ form MnO2 in the presence of carbon. The high electron mobility in the CNTs facilitated the following reaction
| 4MnO4− + 3C + H2O = 4 MnO2 + 2HCO3− + CO32− | (6) |
The above reaction resulted in a MnO2–CNT composite. After casting ∼35 mg cm−2 of the 65 wt% MnO2–CNT composite on the graphite electrode, a high area-capacitance of 5.07 F cm−2 was achieved, which is the highest value for the carbon–metal oxide composites that have been investigated. Moreover, the location of MnO2 on CNT had significant influence on the capacitor performance. It was found that MnO2 in the inner wall of CNT showed much higher capacitance than those decorated on the outer shell (1250 F g−1versus 790 F g−1).87 One of the possible reasons was due to the gap between the inner MnO2 and the CNT, which served as the additional EDCL sites, thus brought extra capacitance by trapping ions inside the gap. Another possible reason was that the CNT contact site was not affected by MnO2 confined inside the nanotubes. Hence a continuous charge transport pathway was retained. Further improvement of the metal oxide–CNT composite materials was realized by introducing a conductive polymer. Hou et al. have fabricated ternary MnO2–CNT–PEDOT-PSS composite. In this composite, the CNT offered conductive pathway and supported the porous MnO2 nanospheres. The introduction of PEDOT-PSS stabilized the MnO2–CNT composite to facilitate the electrode fabrication, improved the conductive interconnection between metal oxide particles and CNT, and offered additional capacitance. A good stability (99% of retention after 1000 cycles) and increased specific capacitance (200 F g−1 at 5 mA cm−2) were observed.89
The metal oxide–CNT composite is also used to construct a symmetric composite hybrid supercapacitor such that the working window in an aqueous electrolyte can be extended, leading to an enhancement in the energy density. It has been reported that a Co–Al hydroxide–MWCNT symmetric hybrid supercapacitor shows a higher power density (6.4 kW kg−1) as compared with the MWCNT–MWCNT capacitor (2.6 kW kg−1). The addition of CNT into the hydroxide can effectively balance the electrode matching and increase the working window up to 1.6 V without decomposition of the electrolyte (6 M KOH).90
It is also worth noting that the metal oxide–CNT composite paste can be injection-printed, which makes it possible to realize a printable wear-resistant solid supercapacitor device. For example, a mixed RuO2 nanowire–SWCNT paste was printed on various substrates by the ink injection method.91 The conductivity of pristine CNT film reached 1562 S cm−1. Specific capacitance of 138 F g−1 and energy density of 18.8 W h kg−1 were achieved in the solid polymer–gel electrolyte (polyvinyl alcohol in phosphoric acid, PVA–H3PO4).
Carbon nanofibers (CNFs): CNFs are inexpensive since they can be massively fabricated by various methods such as electrospinning or vapor growth. Activated electrospun CNF (E-CNF) has been employed as the alternative of activated carbon for supercapacitor electrodes. The pore size and the specific surface area can be adjusted via gas phase activation (H2O vapor or CO2) or chemical activation (KOH and ZnCl2).92,93 The specific surface area of the CNFs could reach 940 m2 g−1 with the optimized pore distribution.93 The activated E-CNF electrode showed a high specific capacitance of 140 F g−1,92 which was comparable to the AC-based electrode.
CNFs are very attractive as the support for metal oxides in the composite electrodes. Typically the metal oxides are coated on the CNF surface to form a core–shell structure in which the CNF not only serves as the physical backbone support but also offers the channel for charge transport, and the metal oxide acts as the redox center that contributes to specific capacitance and energy density. Various metal oxides such as V2O5,94 SnO2,95 RuO2,96 and MnO297,98 have been deposited on the E-CNFs. Amorphous V2O5 was coated on the activated CNF surface using the electrodeposition method to form a coaxial nanostructure.94 The 4 nm thick V2O5 shell on the E-CNF exhibited a specific capacitance of 1308 F g−1 in the KCl electrolyte at a scan rate of 2 mV s−1. The specific capacitance was gradually reduced when the shell became thicker. When the shell thickness increased to 20 nm, the specific capacitance dropped to 560 F g−1. It was ascribed to the surface-limited redox sites and the reduction of the electronic conductivity of the composite. The V2O5–E-CNF composite electrode showed 3 times enhancement in the specific capacitance as compared with pure electrospun V2O5 nanofibers.
MnO2 is one of the best candidates for pseudo-capacitor electrodes because it has high theoretical specific capacitance (∼1380 F g−1) and is environmentally benign and earth-abundant. Unfortunately the poor electrical conductivity of MnO2 limits the full utilization of its high pseudo-capacitance. Recently the carbon–MnO2 coaxial nanofibers have also shown a specific capacitance of 539 F g−1 at 0.38 mg cm−2 of MnO2 loading.97 However, such high capacitance values can be obtained only at a very slow scan rate (2 mV s−1) and at a low metal oxide loading (<10 wt%). Severe loss occurred at a high scan rate or at a high metal oxide loading, which hinders their practical application when both high power and energy densities are needed.
In order to improve the rate capability of the CNF–metal oxide composite electrode, the electron transfer in the composite and the ion accessibility must be enhanced. A recent paper has demonstrated the highly conductive CNF–MnO2 coaxial cables98 (Fig. 2(a)). In the hierarchical MnO2 structure, an around 4 nm thick sheath surrounds the carbon nanofiber (CNF) in a diameter of 200 nm, and nano-whiskers grew radically outward from the sheath in view of the cross-section of the coaxial cables, which yielded a high specific surface area of MnO2 (Fig. 2(b)). The pseudo-capacitance of CNF–MnO2 coaxial cable electrode was mainly contributed by the redox reactions, which took place only on the outermost surface layer of MnO2 in contact with the aqueous Na2SO4 electrolyte:
| MnO2 surface + Na+ + e− = MnO2−Na+surface (surface absorption) | (7) |
| MnO2 bulk + Na+ + e− = MnOONabulk (bulk ion intercalation) | (8) |
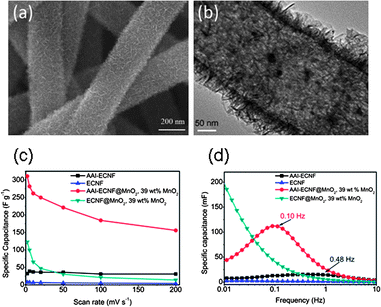 | ||
| Fig. 2 (a) SEM image of the AAI–CNF–MnO2 electrode with 39 wt% MnO2 loading and (b) corresponding TEM image. (c) Specific capacitance of CNF, AAI–CNF, AAI–CNF–MnO2 and regular CNF–MnO2 electrodes at different scan rates, the MnO2 loading was 39 wt% and (d) the Bode plot of the CNF, AAI–CNF, AAI–CNF@MnO2 and CNF–MnO2 electrodes, the time constant was labeled. Adopted with permission from ref. 98. (Copyright (2012) Elsevier). | ||
According to the Nyquist plots of impedance, the capacitance retardation in the CNF@MnO2 samples was ascribed to high series resistance attributed to insufficient conductivity of the electrode and to large charge transfer resistance at the CNF–MnO2 interface that inhibits the Na+ insertion/adsorption process in MnO2, leading to low rate capability. In other words, rapid transfer of Na+ cation and electrons to MnO2 simultaneously is the key to completing the charge storage reaction. Reaction (7) can be achieved by increasing the MnO2 surface area exposed to the electrolyte, which is realized by the high specific surface area of the AAI–CNF support. Also, a thin and porous MnO2 sheath and the nano-whisker structures shorten the path of ion diffusion. Moreover, owing to high surface area of the AAI–CNF backbone and ultra-small thickness of the MnO2 shell, the CNF–MnO2 contact area is large and the interfacial resistance between carbon and MnO2 is very small. This greatly facilitates the transfer of the electron and the ions involved in the redox reactions. Reaction (8) requires the fast electron transport inside the CNF backbone to effectively transport electrons to the MnO2 phase. In this study, iron(III) acetylacetonate (AAI), an iron organic compound, was incorporated in the electrospinning precursor to improve the electronic conductivity when preparing the E-CNFs. The resulting AAI–CNF cable showed much higher electronic conductivity and surface area than the regular CNFs (10 S cm−1versus 1 S cm−1, and 128 m2 g−1versus 28 m2 g−1). The improvement in the electronic conductivity was originated from the larger amount of the ordered graphite phase in the AAI–CNF. As a result, the AAI–CNF–MnO2 electrode showed an impressive specific capacitance of 311 F g−1 for the whole composite electrode and 900 F g−1 for the MnO2 shell at a scan rate of 2 mV s−1 as shown in Fig. 2(c). In contrast, the regular CNF–MnO2 showed only 122 F g−1 for the whole composite and ∼320 F g−1 for the MnO2 under the same test conditions. An improved rate capability was observed for the AAI–CNF@MnO2 hybrid. That is, ∼50% of the AAI–CNF–MnO2 capacitance was retained from 2 mV s−1 to 200 mV s−1 as compared with 11% for the regular CNF–MnO2. The optimal AAI–CNF@MnO2 electrode showed good cycling stability, high energy density (80.2 W h kg−1) and high power density (57.7 kW kg−1) for the active MnO2.
Complex Bode plots of impedance were also used to study the rate capability of the electrodes. The impedance of a supercapacitor device can be expressed as
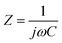 | (9) |
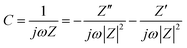 | (10) |
Herein the total capacitance of the device can be written as
| C = C′ − jC′′ | (11) |
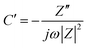 | (12) |
 | (13) |
The real part C′ is defined as the effective capacitance that the device can deliver, and the imaginary part C′′ is related to the irreversible resistivity loss in the device. The frequency f is the character frequency at which C′′ reaches the maximum and τ = 1/f is the time constant of the device, which are characteristic of the rate capability. High power density supercapacitors generally possess high character frequency and smaller τ. A comparison of the Bode plot for the different CNF–MnO2 electrodes is shown in Fig. 2(d). It could be seen that character frequencies for AAI–CNF and AAI–CNF–MnO2 with 39 wt% MnO2 loading were at 0.48 Hz and 0.1 Hz, respectively. It should be noted that the character frequency of AAI–CNF–MnO2 was comparable with that of the activated carbon electrode in which no charge transfer process happened (0.1 Hz, 10 s). However, for the CNF–MnO2 with similar 39 wt% MnO2 loading, the character frequency was far below the lowest testing frequency of 0.01 Hz. This implied that the AAI–CNF–MnO2 electrode was able to deliver energy much faster than its CNF@MnO2 counterparts. This work indicated that high electrical conductivity of the electrode material was crucial to achieving high power and energy density for pseudo-supercapacitors.
The addition of CNTs or graphene into the precursor of the E-CNFs is an alternative and effective method to improve the electronic conductivity.99 The CNF and graphene can be self-aligned inside the fiber along the longitude direction to promote the electron transport. For instance, the addition of 3 wt% of CNTs into the E-CNF increased the electronic conductivity of the E-CNF from 0.42 S cm−1 to 0.98 S cm−1. The specific surface area was also enlarged two times. As a result, for the RuO2–E-CNF composite electrode, the specific capacitance increased from 390 F g−1 to 590 F g−1 based on the RuO2 content.99
Carbon nanofibers network was also used to improve the performance of the battery-like hybrid supercapacitor. The Li4Ti5O12–carbon hybrid nanofibers were prepared by an electrospinning method following by conductive polypyrrole coating. Such an electrode showed 122 mA h g−1 of capacity at 4000 mA g−1. The hybrid supercapacitor exhibited comparable energy density to a lithium ion battery and better power density than an AC–AC supercapacitor, and preserved the good cycling property of Li4Ti5O12.100
1-D carbon nanostructure-based papers: Due to the strong van der Waals force and rich functional groups, sheet- or paper-like CNT films can be obtained using simple filtration and vacuum suction methods. Such resulting papers show excellent mechanical properties. Hence they can be used as flexible supercapacitor electrodes without need of a binder or a current collector. The CuO nanobelt-CNT free-standing mat has been prepared by mixing the CuO nanobelt with SWCNTs as shown in Fig. 3(a) and (b).88 The SEM image in Fig. 3(c) shows that the CuO nanobelt and SWCNT form a rigid and porous structure. This bendable CuO nanobelt–SWCNT supercapacitor electrode was operated at a voltage window of 0–2.5 V in the LiPF6 electrolyte. Such an electrode exhibited a higher specific capacitance (150 F g−1) at a current density of 1 A g−1 than the CuO nanoparticle–carbon black electrode (100 F g−1) and the pure CuO nanobelt electrode (78 F g−1). The enhancement was attributed to the large surface area of the CuO nanobelts and to the conductive CNT matrix, which facilitated the charge transfer and ion diffusion. In addition, a layer-by-layer (LBL) self-assembly method was developed to prepare a CNT film. In such a process, the CNT surface was functionalized with different chemical groups for different surface charge polarities. Alternative change in the surface polarity of the CNT allowed the firm assembly of a multilayer of CNT films on the indium tin oxide (ITO) coated glass. Such a CNT film was used as the support for MnO2 as the electrode.101 The MnO2 component in the CNT–MnO2 composite electrode delivered a specific capacitance of 965 F g−1 at a current density of 0.25 A g−1 in the Na2SO4 electrolyte. The volume capacitance of the composite electrode can reach 200 F cm−3. A linear relationship was observed of the area capacitance versus the film thickness, indicating the potential of scale-up fabrication.
 | ||
| Fig. 3 (a) Image of the flexible CuO–CNT electrode, (b) electrode subjected to bending and (c) SEM image of the CuO nanobelt–CNT electrode. Reprinted with permission from ref. 88. Copyright (2011) American Chemical Society. | ||
Carbon fibers can also form a paper. Liu et al. have recently used carbon fiber paper (CFP) as the backbone to fabricate a CFP/Co3O4 paper electrode.102 The Co3O4 nanonet in the CFP/Co3O4 paper electrode exhibited a specific capacitance of 1190 F g−1 at a current density of 0.25 A cm−2 in the NaOH electrolyte. The CFP/Co3O4 nanonet paper electrode retained 94.4% of its capacitance when the current density increased to 25 A g−1. Such a good rate capability is better than any Co3O4 based electrodes reported so far. One of the reasons for the remarkable specific capacitance and rate capability was attributed to the unique 1-D nanonet structure of the electrodes. Another reason was that the CNF paper played provided excellent electronic conductive channel. The third reason was that CFP/Co3O4 nanonet paper electrode favored the ion diffusion. Li et al. have grown Zn2SnO4 nanoneedles on the CNF paper using the vapor deposition method.103 A carbon layer was further coated on Zn2SnO4 nanoneedles, followed by growth of MnO2. The electrons and ions were effectively transported via the network of MnO2–carbon–Zn2SnO4–CNF, which resulted in a specific capacitance of 565 F g−1 for the MnO2 component at 2 mV s−1. Additionally, the mechanical strength of the CNF paper allowed the electrode to be operated at the bending condition with little performance degradation over 1000 cycles. A similar device was built by growing the WO3−x@Au@MnO2 core–shell nanowires on a CFP. The Au interlayer accelerated the electron transport and improved the charge transfer capability of the electrode. As a result, the IR loss was reduced and a remarkable specific capacitance of 1195 F g−1 was recorded for the active MnO2 mass at a current density of 0.75 A g−1.104
Vertically aligned 1-D nanostructures: 1-D nanostructures can be directly grown on the electrode or the current collector and vertically aligned. Such aligned nanostructures have been proven to greatly promote the charge transport and mass transport in various electrochemical cells. In addition, the aligned 1-D nanostructures are in intimate contact with the current collector, leading to reduced contact resistance at the current collector–CNT interface, which will reduce the IR loss, especially at a high current loading. Recently the vertically aligned NiO nanoplatelet array has been grown on the conductive F-doped tin oxide (FTO) substrate as the supercapacitor electrode105 (Fig. 4(a)). The CV curves exhibited approximately rectangular shape in the 1 M KOH aqueous solution with a three-electrode cell (Fig. 4(b)). The NiO nanoarray electrode exhibited a high specific area capacitance (620 F m−2 equivalent to 312 F g−1) at a current density of 4 mA cm−2. The cyclic stability of such an electrode was excellent and the specific capacitance exhibited no degradation after continuous charge/discharge measurement over 1000 cycles at a current density of 4 mA cm−2 and a voltage range of −0.2 to 0.3 V. Generally, the kinetics of a redox reaction at an electrode is governed by the charge transfer process in the electrode, the surface adsorption of ions on the electrode surface, and the diffusion of ions in the electrolyte. The redox peak current of NiO in Fig. 4(c), which was derived from the CV curves, shows a linear relationship with the scan rate. This indicated that surface adsorption of ions was the rate-limiting step. The single-crystalline nature of the NiO nanoplatelet array facilitated electron transport, leading to small charge transfer resistance, which was further confirmed by the impedance analysis. In addition, the individual vertically aligned nanoplatelets act as ultra-small electrodes, which shortened the distance of ion diffusion to the electrodes in the solution and reduced the IR loss.
 | ||
| Fig. 4 (a) SEM image of the aligned NiO nanoplates array, (b) CV scan of the NiO array electrode at different scan rates, and (c) redox peak current normalized to scan rate. Reproduced from ref. 105 with permission from The Royal Society of Chemistry. | ||
The vertically aligned CNT forest was grown and collected by a directional chemical vapor deposition (CVD) method. The CNT matrix not only served as the metal oxide support but also as the current collector, which was suitable for the supercapacitor electrode.106 Vertically aligned CNT electrodes normally had specific capacitance of less than 100 F g−1 but good rate capability.107 The specific capacitance can be improved by depositing metal oxide on the CNTs. Different metal oxides such as Co3O4, NiO and Mn2O3 were deposited on the CNT forest by thermal decomposition of metal nitrates.106 The resulting Mn2O3–CNT composite electrode showed not only high specific capacitance (508 F g−1 for the whole composite and 800 F g−1 for active Mn2O3 at 10 mV s−1) but also good rate capability (245 F g−1 for the composite at a current density of 155 A g−1). As a result, high energy density (30 W h kg−1) and power density (60 kW kg−1) were achieved. The super performance was attributed to the unique aligned structure, which shortened the charge transfer path of electrons to the metal oxide, as well as the high in-plane conductivity of the CNT sheet. In addition, a hybrid CNT@MnO2 coaxial nanotube array has been prepared.108 The MnO2 nanotubes were first prepared by vacuum filtration into a porous anodic aluminum oxide (AAO) template. The CNTs were then grown inside the MnO2 nanotubes as the conductive pathway. An intimate connection between the working electrode and the current collector was then formed, thus the ohmic drop in the charge/discharge process was negligible. The symmetric MnO2–CNT supercapacitor showed a specific capacitance of 60 F g−1 at a current density of 6 A g−1 in the Na2SO4 electrolyte with long-term stability. Similar vertical aligned CNT–MnO2 electrode showed a specific capacitance of 642 F g−1 for active MnO2 at a scan rate of 10 mV s−1.109 Recently, flower-like MnO2 has been successfully grown on the CNTs. 84% of capacitance was retained when the scan rate increased from 10 mV s−1 to 100 mV s−1, which showed much better rate capability due to smaller charge transfer and diffusion resistance as compared with the electrode made of pure MnO2 nano-flower.110
4 Two-dimensional carbon–metal oxide electrodes
The 2-D carbon nanosheets such as graphene, graphene oxide (GO) and reduced graphene oxide (rGO) have been used for the supercapacitors. Graphene has a large specific surface area and extremely high electronic conductivity, which make it attractable for supercapacitor electrode. Besides graphene, chemically rGO is also attractive as the supercapacitor electrode because of its hydrophilic feature, which is not only good for further compositing with metal oxides but also for wettability in the aqueous electrolyte. In addition, rGO still has high electronic conductivity. Moreover, it is rich in surface functioned groups such as alcohol, epoxide, carbonyl and carboxylic acid. It is also possible to introduce additional functional groups on rGO by further modification.111 The functional groups may serve as the redox centers, which can contribute to pseudo-capacitance. As a result, GO may have higher specific capacitance than graphene.112 Ruoff et al. have produced the activated rGO with a specific surface area of 3100 m2 g−1. The electrode made of such activated rGO exhibited a specific capacitance of 166 F g−1 at a current density of 5.7 A g−1.113The unique planar structure of the graphene family materials makes them good supports for metal oxides. As compared with 1-D carbon nanostructures, 2-D carbon nanostructures are even easier and more flexible to integrate with metal oxides. The formation of rGO (or graphene)–metal oxide composite can be realized via various methods including electrodeposition,114–116 hydrothermal processing,117–119 co-precipitation,120,121 spray pyrolyzing122 and so on. Yu's group has developed a general strategy to prepare rGO–metal oxide core–shell structures, which could be also used to fabricate supercapacitor electrodes.123 So far rGO has been coupled with various metal oxides such as TiO2,27 NiO,114,115,120 Co3O4119 and MnO2116,124,125 as the supercapacitor electrodes. Table 4 lists the representatives of the metal oxide–rGO composite electrodes.
| Synthesis method | Metal oxide–rGO composite | Specific capacitance | References |
|---|---|---|---|
| Electrodepositing | NiO porous film on rGO sheet | 432 F g−1 at 2 A g−1 for NiO, 636 mF cm−2 at 0.5 mA cm−2 | 114 |
| NiO nanoparticles–rGO | 569 F g−1 at 5 A g−1 for NiO | 115 | |
| MnO2 nanoparticle–rGO | 476 F g−1 at 1 A g−1 for MnO2 | 117 | |
| Hydrothermal method | MnO2 nanowire–rGO | 211.2 F g−1 at 0.15 A g−1 for composite | 117 |
| Flower-like NiO–rGO sheet | 346 F g−1 for composite 778.7 F g−1 for NiO at 1.5 A g−1 | 118 | |
| Co3O4 nanoparticle–rGO | 415 F g−1 at 3 A g−1 | 119 | |
| Co-precipitation | Monolayer NiO–rGO | 525 F g−1 at 0.2 A g−1 | 120 |
| MnO2 nanowire–rGO | 176 F g−1 at 5 mV s−1 | 121 |
A synergistic enhancement effect was observed when coupling rGO with metal oxides. For example, the RuO2 nanoparticles were anchored on the rGO surface by the precipitation method.126 The specific capacitance of the RuO2–rGO composite was higher than that of either pure RuO2 or rGO. One reason was that the RuO2 nanoparticles separated the rGO sheets, which increased the EDLC of the rGO. Also, the dispersed rGO prevented the agglomeration of the RuO2 nanoparticles, and offered conductive pathway. Similar results were also observed in the MnO2 nanowires–rGO and Co3O4–rGO composite.127,128 More detailed description of such a synergistic effect can be found in a recent review.129
An additional study of coupling rGO with various kinds of metal oxides as the supercapacitor electrodes was carried out by Ramaprabhu and Mishra.130 It has been found that the rGO-supported metal oxide electrode had higher capacitance as compared with other carbon supports due to the large contact area between rGO and the active metal oxide materials, which suggested the importance of rGO for electrochemical energy storage. It has also been reported that a rGO–MnO2/carbon nanofiber asymmetric supercapacitor exhibited a specific capacitance of 70 F g−1 with an operating window of 2.5 V at 2 mV s−1 scan, which gave a high energy density of 51.5 kW h kg−1.131 Ni(OH)2 nano-flowers were also self-assembled on the rGO to form the rGO–Ni(OH)2 composite electrode, which delivered a power density of 15.2 kW kg−1 at the energy density of 13.5 W h kg−1.132 When rGO was used as the metal oxide support, the specific surface area of rGO can be further increased by an activation process. Zhao et al. prepared a series of KOH-activated microwave-expanded graphite oxide and then coupled such graphite oxide with MnO2 to form a composite. The specific surface area of the activated graphite oxide reached 2690 m2 g−1. After deposition of MnO2, the pore size showed a negligible change (constant 1.8 nm). The asymmetric hybrid supercapacitor with the activated graphite oxide as the positive electrode and the composite as the negative electrode showed a specific capacitance of 175 F g−1 at 0.25 A g−1 with a working window of 2 V.133
Recently the single-crystalline TiO2 nanobelts have been deposited on the rGO sheets to form the rGO–TiO2 nanobelt composite as the supercapacitor electrode (Fig. 5).134 A mixture of GO and TiO2 nanobelts (NBs) was subject to mild hydrothermal processing in the ethanol solution, which reduced GO to rGO and incorporated the TiO2 nanobelts into the rGO sheets simultaneously. For a comparative study, the rGO–TiO2 nanoparticle (NP) composite was also prepared. The results have shown that the electrochemical performance of the rGO–TiO2 composite was much better than that of any individual components, indicating the synergistic effect. The monolithic rGO electrode had a specific capacitance of 40 F g−1. The TiO2 nanobelt and nanosphere electrodes showed capacitances of 17 F g−1 and 11 F g−1 in the Na2SO4 electrolyte at a scan rate of 2 mV s−1, respectively. After rGO was coupled with TiO2 nanobelts or nanoparticles, the specific capacitance significantly increased. The optimized rGO/TiO2 mass ratio was 7:3 for both the rGO–TiO2 nanoblets and the rGO–TiO2 nanoparticles, which resulted in 200 F g−1 and 60 F g−1 of specific capacitance, respectively. Interestingly, the shape of the TiO2 nanomaterial had significant influence on the performance of the composite electrode. The rGO–TiO2 nanobelt composite electrode showed much higher specific capacitance, energy density and power density than its rGO–TiO2 nanosphere counterpart (Fig. 5). One of the reasons was that the TiO2 nanobelts favored the charge mobility as compared with the TiO2 nanoparticles. In addition, the TiO2 nanobelts were ∼10 nm thick, which was close to the theoretical ion intercalation depth of TiO2. Therefore the whole TiO2 material in the nanobelts was actively involved in the charge storage process while only the outermost surface layer (∼10 nm thick) was active in the TiO2 nanoparticles in a diameter of 150 nm. Furthermore, the TiO2 nanoparticles had a point contact with the rGO sheet while The TiO2 nanobelts had planar contact with the rGO sheet. This resulted in a much higher contact area at the rGO–TiO2 interface, which improved the charge transfer and the charge storage ability of the electrode. Impedance spectra analysis also confirmed the rGO–TiO2 nanobelt composite electrode had fast time constant as compared with the rGO–TiO2 nanosphere electrode.
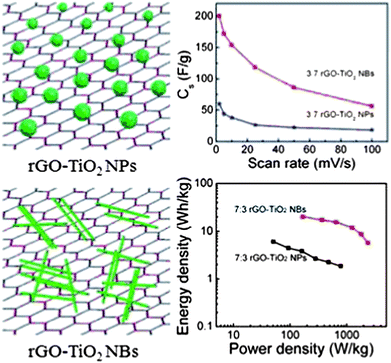 | ||
| Fig. 5 Schematic of the rGO–TiO2 composite with different TiO2 morphologies and corresponding electrochemical performance. Reproduced from ref. 134 with permission from The Royal Society of Chemistry. | ||
It is worth noting that a MnO2–rGO composite electrode has been prepared by immersing the rGO sheets in the KMnO4 solution.128 Nanoscale MnO2 was produced on the rGO surface by a self-limited electroless deposition process. With 78 wt% of MnO2 loading, the specific capacitance of the composite electrode reached 310 F g−1 at a scan rate of 2 mV s−1. Moreover, 74% of the capacitance was retained even when the scan rate increased to 500 mV s−1, which implied excellent rate capability. Such impressive performance was beneficial from the high specific surface area of the composite electrode (287 m2 g−1) and the high electronic conductivity of the composite electrodes. As the metal oxide support, the specific surface area of rGO can be further increased by an activation process. A recent report shows that the addition of CNT into the rGO–MnO2 composite can further enhance the conductivity of electrode, leading to the improvement in the rate capability.135
Another advantage of graphene and rGO based supercapacitor electrodes is their high mechanical strength. rGO can form a robust film or a free-standing paper using a simple filtration and vacuum suction process. The Young's modulus of graphene can reach as high as 1.0 TPa, which is more resilient than other carbon materials based papers.49 The electrode volume change of metal oxide supported on such a flexible matrix during the charge–discharge process can be migrated, which can further improve the long-term stability. Fig. 6 shows the fabrication process of the supercapacitor electrode based on the flexible rGO–MnO2 paper. Such an electrode exhibited a specific capacitance of 256 F g−1 at a discharge current of 0.5 A g−1.132
 | ||
| Fig. 6 Schematic of the fabrication process of the rGO–MnO2 paper electrode. Reproduced from ref. 132 with permission from The Royal Society of Chemistry. | ||
5 Three-dimensional porous carbon–metal oxide electrodes
High specific capacitance, energy density and power density are always desirable for a supercapacitor. On the other hand, the total energy and power stored in the whole device are sought to be maximized without increasing the overall footprint of device. Hence, each electrode is required to load active materials as much as possible to store a total amount of capacitance, energy and power as high as possible. It is a challenge to load a large amount of material on each electrode without undermining the electrochemical performance in terms of specific capacitance, rate capability, energy density and power density. 3-D porous carbon–metal oxide nano-architectures are being developed to address this challenge. Thus many carbon nanostructures are assembled with the building blocks of lower dimensional carbon nanostructures. Such building blocks form a hierarchical structure in the 3-D space but the individual building blocks can still keep their original merits.3-D carbon nanofoams are promising candidates for supporting metal oxides. One of the fabrication methods is the infiltration of carbon fiber paper with polymer resorcinol and subsequent pyrolysis at high temperatures. 3-D carbon nanofoams had a “through-connected” pore network and high electronic conductivity (∼1 S cm−1).136,137 The 3-D nanofoam structures had large specific surface areas to allow high metal oxide loading (∼50 wt%). FeOx and MnO2 were then electrolessly deposited on the carbon nanofoam via in situ reduction of high-valence Mn and Fe ions by carbon, respectively. The carbon–MnO2 and carbon–FeOx nanofoam electrodes showed high area specific capacitance of 1.5 F cm−2 and 0.85 F cm−2, respectively.
It was reported that graphene was directly grown on a Ni foam by the CVD process, in which the Ni foam served not only as the template but also as the catalyst. Subsequently the Ni foam template was dissolved to form a porous, highly conductive 3-D graphene network.138 Such a highly interconnected graphene foam had conductivity of over 10 S cm−1 and the specific surface was close to the theoretical value for single-layer graphene. The graphene nanofoam alone can act as good supercapacitor electrode material, which exhibited a specific capacitance of ∼100 F g−1 with good rate capability. Incorporation of metal oxides such as Co3O4 and ZnO into this graphene foam can further improve the specific capacitance as the metal oxides serve as the redox centers. For example, the Co3O4 nanowires were deposited to the graphene foam to form a Co3O4–graphene nanofoam electrode (Fig. 7),139 which showed a specific capacitance of 765 F g−1. 80% of capacitance was retained when the scan rate increased from 2 mV s−1 to 100 mV s−1. Recently ZnO has been hydrothermally grown on the graphene foam and used as the supercapacitor electrode.140 Clear redox peaks of ZnO were observed in the KOH electrolyte. A 211 F g−1 of specific capacitance was obtained at a current density of 33.3 A g−1. The results indicated that the enlarged surface area as well as the fast charge transfer ability brought by the graphene foam was the key to high performance of supercapacitor electrodes.
 | ||
| Fig. 7 (a) 3-D graphene nanofoam, (b) Co3O4 nanowires incorporated into graphene nanofoam, (c) the charge–discharge plots of the electrode at different current density and (d) the long term stability of the Co3O4 nanowires-3-D graphene foam electrode. Reprinted with permission from ref. 139. Copyright (2012) American Chemical Society. | ||
Cui's group has developed a facile method to immobilize rGO on a flexible substrate such as the polymer texture.141 Such a mesh-like rGO network was flexible and conductive, and can survive after mechanical bending. MnO2 was then grown on such a scaffold with the hydrothermal method. The resulting MnO2–rGO-mesh composite electrode exhibited a specific capacitance of 315 F g−1 for the active MnO2 at a scan rate of 2 mV s−1. In addition, a 3-D rGO framework has been prepared by using polystyrene spheres as the sacrificial templates.142 Such 3-D nanostructures not only offered high surface area but also preserved good electronic conductivity (1204 S m−1). The 3-D MnO2–rGO framework composite electrode exhibited a specific capacitance of 389 F g−1 at a current density of 1 A g−1. Moreover, 97.7% of capacitance was retained when the current density increased to 35 A g−1.
Besides graphene and rGO foams, CNT foams (or sponges) have also been fabricated as the supercapacitor electrodes in a similar way. The porous structure allows for the accessibility of the ions to the CNT electrode surface.143 The interconnected network provides continuous channels for electron transport. The CNT sponge electrode has been demonstrated to be capable of operation at a scan rate as high as 200 V s−1. After coating MnO2 on the CNT sponge, the power density and the energy density were further improved and reached 63 kW kg−1 and 31 W h kg−1, respectively. The specific capacitance of the active MnO2 component in the composite electrode reached 1270 F g−1, which was close to the theoretical value.
6 Summary and future direction
This paper has reviewed the research progress in the carbon–metal oxide composites for supercapacitor electrodes. In such a composite electrode, the metal oxide provides the source of high specific capacitance and high energy density; and the carbon nanostructure ensures good rate capability and high power density at a large current (or at a high scan rate). Moreover, the carbon nanostructures at different dimensions have their own unique features, which provide the flexibility to tailor the microstructure and properties of carbon–metal oxide composite electrodes. Each type of carbon nanostructure has its advantages as follows:• 0-D carbon nanoparticles provide the flexibility for tuning the porosity, which can be optimized to be suitable for different electrolytes.
• 1-D carbon nanostructures can facilitate the charge transport in the supercapacitor electrode. Although they have relatively smaller specific surface area as compared with 0-D carbon nanomaterials, the high conductivity of 1-D carbon nanostructures is greatly beneficial to the redox activities of the metal oxides, which enhances the pseudo-capacitance and improves the rate capability.
• 2-D carbon nanostructures combine the high surface area, high electronic conductivity and high mechanical strength. Such properties are very attractive for flexible energy-storage devices and for improvement in the charge/discharge reaction kinetics of supercapacitor electrodes.
• 3-D carbon nanostructures result from the assembly of low-dimensional carbon nanomaterials while retaining the advantages of the building blocks, which enlarges the total active volume of the supercapacitor electrodes without increasing the overall footprint of the device. This will result in compact energy storage devices.
The global market for supercapacitors is $470 million in the world in 2010.144 The market is projected to grow at an annual rate of 21.4%144 and reach $1.2 billion by 2015.145 Taking 5% of the battery market, the supercapacitor market is projected to be $3.5 billion in 2020.146 The increasing market share will further stimulate the research and development of supercapacitors. It is essential to further improve the performance including specific capacitance, energy density, power density, rate capability, long-term cyclic stability and performance/price ratio in order to meet the commercial need of supercapacitors. In particular, it remains a significant challenge to overcome the barrier of insufficient rate capability of carbon–metal oxide composite electrodes. The critical factors governing the performance of carbon–metal oxide composite supercapacitor electrodes should be considered in the future research:
Optimizing the dimension, shape, orientation and porosity of carbon nanostructures: since the carbon nanostructure serves as the physical support of metal oxides, its structure determines the architecture of the whole carbon–metal oxide composite. High specific surface area of the carbon support is the prerequisite to maximize the loading of metal oxides and to achieve large area of the carbon–metal interface. Small dimension and optimized porosity will shorten the distance of the ion diffusion to the electrode surface.
Improving the electronic conductivity of carbon nanostructures: the rate capability of a supercapacitor is dependent heavily on the electronic conductivity of electrodes. Since metal oxides generally have very low electronic conductivity, the carbon nanostructure is the only option of tuning the electronic conductivity. The intimate contact between the carbon nanostructure and the current collector is also the key to minimizing the interfacial resistance.
Enlarging the interfacial carbon–oxide area and controlling the thickness of metal oxide: intimate contact at the carbon–oxide interface and large carbon–oxide interfacial area are the important factors that affect the charge transfer ability of the composite electrode. Since the redox reactions only occur on the outermost surface layer of metal oxide for pseudo-supercapacitors, the thickness of metal oxides supported on the carbon nanostructure should be thin enough (comparable to the ion insertion depth) to allow the full utilization of electro-active materials. On the other hand, since the electrons produced in the Faradaic process have to be transferred across the carbon–metal oxide interface, the thickness of metal oxide should also be comparable to the electron diffusion length in the metal oxide so that the electrons involved in the redox reactions can be transferred to the current collector.
Developing hybrid supercapacitors: current symmetric supercapacitor has a low energy density. To improve the energy density, one of the effective routes is to develop hybrid supercapacitors. For example, a hybrid supercapacitor with the nanostructured NiO cathode and the carbon anode exhibited a high energy density of 10 W h kg−1 at a high power density of 10 kW kg−1.147 Since the energy density is directly proportional to the square of the operating voltage window, the non-aqueous electrolyte is desirable for supercapacitors. It is recommended that the metal oxide–carbon composite electrode to be used in hybrid supercapacitors with a large operating voltage window.
Exploring sustainable materials for supercapacitors: we must respond to an unprecedented rise in the demand for reduction of raw material consumption and minimizing the environmental impact of manufacturing process. Therefore there is strong incentive to develop “green materials”, which are earth-abundant, renewable, recyclable and non-toxic.
Establishing the testing standards: it is necessary to establish the standards for testing supercapacitors in order to compare the performance data taken from different laboratories.16,148 In many previous papers, the specific capacitance data were only taken from the metal oxide component. In practical application, it is important that the overall performance data are given for composite electrodes.
Notes and references
- U. Eberle and R. von Helmolt, Energy Environ. Sci., 2010, 3, 689–699 CAS
.
- J. T. Zhang and X. S. Zhao, ChemSusChem, 2012, 5, 818–841 CrossRef CAS
- G. P. Wang, L. Zhang and J. J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS
- M. Jayalakshmi and K. Balasubramanian, Int. J. Electrochem. Sci., 2008, 3, 1196–1217 CAS
- S. Mallika and R. S. Kumar, Int. J. Eng. Technol., 2011, 3, 37–43 Search PubMed
- A. Kuperman and I. Aharon, Renewable Sustainable Energy Rev., 2011, 15, 981–992 CrossRef
- Y. Zhang, H. Feng, X. B. Wu, L. Z. Wang, A. Q. Zhang, T. C. Xia, H. C. Dong, X. F. Li and L. S. Zhang, Int. J. Hydrogen Energy, 2009, 34, 4889–4899 CrossRef CAS
- X. Zhao, B. M. Sánchez, P. J. Dobson and P. S. Grant, Nanoscale, 2011, 3, 839–855 RSC
- S. Bose, T. Kuila, A. K. Mishra, R. Rajasekar, N. H. Kim and J. H. Lee, J. Mater. Chem., 2012, 22, 767–784 RSC
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC
- B. E. Conway, Electrochemical Supercapacitors: Scientific Fundamentals and Technological Applications, 1999, Plenum Pub Corp, p. 222 Search PubMed
- H. Jiang, J. Ma and C. Z. Li, Adv. Mater., 2012, 24, 4197–4202 CrossRef CAS
- C. D. Lokhande, D. P. Dubal and O. S. Joo, Curr. Appl. Phys., 2011, 11, 255–270 CrossRef
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci, 2010, 3, 1294–1301 CAS
- B. Andrea, D. Romain, T. Pierre-Louis, S. Patrice, P. Dominique, M. Marina and P. Stefano, J. Power Sources, 2007, 165, 922–927 CrossRef
- V. R. Koch, Recent Advances in Electrolyte for Electrochemical Double Layer Capacitors, 2005 Search PubMed.
- M. D. Stoller, C. W. Magnuson, Y. W. Zhu, S. Murali, J. W. Suk, R. Piner and R. S. Ruoff, Energ. Environ. Sci., 2011, 4, 4685–4689 RSC
- E. Frackowiak and F. Beguin, Carbon, 2001, 39, 937–950 CrossRef CAS
- http://www.mpoweruk.com/performance.htm.
- V. D. Patake, C. D. Lokhande and O. S. Joo, Appl. Surf. Sci., 2009, 255, 4192–4196 CrossRef CAS
- D. L. Yan, Z. L. Guo, G. S. Zhu, Z. Z. Yu, H. R. Xu and A. B. Yu, J. Power Sources, 2012, 199, 409–412 CrossRef CAS
- U. M. Patil, R. R. Salunkhe, K. V. Gurav and C. D. Lokhande, Appl. Surf. Sci., 2008, 255, 2603–2607 CrossRef CAS
- B. R. Duan and Q. Cao, Electrochim. Acta, 2012, 64, 154–161 CrossRef CAS
- T. Lu, Y. P. Zhang, H. B. Li, L. K. Pan, Y. L. Li and Z. Sun, Electrochim. Acta, 2010, 55, 4170–4173 CrossRef CAS
- X. Sun, M. Xie, G. K. Wang, H. T. Sun, A. S. Cavanagh, J. J. Travis, S. M. George and J. Lian, J. Electrochem. Soc., 2012, 159, A364–A369 CrossRef CAS
- X. Y. Chen, E. Pomerantseva, P. Banerjee, K. Gregorczyk, R. Ghodssi and G. Rubloff, Chem. Mater., 2012, 24, 1255–1261 CrossRef CAS
- J. S. Shaikh, R. C. Pawar, R. S. Devan, Y. R. Ma, P. P. Salvi, S. S. Kolekar and P. S. Patil, Electrochim. Acta, 2011, 56, 2127–2134 CrossRef CAS
- P. M. Kulal, D. P. Dubal, C. D. Lokhande and V. J. Fulari, J. Alloys Compd., 2011, 509, 2567–2571 CrossRef CAS
- X. B. Ren, H. Y. Lu, H. B. Lin, Y. N. Liu and Y. Xing, Russ. J. Electrochem., 2010, 46, 77–80 CrossRef CAS
- P. Lu, D. Xue, H. Yang and Y. Liu, Int. J. Smart Nano Mater., 2012, 1, 1–25 CrossRef
- X. Y. Lang, A. Hirata, T. Fujita and M. W. Chen, Nat. Nanotechnol., 2011, 6, 232–236 CrossRef CAS
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2004, 16, 3184–3190 CrossRef CAS
- D. Bélanger, T. Brousse and J. W. Long, Electrochem. Soc. Interface, 2008, 17, 49–52 Search PubMed
- D. S. Kong, J. M. Wang, H. B. Shao, J. Q. Zhang and C. N. Cao, J. Alloys Compd., 2011, 509, 5611–5616 CrossRef CAS
- S. Nandy, U. N. Maiti, C. K. Ghosh and K. K. Chattopadhyay, J. Phys.: Condens. Matter, 2009, 21, 115804 CrossRef CAS
- R. B. Rakhi, W. Chen, D. K. Cha and H. N. Alshareef, Nano Lett., 2012, 12, 2559–2567 CrossRef CAS
- C. Z. Yuan, L. Yang, L. R. Hou, L. F. Shen, X. G. Zhang and X. W. Lou, Energy Environ. Sci., 2012, 5, 7883–7887 CAS
- M. Hamdani, R. N. Singh and P. Chartier, Int. J. Electrochem. Sci., 2010, 5, 556–577 CAS
- W. Pan and J. H. Gong, Key Eng. Mater., 2007, 336–338, 2134–2137 Search PubMed
- C. C. Hu, W. C. Chen and K. H. Chang, J. Electrochem. Soc., 2004, 151, A281–A290 CrossRef CAS
- A. Adeyemoa, G. Hunterb and P. K. Dutta, Sens. Actuators, B, 2011, 152, 307–315 CrossRef
- K. Liang, X. Z. Tang and W. C. Hu, J. Mater. Chem., 2012, 22, 11062–11067 RSC
- V. V. Pokropivny and V. V. Skorokhod, Mater. Sci. Eng., C, 2007, 27, 990–993 CrossRef CAS
- J. N. Tiwari, R. N. Tiwari and K. S. Kim, Prog. Mater. Sci., 2012, 57, 724–803 CrossRef CAS
- Y. Zou and B. X. Han, Energy Fuels, 2001, 15, 1383–1386 CrossRef CAS
- G. P. Meisner and Q. Y. Hu, Nanotechnology, 2009, 20, 204023 CrossRef
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS
- O. Barbieri, M. Hahn, A. Herzog and R. Kötz, Carbon, 2005, 43, 1303–1310 CrossRef CAS
- K. Lota, A. Sierczynska and G. Lota, Int. J. Electrochem., 2011, 2011, 321473 Search PubMed
- C. Lin, J. A. Ritter and B. N. Popov, J. Electrochem. Soc., 1999, 146, 3155–3160 CrossRef CAS
- A. B. Yuan and Q. L. Zhang, Electrochem. Commun., 2006, 8, 1173–1178 CrossRef CAS
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater, 2010, 22, E28–E62 CrossRef CAS
- J. H. Bae, J. H. Han and T. D. Chung, Phys. Chem. Chem. Phys., 2012, 14, 448–463 RSC
- H. Yamada, H. Nakamura, F. Nakahara, I. Moriguchi and T. Kudo, J. Phys. Chem. C, 2007, 111, 227–233 CAS
- E. Raymundo-PiMero, K. Kierzek, J. Machnikowski and F. BKguin, Carbon, 2006, 44, 2498–2507 CrossRef
- G. Gryglewicza, J. Machnikowski, E. Lorenc-Grabowska, G. Lota and E. Frackowiak, Electrochim. Acta, 2005, 50, 1197–1206 CrossRef
- H. Q. Wang, Z. S. Li, Y. G. Huang, Q. Y. Li and X. Y. Wang, J. Mater. Chem., 2010, 20, 3883–3889 RSC
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2008, 47, 373–376 CrossRef CAS
- X. P. Dong, W. H. Shen, J. L. Gu, L. M. Xiong, Y. F. Zhu, H. Li and J. L. Shi, J. Phys. Chem. B, 2006, 110, 6015 CrossRef CAS
- M. W. Xua, W. Jia, S. J. Bao, Z. Su and B. Dong, Electrochim. Acta, 2010, 55, 5117–5122 CrossRef
- H. Jiang, T. Sun, C. Z. Li and J. Ma, RSC Adv., 2011, 1, 954–957 RSC
- Z. B. Lei, J. T. Zhang and X. S. Zhao, J. Mater. Chem., 2012, 22, 153–160 RSC
- Y. Wang, S. F. Yu, C. Y. Sun, T. J. Zhu and H. Y. Yang, J. Mater. Chem., 2012, 22, 17584–17588 RSC
- J. F. Ni, L. X. Yang, H. B. Wang and L. J. Gao, J. Solid State Electrochem., 2012, 12, 2791–2796 CrossRef
- J. B. Baxter and E. S. Aydil, Appl. Phys. Lett., 2005, 86, 053114 CrossRef
- M. Zhi, N. Mariani, R. Gemmen, K. Gerdes and N. Q. Wu, Energy Environ. Sci., 2011, 4, 417–420 CAS
- M. Zhi, S. Lee, N. Miller, N. H. Menzler and N. Q. Wu, Energy Environ. Sci., 2012, 5, 7066–7071 CAS
- M. Yang, J. Wang, H. Li, J.-G. Zheng and N. Q. Wu, Nanotechnology, 2008, 19, 075502 CrossRef
- N. Q. Wu, J. Wang, D. Tafen, H. Wang, J.-G. Zheng, J. P. Lewis, X. Liu, S. S. Leonard and A. Manivannan, J. Am. Chem. Soc., 2010, 132, 6679–6685 CrossRef CAS
- D. Wang, H. Zhao, N. Q. Wu, A. El Khakani and D. Ma, J. Phys. Chem. Lett., 2010, 1, 1030–1035 CrossRef CAS
- H. Pan, J. Y. Li and Y. P. Feng, Nanoscale Res. Lett., 2010, 5, 654–668 CrossRef CAS
- R. R. Bi, X. L. Wu, F. F. Cao, L. Y. Jiang, Y. G. Guo and L. J. Wan, J. Phys. Chem. C, 2010, 114, 2448–2451 CAS
- R. Vellacheri, V. K. Pillai and S. Kurungot, Nanoscale, 2012, 4, 890–896 RSC
- J. B. Mu, B. Chen, Z. C. Guo, M. Y. Zhang, Z. Y. Zhang, P. Zhang, C. L. Shao and Y. C. Liu, Nanoscale, 2011, 3, 5034–5040 RSC
- J. G. Wang, Y. Yang, Z. H. Huang and F. Y. Kang, Mater. Lett., 2012, 72, 18–21 CrossRef CAS
- R. B. Rakhi, D. Y. Cha, W. Chen and H. N. Alshareef, J. Phys. Chem. C, 2011, 115, 14392–14399 CAS
- P. C. Chen, G. Shen, S. Sukcharoenchoke and C. Zhou, Appl. Phys. Lett., 2009, 94, 043113 CrossRef
- J. H. Park, J. M. Ko and O. O. Park, J. Electrochem. Soc., 2003, 150, A864–A867 CrossRef CAS
- J. Yan, Z. J. Fan, T. Wei, J. Cheng, B. Shao, K. Wang, L. P. Song and M. L. Zhang, J. Power Sources, 2009, 194, 1202–1207 CrossRef CAS
- R. Amade, E. Jover, B. Caglar, T. Mutlu and E. Bertran, J. Power Sources, 2011, 196, 5779–5783 CrossRef CAS
- M. Jayalakshmi, M. M. Rao, N. Venugopal and K. B. Kim, J. Power Sources, 2007, 166, 578–583 CrossRef CAS
- J. Y. Lee, K. Liang, K. H. An and Y. H. Lee, Synth. Met., 2005, 150, 153–157 CrossRef CAS
- Z. Chen, Y. C. Qin, D. Weng, Q. F. Xiao, Y. T. Peng, X. L. Wang, H. X. Li, F. Wei and Y. F. Lu, Adv. Funct. Mater., 2009, 19, 3420–3426 CrossRef CAS
- X. B. Jin, W. Z. Zhou, S. W. Zhang and G. Z. Chen, Small, 2007, 3, 1513–1517 CrossRef CAS
- W. Chen, Z. L. Fan, L. Gu, X. H. Bao and C. L. Wang, Chem. Commun., 2010, 46, 3905–3907 RSC
- X. Zhang, W. Shi, J. Zhu, D. J. Kharistal, W. Zhao, B. S. Lalia, H. H. Hng and Q. Yan, ACS Nano, 2011, 5, 2013–2019 CrossRef CAS
- Y. Hou, Y. W. Cheng, T. Hobson and J. Liu, ACS Nano, 2012, 6, 5404–5412 CrossRef
- L. H. Su, X. G. Zhang, C. Z. Yuan and B. Gao, J. Electrochem. Soc., 2008, 155, A110–A114 CrossRef CAS
- P. C. Chen, H. T. Chen, J. Qiu and C. W. Zhou, Nano Res., 2010, 3, 594–603 CrossRef CAS
- C. Kim, B. T. N. Ngoc, K. S. Yang, M. Kojima, Y. A. Kim, Y. J. Kim, M. Endo and S. C. Yang, Adv. Mater., 2007, 19, 2341–2346 CrossRef CAS
- C. Kim, Y. I. Jeong, B. T. N. Ngoc, K. S. Yang, M. Kojima, Y. A. Kim, M. Endo and J. Lee, Small, 2007, 3, 91–95 CrossRef CAS
- A. Ghosh, E. J. Ra, M. H. Jin, H. K. Jeong, T. H. Kim, C. Biswa and Y. H. Lee, Adv. Funct. Mater., 2011, 13, 2541–2547 CrossRef
- J. B. Mu, B. Chen, Z. C. Guo, M. Y. Zhang, Z. Y. Zhang, C. L. Shao and Y. C. Liu, J. Colloid Interface Sci., 2011, 356, 706–712 CrossRef CAS
- C. M. Chuang, C. W. Huang, H. Teng and J. M. Ting, Compos. Sci. Technol., 2012, 72, 1524–1529 CrossRef CAS
- J. G. Wang, Y. Yang, Z. H. Huang and F. Y. Kang, Electrochim. Acta, 2011, 56, 9240 CrossRef CAS
- M. Zhi, A. Manivannan, F. Meng and N. Q. Wu, J. Power Sources, 2012, 208, 345–353 CrossRef CAS
- K. S. Yang, Y. W. Joo, T. Kim, J. H. Kim and W. J. Lee, Proceedings of the 3rd WSEAS/IASME Int. Conf. on Electroscience and Technology for Naval Engineering, Greece, July 14–16, 2006, pp. 6–10 Search PubMed
- H. S. Choi, T. H. Kim, J. H. Im and C. R. Park, Nanotechnology, 2011, 22, 405402 CrossRef
- S. W. Lee, J. Y. Kim, S. Chen, P. T. Hammond and Y. Shao-Horn, ACS Nano, 2010, 4, 3889–3896 CrossRef CAS
- L. Yang, S. Cheng, Y. Ding, X. B. Zhu, Z. L. Wang and M. L. Liu, Nano Lett., 2012, 12, 321–325 CrossRef CAS
- L. H. Bao, J. F. Zang and X. D. Li, Nano Lett., 2011, 11, 1215–1220 CrossRef CAS
- X. H. Lu, T. Zhai, X. H. Zhang, Y. Q. Shen, L. Y. Yuan, B. Hu, L. Gong, J. Chen, Y. H. Gao, J. Zhou, Y. X. Tong and Z. L. Wang, Adv. Mater., 2012, 24, 938–944 CrossRef CAS
- J. Li, W. Zhao, F. Huang, A. Manivannan and N. Q. Wu, Nanoscale, 2011, 3, 5103–5109 RSC
- R. F. Zhou, C. Z. Meng, F. Zhu, Q. Q. Li, C. H. Liu, S. S. Fan and K. L. Jiang, Nanotechnology, 2010, 21, 345701 CrossRef
- L. Basirićo and G. Lanzara, Nanotechnology, 2012, 23, 305401 CrossRef
- A. L. M. Reddy, M. M. Shaijumon, S. R. Gowda and P. M. Ajayan, J. Phys. Chem. C, 2010, 114, 658–663 CAS
- R. Amade, E. Jover, B. Caglar, T. Mutlu and E. Bertran, J. Power Sources, 2011, 196, 5779–5783 CrossRef CAS
- H. Xia, Y. Wang, J. Y. Lin and L. Lu, Nanoscale Res. Lett., 2012, 7, 33 CrossRef CAS
- S. F. Pei and H. M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS
- B. Xu, S. F. Yue, Z. Y. Sui, X. T. Zhang, S. S. Hou, G. P. Cao and Y. P. Yang, Energy Environ. Sci., 2011, 4, 2826–2830 CAS
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS
- X. H. Xia, J. P. Tu, Y. J. Mai, R. Chen, X. L. Wang, C. D. Gu and X. B. Zhao, Chem.–Eur. J., 2011, 17, 10898–10905 CrossRef CAS
- M. S. Wu, Y. P. Lin, C. H. Lin and J. T. Lee, J. Mater. Chem., 2012, 22, 2442–2448 RSC
- Y. Q. Zhao, D. D. Zhao, P. Y. Tang, Y. W. Wang, C. L. Xu and H. L. Li, Mater. Lett., 2012, 76, 127–130 CrossRef CAS
- S. Chen, J. W. Zhu, X. D. Wu, Q. F. Han and X. Wang, ACS Nano, 2010, 4, 2822–2830 CrossRef CAS
- C. Y. Ge, Z. H. Hou, B. H. He, F. Y. Zeng, J. G. Cao, Y. M. Liu and Y. F. Kuang, J. Sol-Gel Sci. Technol., 2012, 63, 146–152 CrossRef CAS
- G. Y. He, J. H. Li, H. Q. Chen, J. Shi, X. Q. Sun, S. Chen and X. Wang, Mater. Lett., 2012, 82, 61–63 CrossRef CAS
- B. Zhao, J. S. Song, P. Liu, W. W. Xu, T. Fang, Z. Jiao, H. J. Zhang and Y. Jiang, J. Mater. Chem., 2011, 21, 18792–18798 RSC
- R. R. Jiang, T. Huang, J. L. Liu, J. H. Zhuang and A. S. Yu, Electrochim. Acta, 2009, 54, 3047–3052 CrossRef CAS
- A. Chidembo, S. H. Aboutalebi, K. Konstantinov, M. Salari, B. Winton, S. A. Yamini, I. P. Nevirkovets and H. K. Liu, Energy Environ. Sci., 2012, 5, 5236–5240 CAS
- W. W. Zhou, J. X. Zhu, C. W. Cheng, J. P. Liu, H. P. Yang, C. X. Cong, C. Guan, X. T. Jia, H. J. Fan, Q. Y. Yan, C. M. Li and T. Yu, Energy Environ. Sci., 2011, 4, 4954–4961 CAS
- J. Yan, Z. J. Fan, T. Wei, W. Z. Qian, M. L. Zhang and F. Wei, Carbon, 2010, 48, 3825–3833 CrossRef CAS
- H. J. Huang and X. Wang, Nanoscale, 2011, 3, 3185–3191 RSC
- Z. S. Wu, D. W. Wang, W. C. Ren, J. P. Zhao, G. M. Zhou, F. Li and H. M. Cheng, Adv. Funct. Mater., 2010, 20, 3595–3602 CrossRef CAS
- Z. S. Wu, W. C. Ren, D. W. Wang, F. Li, B. L. Liu and H. M. Cheng, ACS Nano, 2010, 4, 5835–5842 CrossRef CAS
- Z. S. Wu, W. C. Ren, L. Wen, L. B. Gao, J. P. Zhao, Z. P. Chen, G. M. Zhou, F. Li and H. M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS
- Z. S. Wu, G. M. Zhou, L. C. Yin, W. C. Ren, F. Li and H. M. Cheng, Nano Energy, 2012, 1, 107–131 CrossRef
- A. K. Mishra and S. Ramaprabhu, J. Phys. Chem. C, 2011, 115, 14006–14013 CAS
- Z. J. Fan, J. Yan, T. Wei, L. J. Zhi, G. Q. Ning, T. Y. Li and F. Wei, Adv. Funct. Mater., 2011, 21, 2366–2375 CrossRef CAS
- Z. P. Li, Y. J. Mi, X. H. Liu, S. Liu, S. R. Yang and J. Q. Wang, J. Mater. Chem., 2011, 21, 14706–14711 RSC
- X. Zhao, L. L. Zhang, S. Murali, M. D. Stoller, Q. H. Zhang, Y. W. Zhu and R. S. Ruoff, ACS Nano, 2012, 6, 5404–5412 CrossRef CAS
- C. Xiang, M. Li, M. Zhi, A. Manivannan and N. Q. Wu, J. Mater. Chem., 2012, 22, 19161–19167 RSC
- Y. W. Cheng, S. T. Lu, H. B. Zhang, C. V. Varanasi and J. Liu, Nano Lett., 2012, 12, 4206–4211 CrossRef CAS
- M. B. Sassin, A. N. Mansour, K. A. Pettigrew, D. R. Rolison and J. W. Long, ACS Nano, 2010, 4, 4505–4514 CrossRef CAS
- A. E. Fischer, K. A. Pettigrew, D. R. Rolison, R. M. Stroud and J. W. Long, Nano Lett., 2007, 7, 281–286 CrossRef CAS
- Z. P. Chen, W. C. Ren, L. B. Gao, B. L. Liu, S. F. Pei and H.-M. Cheng, Nat. Mater., 2011, 10, 424–428 CrossRef CAS
- X. C. Dong, H. Xu, X. W. Wang, Y. X. Huang, M. B. Chan-Park, H. Zhang, L. H. Wang, W. Huang and P. Chen, ACS Nano, 2012, 6, 3206–3213 CrossRef CAS
- X. C. Dong, Y. F. Cao, J. Wang, M. B. Chan-Park, L. h. Wang, W. Huang and P. Chen, RSC Adv., 2012, 2, 4364–4369 RSC
- L. B. Hu, W. Chen, X. Xie, N. Liu, Y. Yang, H. Wu, Y. Yao, M. Pasta, H. N. Alshareef and Y. Cui, ACS Nano, 2011, 5, 8904–8913 CrossRef CAS
- B. G. Choi, M. Yang, W. H. Hong, J. W. Choi and Y. S. Huh, ACS Nano, 2012, 6, 4020–4028 CrossRef CAS
- W. Chen, R. B. Rakhi, L. B. Hu, X. Xie, Y. Cui and H. N. Alshareef, Nano Lett., 2011, 11, 5165–5172 CrossRef CAS
- http://gigaom.com/cleantech/supercapacitor-market-to-surge-to-877m-by-2014/, accessed 24 July 2012.
- http://www.marketresearchmedia.com/2012/03/16/ultracapacitor-market/, accessed 24 July 2012.
- http://www.reportlinker.com/p0363454/Supercapacitors-Technology-Developments-and-Global-Markets.html, accessed 24 July 2012.
- D. W. Wang, F. Li and H. M. Cheng, J. Power Sources, 2008, 185, 1563–1568 CrossRef CAS
- Y. Gogotsi and P. Simon, Science, 2011, 18, 917–918 CrossRef
| This journal is © The Royal Society of Chemistry 2013 |