A general and convergent synthesis of diverse glycosylphosphatidylinositol glycolipids†
Yu-Hsuan
Tsai
ab,
Sebastian
Götze
ab,
Ivan
Vilotijevic
a,
Maurice
Grube
a,
Daniel Varon
Silva
*ab and
Peter H.
Seeberger
*ab
aDepartment of Biomolecular Systems, Max Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: seeberger@mpikg.mpg.de; daniel.varon@mpikg.mpg.de; Fax: +49 3083859302; Tel: +49 3083859301
bInstitute of Chemistry and Biochemistry, Free University of Berlin, Arnimallee 22, 14195 Berlin, Germany
First published on 1st November 2012
Abstract
Glycosylphosphatidylinositol (GPI) glycolipids anchor a large number of proteins in the cell membrane of eukaryotic cells. Their conserved pseudopentasaccharide core carries additional phosphoethanolamine, saccharide and lipid substituents. These structural variations are characteristic for a species or a tissue but their functional significance remains largely unknown. Studies that would link a specific function to a structurally unique GPI rely on availability of homogeneous samples of these glycolipids. To address this need we have developed a general synthetic route to GPI glycolipids. Our convergent synthesis starts from common building blocks and relies on a fully orthogonal set of protecting groups that enables the regioselective introduction of phosphodiesters and efficient assembly of the GPI glycans. Here, we report on the development of this synthetic strategy, evaluation of the set of protecting groups with respect to phosphorylation methods, evaluation of the assembly plan for the GPI glycan, optimization of the glycosylation reactions, and the application of this strategy to the total syntheses of four structurally diverse branched GPI glycolipids.
Introduction
Glycosylphosphatidylinositols (GPIs) are complex glycolipids that are found in eukaryotic cells either attached to the C-terminus of proteins or in free form.1 These complex glycolipids feature a phosphoethanolamine unit connecting the C-terminus of the protein to the glycan, a conserved α-Man-(1→2)-α-Man-(1→6)-α-Man-(1→4)-α-GlcN-(1→6)-myo-Inositol pseudopenta-saccharide core and a lipid attached to the core glycan via a phosphodiester at the C1 position of myo-inositol (Fig. 1). The conserved GPI structure can be further decorated by various substituents including additional phosphoethanolamine units, an additional fatty acid ester at the C2 position of myo-inositol and oligosaccharide branches at the C3 or C4 position of ManI. The constitutive identity of the lipid subunit is variable and may include diacylglycerol, alkylacylglycerol or a ceramide, with chains of different length and varying degrees of unsaturation. When GPIs are isolated from natural sources they are often obtained as heterogeneous mixtures especially in respect to the lipid subunit.2 GPIs isolated from different species and, in some cases, from different tissues of the same organism, feature significant structural differences (Fig. 1).1,3 The functional significance of these variations remains largely unknown.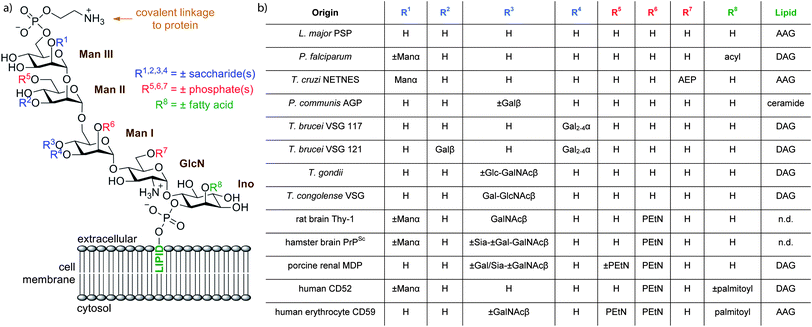 | ||
| Fig. 1 (a) The conserved core structure of GPIs and possible substituents. (b) Structural modifications of selected GPIs.15 PEtN = phosphoethanolamine; AEP = 2-aminoethylphosphonate; AAG = 1-alkyl-2-acylglycerol; DAG = diacylglycerol; AG = 1-alkyl-2-lysoglycerol; n.d. = not determined. | ||
The primary biological role of GPIs is to localize the attached protein to the outer surface of the plasma membrane bilayer.3,4 It is suggested that GPIs play a role in the association of anchored proteins with lipid rafts and are, thereby, involved in diverse processes such as regulation of innate immunity, protein trafficking, and antigen presentation.5 The complexity of GPI structure and constitution suggests that additional biological functions may be attributed to GPIs themselves; however, beyond the membrane anchoring role, little is known about their involvement in biological processes.3,6
Deciphering and evaluating both the biological functions and biophysical properties of GPIs require a systematic approach to link the structures of different GPI molecules to specific biological activities and physical properties. Such biochemical and biophysical studies rely on the availability of a diverse set of homogeneous GPI structures.7 Due to heterogeneity of GPIs isolated from biological samples and their amphiphilic character, which renders purification of GPI structures challenging, homogeneous samples of these glycolipids are only accessible via chemical synthesis. The capacity of synthetic GPIs to serve as powerful tools for biological research8 has inspired a number of elegant syntheses of GPIs9 and their fragments.10 These routes have served as platforms for the development of new methods9e and technologies10j for oligosaccharide synthesis.11
In most of the reported synthetic routes to GPIs the target molecules are built from individual monosaccharides in a linear stepwise manner (Fig. 2). The oligomannoside, in particular, has often been constructed in a linear fashion applying a participating group at the C2 position to facilitate stereoselective α-mannosylations (Sections a, b, c and e, Fig. 2). Even the branched structures have been predominantly produced in a linear fashion adding one monosaccharide to the growing glycan at a time.12 Although convergent synthetic routes require less protecting group manipulations and transformations that include large oligosaccharide intermediates, only a few such routes have been reported. The notable work in this area was reported by Guo,9f,m Ley9d,e and Fraser-Reid10n typically using the disconnections around ManI to achieve convergence. With no exception, the reported synthetic routes to GPIs are target oriented and although such approaches typically result in shorter synthetic schemes, these routes are not readily amenable to modifications that would allow for efficient production of analogues or other GPIs.13 Despite considerable work in the field, GPIs with varied structures have remained difficult to access in appreciable quantities. With this in mind, we initiated a synthetic program to address the need for a diverse set of homogeneous GPI glycolipids and their analogues as a basis for structure–activity studies.14 Here, we describe the development of a general convergent strategy for the synthesis of branched GPI structures and its application to the syntheses of several structurally distinct GPIs.
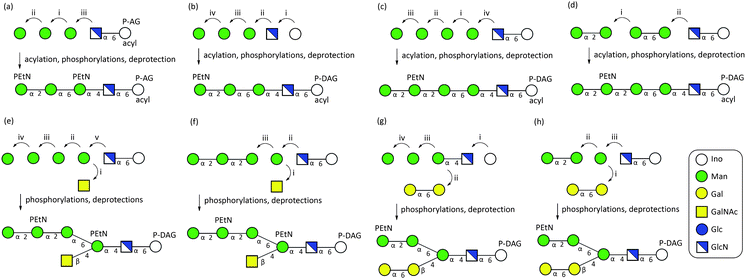 | ||
| Fig. 2 Synthetic strategies for the preparation of GPIs: (a) synthesis of CD52 GPI by the Guo group;9m (b) synthesis of P. falciparum GPI by the Fraser-Reid group;9h (c) synthesis of P. falciparum GPI by the Seeberger group;9k (d) synthesis of P. falciparum GPI by the Vishwakarma group;9p (e) synthesis of Thy-1 GPI by the Schmidt group;16 (f) synthesis of Thy-1 GPI by the Fraser-Reid group;9c,10n (g) synthesis of T. brucei GPI by the Ogawa group;9a (h) synthesis of a T. brucei GPI by the Ley group.9d,e | ||
Results and discussion
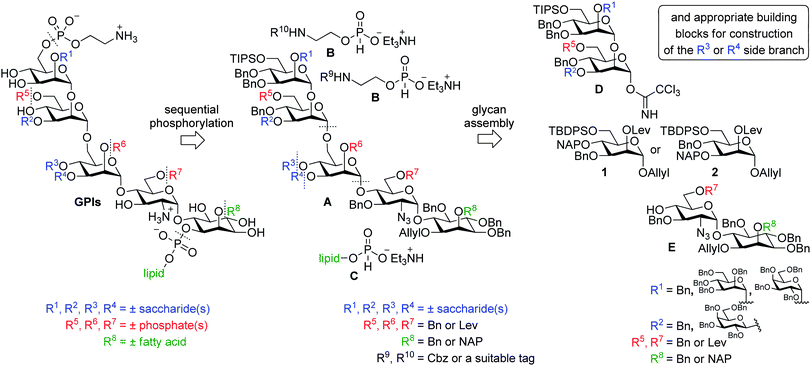
The retrosynthetic analysis identified phosphodiesters as the natural point for initial bond disconnections. The late stage phosphorylations that would install any desired GPI phosphorylation pattern, thus, emerged as one of the key challenges that served as a template for the design of a comprehensive protecting group strategy with special emphasis on the orthogonal protection of phosphorylation sites. If all possible sites of phosphorylation in GPIs17 are taken into account (Fig. 1), a conservative retrosynthetic analysis would require orthogonal protection in five positions. This, however, may not be necessary considering the identity and arrangement of the phosphate substituents along the backbone of known GPIs. The only substituents found at C6 of ManII (R5) and C2 of ManI (R6) are the phosphoethanolamine units that can be installed simultaneously if the same protecting group is used at both sites.9c Although such analysis could also incorporate the phosphoethanolamine at the C6 position of ManIII, this avenue was not taken because it would have prevented the installation of the phosphoethanolamine analogues suitable for ligation of the synthetic GPI with peptides and proteins.18 The (2-aminoethyl)phosphonate unit is only present at the C6 position of glucosamine (R7) in GPIs of Trypanosoma cruzi. Fortuitously, this is also the only additional phosphorylation site of the T. cruzi GPIs. In light of this analysis, only three orthogonal protecting groups are sufficient to introduce any of the reported GPI phosphorylation patterns if arranged in an appropriate, target dependent manner: one protecting group for the C6 position of ManIII, one for the C1 position of myo-inositol and one for the positions carrying additional ethanolamine or (2-aminoethyl)phosphonate substituents (R5, R6 and R7). In addition to creating a coherent set, these groups are, naturally, required to be: orthogonal to the benzyl ethers chosen for permanent protection of hydroxyls,19 stable under the phosphorylation conditions, and removable in the presence of already introduced phosphodiesters. To allow for more flexibility in the design of the synthetic route, the groups that are labile only under a specific set of conditions were selected.20 A levulinoyl ester (Lev) that is known to be relatively stable under mildly basic conditions but is easily cleaved in the presence of other esters via hydrazinolysis was chosen over generally base sensitive groups such as acetyl or benzoyl esters. For similar reasons, allyl and triisopropylsilyl (TIPS) ethers were selected to complete the fully orthogonal set of protecting groups, on which further development of the general synthetic strategy for GPIs was based.The specific pattern for protecting groups placement within the fully protected glycan (A series, Fig. 3) was created in a way that either enables these substituents to control stereoselectivity of glycosylation reactions during the assembly of the glycan (e.g., Lev at C2 of ManI) or simplifies the synthesis of the required building blocks (e.g., TIPS ether at C6 of ManIII). A placeholder was left at the C2 position of myo-inositol for an oxidatively labile group, such as 2-naphtylmethyl ether (NAP) that may add one more level of orthogonality needed for the introduction of an additional fatty acid ester, e.g., long chain acyl substituents in the Plasmodium falciparum GPIs.15b,21 Depending on the specific GPI target and the position of phosphorylation sites within the molecule, a Lev group may also be necessary at C6 of ManII (R5) or C6 of the glucosamine (R7).
 | ||
| Fig. 3 General retrosynthetic analysis of known GPI structures starting from interchangeable common building blocks. Monosaccharide units in the building blocks and intermediates are referred to by their position in the target GPI, e.g., mannose at the non-reducing end of D is referred to as ManIII. | ||
The key tactical consideration in further retrosynthetic analysis was efficient deconstruction of the protected GPI glycan into simple building blocks. A synthetic pathway that would achieve maximum diversity starting from a minimal number of common building blocks was seen as a superior solution for rapid synthesis of diverse GPI targets.22 Since the identity and connectivity of the side branch varies across the reported GPIs and likely requires a unique strategy and building blocks for each GPI target molecule, the next logical disconnection was at the oligosaccharide branch at either the C3 or C4 position of ManI (R3 and R4, Fig. 3). The frequency with which additional saccharide units at C2 of ManIII (R1) and C3 of ManII (R2) appear in GPIs, and their relative homogeneity across various species, suggest that this problem would be best addressed by a modular approach, using different building blocks.
The orthogonal protecting group strategy designed for late stage phosphorylations was fully integrated into the plan for assembly of the GPI glycans. A simple matching of the protecting groups between the position of the glycosyl acceptor and the corresponding glycosylating agent was followed in order to avoid coexistence of multiple temporary protecting groups of the same type in the growing glycan. Incorporation of all protecting groups into a single, central building block would simplify synthesis of the building blocks and result in the most convergent route. Following this logic, the next two bond disconnections were made at the glycosidic bonds around the ManI (Fig. 3) giving rise to the fully orthogonally protected central mannose building blocks 1 and 2,23 and the simple set of interchangeable building blocks which served to introduce the top oligomannoside (D series) and GlcN-myo-inositol subunits (E series, Fig. 3) together with the respective saccharide branches and protecting groups. These disconnections emerged as a reasonable choice considering the variety of glycosylation protocols that reliably produce α-mannosides and the appropriate positioning of the directing protecting groups.9d,e,10n The collection of protected GPI fragments listed in Fig. 3 represents a complete set of building blocks that serves to access any phosphorylated GPI glycan characterized to date.
Synthesis of the GPI anchor of Toxoplasma gondii
For the initial evaluation of the outlined synthetic strategy, a simple branched GPI anchor of the T. gondii parasite (3, Fig. 4) was selected as a target molecule. In addition to the conserved core, this GPI features a β-galactosamine branch at the C4 position of ManI. This structure appears as a common motif in many GPI anchors and, as such, it was seen as an attractive platform for testing several aspects of the proposed synthetic route such as the syntheses of building blocks,23 the order of glycan assembly, reliability of the key glycosylation protocols with respect to yield and stereoselectivity, and feasibility of the protecting group strategy.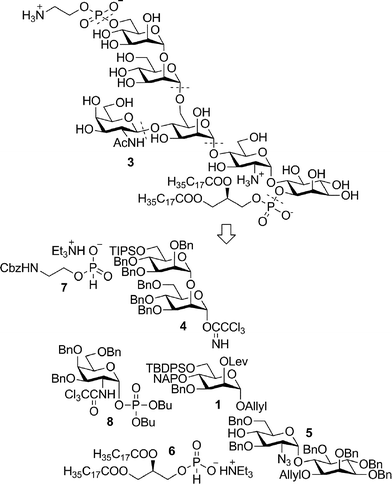 | ||
| Fig. 4 Retrosynthetic analysis of T. gondii GPI anchor 3. | ||
According to the general retrosynthetic scheme, the fully protected direct precursor of T. gondii (Fig. 4) can be prepared via sequential phosphorylation using H-phosphonates 7 and 6, and the fully protected GPI glycan. The glycan was envisioned to arise from the building blocks 1, 4,145,9e and 8 (ref. 9j) via sequential deprotection–glycosylation cycles. The order of phosphorylation reactions is inconsequential owing to the fully orthogonal character of the protecting group set. In contrast, the order in which the saccharide building blocks are assembled may have far reaching consequences with respect to the efficacy of this route in generating diverse GPI structures. Efforts required for the synthesis of building blocks and/or common late stage intermediates that lead to various GPIs could be significantly reduced if the point of divergence appears at the latest possible stage of the synthesis. Since the C3/C4 saccharide branches constitute the most variable portion of GPI molecules, the glycosylation reaction that appends the saccharide branch to the core glycan is desirable as the final step in the assembly sequence. In the case of the T. gondii GPI, this translates into the [1 + 5] glycosylation strategy where the building blocks 1, 4 and 5 are joined together to form the pseudopentasaccharide core prior to the final glycosylation to append the side branch with the building block 8.
![Assembly of the T. gondii GPI glycan via a [1 + 5] glycosylation strategy. Reagents and conditions: (a) HF·pyridine, THF, 96%; (b) 4, TBSOTf, thiophene, 4 Å MS, CH2Cl2, 92%; (c) (i) [Ir(COD)(PPh2Me)2]PF6, H2, THF; (ii) HgCl2, HgO, H2O, acetone; (iii) Cl3CCN, DBU, CH2Cl2, 55%; (d) 5, TMSOTf, PhMe, −40 °C, 98%; (e) DDQ, H2O, CH2Cl2, 85%; (f) 8 or 13, TMSOTf, CH2Cl2, −40 °C, no reaction.](/image/article/2013/SC/c2sc21515b/c2sc21515b-s1.gif) | ||
| Scheme 1 Assembly of the T. gondii GPI glycan via a [1 + 5] glycosylation strategy. Reagents and conditions: (a) HF·pyridine, THF, 96%; (b) 4, TBSOTf, thiophene, 4 Å MS, CH2Cl2, 92%; (c) (i) [Ir(COD)(PPh2Me)2]PF6, H2, THF; (ii) HgCl2, HgO, H2O, acetone; (iii) Cl3CCN, DBU, CH2Cl2, 55%; (d) 5, TMSOTf, PhMe, −40 °C, 98%; (e) DDQ, H2O, CH2Cl2, 85%; (f) 8 or 13, TMSOTf, CH2Cl2, −40 °C, no reaction. | ||
Oxidative removal of the NAP ether from 11 set the stage for the final [1 + 5] glycosylation en route to the T. gondii GPI pseudohexasaccharide 14 (Scheme 1). Despite considerable efforts to identify reaction conditions to render this transformation feasible, the glycosylation reactions of 12 with simple GalNAc trichloroacetimidate building block 13 or the corresponding phosphate 8 could not reliably produce the desired glycan 14. While it is conceivable that the glycosylating agents 8 and 13 are inherently not sufficiently reactive, it is more likely that the steric crowding in pseudopentasaccharide renders the C4 hydroxyl of ManI in 12 only weakly nucleophilic which allows for competitive decomposition of the activated glycosylating agent during the reactions. To test this hypothesis, the C4 hydroxyl of ManI in a corresponding monosaccharide was glycosylated with glycosyl phosphate 8. (Scheme 2). A suitable nucleophile, alcohol 15, was prepared by cleaving the NAP ether of mannoside 1 using DDQ. Glycosylation of 15 with phosphate 8 in the presence of TMSOTf in dichloromethane at −40 °C went smoothly to produce the corresponding disaccharide 16 in 82% yield. Based on this result the synthetic plan was revised and the synthesis of the desired pseudohexasaccharide was continued utilizing a [2 + 2 + 2] glycosylation strategy.
![Synthesis of the T. gondii GPI anchor based on the general synthetic strategy for GPIs. Reagents and conditions: (a) DDQ, H2O, CH2Cl2, 85%; (b) 8, TMSOTf, CH2Cl2, 4 Å MS, −40 °C, 82%; (c) HF·pyridine, THF, 80%; (d) 4, TBSOTf, Et2O, 4 Å MS, 0 °C, 74%; (e) Zn, AcOH, 55 °C, 74% (f) (i) [Ir(COD)(PPh2Me)2]PF6, H2, THF; (ii) HgCl2, HgO, H2O, acetone 69% (two steps); (iii) Cl3CCN, DBU, CH2Cl2, 89%; (g) 5, TBSOTf, Et2O, 4 Å MS, 0 °C, 81%; (h) (i) [Ir(COD)(PPh2Me)2]PF6, H2, THF; (ii) HgCl2, HgO, H2O, acetone, 82% (two steps); (i) (i) 6, PivCl, pyridine; (ii) I2, H2O, 91% (two steps); (iii) Sc(OTf)3, MeCN, CHCl3, 71%; (j) (i) 7, PivCl, pyridine; (ii) I2, H2O; (iii) H2NNH2, AcOH, pyridine, CHCl3, 80% (three steps); (k) H2, Pd/C, CHCl3–MeOH–AcOH–H2O, 89%.](/image/article/2013/SC/c2sc21515b/c2sc21515b-s2.gif) | ||
| Scheme 2 Synthesis of the T. gondii GPI anchor based on the general synthetic strategy for GPIs. Reagents and conditions: (a) DDQ, H2O, CH2Cl2, 85%; (b) 8, TMSOTf, CH2Cl2, 4 Å MS, −40 °C, 82%; (c) HF·pyridine, THF, 80%; (d) 4, TBSOTf, Et2O, 4 Å MS, 0 °C, 74%; (e) Zn, AcOH, 55 °C, 74% (f) (i) [Ir(COD)(PPh2Me)2]PF6, H2, THF; (ii) HgCl2, HgO, H2O, acetone 69% (two steps); (iii) Cl3CCN, DBU, CH2Cl2, 89%; (g) 5, TBSOTf, Et2O, 4 Å MS, 0 °C, 81%; (h) (i) [Ir(COD)(PPh2Me)2]PF6, H2, THF; (ii) HgCl2, HgO, H2O, acetone, 82% (two steps); (i) (i) 6, PivCl, pyridine; (ii) I2, H2O, 91% (two steps); (iii) Sc(OTf)3, MeCN, CHCl3, 71%; (j) (i) 7, PivCl, pyridine; (ii) I2, H2O; (iii) H2NNH2, AcOH, pyridine, CHCl3, 80% (three steps); (k) H2, Pd/C, CHCl3–MeOH–AcOH–H2O, 89%. | ||
With pseudohexasaccharide 20 in hand, the TIPS/Lev/Allyl set of protecting groups was tested in the sequential phosphorylation reactions. Cleavage of the allyl ether at the C2 position of myo-inositol in 20via the Ir-catalyzed isomerization, followed by the mercury(II) promoted hydrolysis of the resulting enol ether, unveiled the site for conjugation with the lipid (Scheme 2). Removal of the allyl ether could also be achieved by a one-step palladium(II) catalyzed isomerization/in situ hydrolysis sequence. This protocol, however, gave consistently lower yields due to competitive Wacker oxidation or cleavage of the silyl ether when reactions were carried out under acidic conditions. Formation of the H-phosphonate diester from the in situ generated mixed anhydride of 1,2-distearoyl-sn-glycerol H-phosphonate 6 and pivalic acid followed by oxidation produced the corresponding phosphodiester. Removal of the silyl group from the C6 position of ManIII required specific reaction conditions due to the presence of lipid phosphodiester in the molecule. When a mild Lewis acid such as Sc(OTf)3 was used, the silyl ether was successfully cleaved and the desired primary alcohol 21 was isolated in good yield (Scheme 2). Formation of the H-phosphonate diester of 7 and 21 followed by oxidation afforded the corresponding diphosphate. Removal of the Lev ester could not be selectively achieved under the typical, strongly basic conditions due to the presence of fatty acid esters. Treatment with acetic acid-buffered hydrazine acetate, however, selectively removed the levulinate to produce 22 in 80% yield over three steps starting from 21. Global deprotection via palladium catalyzed hydrogenolysis produced 3, the fully lipidated and phosphorylated GPI anchor of T. gondii.
Synthesis of the T. gondii low molecular weight antigen
Success with the construction of glycosidic bonds around ManI and the late stage phosphorylation in the synthesis of 3 instilled confidence that the original bond disconnections were also well suited for the low molecular weight antigen of T. gondii (23, Fig. 5). A retrosynthetic scheme was created based on the general retrosynthetic scheme with modifications that incorporate the lessons learned from the previous syntheses. The order of glycan assembly had to be changed due to problems associated with the [n + 5] approach. The fully protected pseudoheptasaccharide was envisioned to arise from the branch containing trisaccharide 24 and the common building blocks 4 and 5 (Fig. 5) via a [2 + 3 + 2] glycosylation sequence.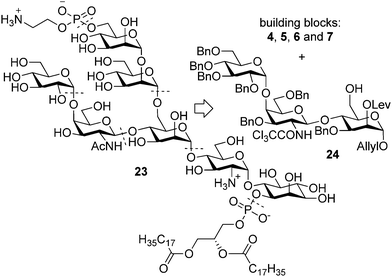 | ||
| Fig. 5 Retrosynthetic analysis of T. gondii low molecular weight antigen. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β = 3:1). Significant quantities of phenyl 2,3,4,6-tetra-O-benzyl-1-phenylseleno-D-glucopyranoside were isolated as the major side product, possibly arising from aglycon transfer.27 Attempts to further optimize this process were met with failure. Since problems associated with glycosylations of selenoglycosides with thioglycosides are well documented,28 the sulfoxide 28 (ref. 29) was used instead but this strategy appeared to be ineffective under a variety of conditions.
:β = 3:1). Significant quantities of phenyl 2,3,4,6-tetra-O-benzyl-1-phenylseleno-D-glucopyranoside were isolated as the major side product, possibly arising from aglycon transfer.27 Attempts to further optimize this process were met with failure. Since problems associated with glycosylations of selenoglycosides with thioglycosides are well documented,28 the sulfoxide 28 (ref. 29) was used instead but this strategy appeared to be ineffective under a variety of conditions.
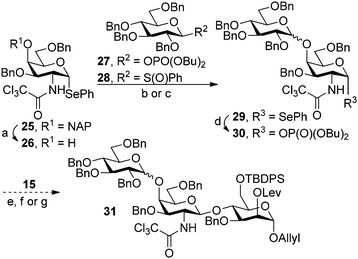 | ||
| Scheme 3 Synthesis of the trisaccharide 31 starting from the non-reducing end. Reagents and conditions: (a) DDQ, H2O, CH2Cl2, 87%; (b) 27, TMSOTf, PhMe, −40 °C, 79% (α:β = 3:1); (c) 28, Tf2O, DTMP, PhMe, −78 °C, no reaction; (d) dibutyl phosphate, NIS, CH2Cl2, 43%; (e) 29, MeOTf, CH2Cl2, no reaction; (f) 29, N-(phenylthio)-ε-caprolactam, Tf2O, TTBP, CH2Cl2, 0%; (g) 30, TMSOTf, CH2Cl2, −40 °C, up to 10%. | ||
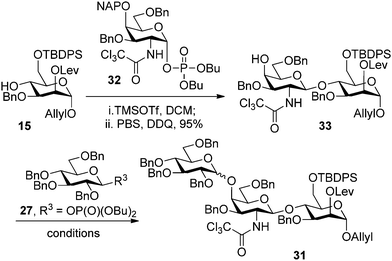
Glycosylation of the C4 position in the ManI building block 15 also proved to be challenging (Scheme 3). Initial attempts relied on selenoglycoside 29 for a direct glycosylation of 15. The presence of an allyl ether in the nucleophile precluded the use of more common NIS and TfOH activation conditions30 and, therefore, the reactions were carried out in the presence of MeOTf31 or N-(phenylthio)-ε-caprolactam and triflic anhydride.32 Failure of these conditions to produce any of the desired trisaccharide 31 triggered a change in the choice of glycosylating agent. Selenoglycoside 29 was converted to the corresponding glycosyl phosphate in the presence of NIS and dibutylphosphate and was used for the glycosylation of 15. Although a similar set of reactants (8 and 15, Scheme 2) was used previously to form the desired α-glycosidic linkage, but this reaction was inefficient when glycosyl phosphate 30 was used, producing only trace amounts of the desired trisaccharide 31.
At this junction it became apparent that a change in glycosylation strategy may be required for the synthesis of trisaccharide 24. The order of glycosylation reactions was reversed and the synthesis was carried out from the reducing end, starting from mannoside 15. Attempts to glycosylate the C4 position of the ManI building block using selenogalactoside 25 (ref. 17) in the presence of either MeOTf31 or AgOTf33 were not successful. Exchanging the selenide 25 for the corresponding dibutylphosphate 32 (ref. 23) emerged as a key improvement towards making the desired glycosidic bond. TMSOTf catalyzed glycosylation in dichloromethane followed by the oxidative removal of the NAP group, executed as a one-pot procedure, produced the desired disaccharide 33 in 95% yield. The initial attempts to construct the trisaccharide by glycosylating 33 with phosphate 27 in the presence of TBSOTf produced the desired material in only modest yield and low diastereoselectivity. The mixture of diastereomers of 31 could not be separated by standard methods. Using a toluene–dioxane mixture as the reaction solvent significantly improved the stereoselectivity of the glycosylation (Table 1). The yields were generally improved in the presence of molecular sieves. The α-selectivity was further improved when thiophene was present in the reaction mixture as an additive with an appropriate Lewis acid.24 Increasing the amount of thiophene in the reaction mixture and using it as a solvent resulted in higher yields but reduced the preference for α product. Using dimethylformamide to improve the α-selectivity, as reported for glycosylations with thioglycosides,34 appeared to be ineffective for glycosyl phosphate 27 with selectivities remaining lower than 3:1.
|
|
|||||
|---|---|---|---|---|---|
| Activator (equiv.) | Additive (equiv.) | Solvent | t/h | Yield (%, α/β) | |
|
a
General conditions: 27 (0.036 mmol, 1.2 equiv.), 33 (0.030 mmol) in 1 mL of solvent (A = PhMe–dioxane 1:3, B = PhMe–thiophene 2:1) in the presence of 4 Å MS at 0 °C. The α/β ratio was determined by 1H NMR of the crude product mixture.
b MS not used.
c Preactivation.
d 1.5 Equiv. of phosphate.
e Acid washed MS. NR = no reaction.
|
|||||
| 1b | TBSOTf (1.3) | — | A | 1 | 50, 3.4:1 |
| 2b | TBSOTf (1.4) | Thiophene (24) | A | 1 | 66, 4.9:1 |
| 3 | Sc(OTf)3 (1.4) | Thiophene (24) | A | 24 | NR |
| 4 | TMSOTf (1.4) | Thiophene (24) | A | 1 | 76, 5.4:1 |
| 5 | TMSOTf (1.4) | — | Thiophene | 1 | 73, 4.3:1 |
| 6 | TMSOTf (1.4) | — | B | 1 | 91, 3.5:1 |
| 7 | TBSOTf (1.4) | — | B | 1 | 89, 3.3:1 |
| 8 | TMSOTf (3) | DMF (6) | CH2Cl2 | 1 | NR |
| 9c,d | TMSOTf (7.5) | DMF (6) | CH2Cl2 | 1 | 16, 2.0:1 |
| 10 | TMSOTf (7) | DMF (6) | CH2Cl2 | 2 | 68, 3.0:1 |
| 11d,e | TBSOTf (7.5) | DMF (6) | CH2Cl2 | 2 | 62, 2.4:1 |
| 12 | TBSOTf (7) | DMF (6) | A | 1 | 32, 3.0:1 |
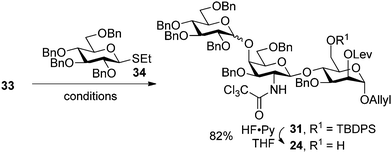
Despite reports on NIS being ineffective in glycosylation of allyl containing nucleophiles,30 glycosylation of 33 with thioglycoside 34 (ref. 35) in the presence of DMF produced the desired trisaccharide with selectivities higher than 8:1 (entry 1, Table 2) when the original NIS and TMSOTf activation protocol34 was followed. Further optimization of the reaction solvent, the amount of additive and the protocol helped to identify conditions that afford the desired trisaccharide in yields of up to 97% (entry 7, Table 2) and selectivities as high as 26:1 (entry 8, Table 2). Preactivation of the glycosylating agent that was reported to be an important factor in achieving high selectivities, appeared to have little effect on the stereochemical outcome in these reactions.36
|
|
||||
|---|---|---|---|---|
| Additive (equiv.) | Solvent | t/h | Yield (%, α/β) | |
| a General conditions: 34 (0.045 mmol, 1.5 equiv.), NIS (0.045 mmol, 1.5 equiv.), TMSOTf (0.045 mmol, 1.5 equiv.), 33 (0.030 mmol, 1.0 equiv.) in 1 mL of solvent in presence of 4 Å MS at 0 °C in a sonicator. The α/β ratio was determined by 1H NMR of the crude product mixture. b Acid washed MS. c No sonication. d No preactivation. e 3.0 Equiv. of reagent, NIS and TMSOTf. f Starting with 0.150 mmol of 33. | ||||
| 1b,c | DMF (6) | CH2Cl2 | 22 | 12, 8:1 |
| 2 | DMF (6) | CH2Cl2 | 22 | 54, 12:1 |
| 3d | DMF (6) | CH2Cl2 | 22 | 48, 11:1 |
| 4e | DMF (12) | CH2Cl2 | 16 | 70, 11:1 |
| 5 | DMF (6) | Et2O | 20 | 70, 18:1 |
| 6 | DMF (6) | PhMe | 16 | 83, 19:1 |
| 7d,f | DMF (9) | PhMe | 16 | 97, 18:1 |
| 8f | DMF (6) | PhMe–thiophene 2:1 |
12 | 82, 26:1 |
![Synthesis of the low molecular weight antigen of T. gondii. Reagents and conditions: (a) 4, TBSOTf, 4 Å MS, thiophene–PhMe (2 : 1), 85%; (b) Zn, AcOH, 55 °C, 94%; (c) (i) [Ir(COD)(PPh2Me)2]PF6, H2, THF, (ii) HgCl2, HgO, H2O, acetone; (iii) Cl3CCN, DBU, CH2Cl2, 83% (three steps); (d) 5, TMSOTf, PhMe, −40 °C, 80%; (e) (i) [Ir(COD)(PPh2Me)2]PF6, H2, THF (ii) HgCl2, HgO, H2O, acetone, 95% (two steps); (f) (i) 6, PivCl, pyridine; (ii) I2, H2O; Dowex 50WX8 (Na+), CHCl3, MeOH; (iii) Sc(OTf)3, MeCN, CHCl3, 64% (three steps); (g) (i) 7, PivCl, pyridine; (ii) I2, H2O; (iii) H2NNH2, AcOH, pyridine, CHCl3, 94% (three steps); (h) H2, Pd/C, CHCl3–MeOH–H2O (3 : 3 : 1), 62%.](/image/article/2013/SC/c2sc21515b/c2sc21515b-s4.gif) | ||
| Scheme 4 Synthesis of the low molecular weight antigen of T. gondii. Reagents and conditions: (a) 4, TBSOTf, 4 Å MS, thiophene–PhMe (2:1), 85%; (b) Zn, AcOH, 55 °C, 94%; (c) (i) [Ir(COD)(PPh2Me)2]PF6, H2, THF, (ii) HgCl2, HgO, H2O, acetone; (iii) Cl3CCN, DBU, CH2Cl2, 83% (three steps); (d) 5, TMSOTf, PhMe, −40 °C, 80%; (e) (i) [Ir(COD)(PPh2Me)2]PF6, H2, THF (ii) HgCl2, HgO, H2O, acetone, 95% (two steps); (f) (i) 6, PivCl, pyridine; (ii) I2, H2O; Dowex 50WX8 (Na+), CHCl3, MeOH; (iii) Sc(OTf)3, MeCN, CHCl3, 64% (three steps); (g) (i) 7, PivCl, pyridine; (ii) I2, H2O; (iii) H2NNH2, AcOH, pyridine, CHCl3, 94% (three steps); (h) H2, Pd/C, CHCl3–MeOH–H2O (3:3:1), 62%. | ||
Removal of the allyl ether from glycan 38 provided alcohol 39 in 95% yield. Phosphonylation via the mixed anhydride prepared in situ from 1,2-distearoyl-sn-glycerol H-phosphonate 6 and pivaloyl chloride followed by the iodine/water mediated oxidation and subsequent removal of the TIPS group by treatment with a Lewis acid gave glycolipid 40 in 64%. The second phosphorylation using the Cbz protected ethanolamine H-phosphonate 7 and subsequent hydrazinolysis of the levulinate ester afforded protected glycolipid 41. Hydrogenolysis of 41 in the presence of palladium on charcoal in a mixture of methanol, chloroform, water and acetic acid provided GPI 23 in 58% yield over four steps, starting from 40.
The syntheses of GPIs 3 and 23 largely served as a testing and optimization platform for the forward synthetic route based on the general retrosynthetic analysis outlined in Fig. 3. Although the protecting group strategy for phosphorylations and glycan assembly may appear to be two isolated issues, they are intertwined on both the strategic and experimental level. Glycosylation reactions used in convergent construction of the GPI glycans are shaped by the position of temporary protecting groups within the common building blocks because this determines the coupling partners. Since positioning of the temporary protecting groups remains the same across the interchangeable building blocks used in this synthetic route, it is reasonable to suggest that the optimized conditions for analogous glycosylation reactions will be transferable between the syntheses of different GPIs. These conditions have been optimized in the initial syntheses and will be applied in the syntheses of other GPIs. With a better insight into the details of protecting group manipulations, related glycosylations and the order of building block assembly into the GPI structures, we were poised to tackle new targets.
Synthesis of the Trypanosoma congolense VSG GPI anchor
The GPI anchor of T. congolense variant surface glycoprotein (VSG) features a β-Gal-(1 → 6)-β-GlcNAc-(1 → 4)-α-ManI side branch that is distinctly different the previously prepared GPI anchors as is its 1,2-dimyristoyl-sn-glycerol lipid. All of the bond disconnections from the original retrosynthetic scheme were implemented in the retrosynthetic analysis of the T. congolense VSG GPI anchor but the order of glycosylations was adjusted to reflect the insights gained during the synthesis of the low molecular weight antigen of T. gondii (Fig. 6). In short, the target GPI would be prepared from the protected pseudoheptasaccharide that will arise from the building blocks 4, 5 and the branch-containing trisaccharide via a [2 + 3 + 2] glycosylation strategy. The trisaccharide building block was envisioned to be derived from central ManI building block 15 and glycosylating agents 44 (ref. 37) and 45 (ref. 38) with the order of assembly adopted from the synthesis of 23.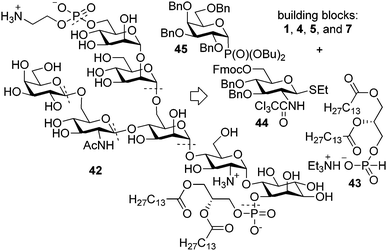 | ||
| Fig. 6 Retrosynthetic analysis of the GPI anchor of T. congolense VSG. | ||
Glycosylation of mannoside 15 with thioglycoside 44 followed by deprotection of the Fmoc group provided disaccharide 46 in 95% yield (Scheme 5). Subsequent installation of the β-galactoside was more troublesome than expected. Glycosylation of 46 with per-O-acetylated galactosyl phosphate39 resulted in partial acetylation of the disaccharide. Per-O-benzylated galactosylating reagents that have been shown to be competent in the synthesis of β-galactosides, were used to circumvent acyl migration.40 Glycosylation with per-O-benzylated galactosyl trichloroacetimidate41 using Pd(CH3CN)4(BF4)2 as a catalyst in dichloromethane or acetonitrile, however, suffered from low conversion. With the same set of coupling partners using TMSOTf as the activating reagent at low temperatures in a 1:2:1 CH2Cl2–MeCN–EtCN solvent mixture,42 reactions also proceeded sluggishly and significant production of the rearranged imidate side product was observed. In an attempt to eliminate the possibility of glycosylating agent rearrangement per-O-benzylated galactosyl phosphate 45 was employed. In the presence of TMSOTf in a 60 mM propionitrile solution at −78 °C, 45 and 46 provided the desired trisaccharide 47 in 82% yield. The TBDPS group was removed from 47 by treatment with HF·pyridine to produce the alcohol that was glycosylated with dimannosyl trichloroacetimidate 4 to form pentasaccharide 49 in 84% yield.
![Synthesis of the GPI anchor of T. congolense VSG. Reagents and conditions: (a) (i) 44, NIS, TMSOTf, 4 Å MS, CH2Cl2, −30 °C to 0 °C; (ii) NEt3, 95% (two steps); (b) 45, TMSOTf, EtCN, −78 °C, 81%; (c) HF·pyridine, THF, 89%; (d) 4, TBSOTf, 4 Å MS, thiophene, Et2O, 0 °C, 84%; (e) Zn, AcOH, 55 °C, 87%; (f) (i) [Ir(COD)(PPh2Me)2]PF6, H2, THF; (ii) HgCl2, HgO, H2O, acetone; (iii) Cl3CCN, DBU, CH2Cl2, 56% (three steps); (g) 5, TMSOTf, thiophene, PhMe, −40 °C, 87%; (h) (i) [Ir(COD)(PPh2Me)2]PF6, H2, THF (ii) HgCl2, HgO, H2O, acetone, 79% (two steps); (i) (i) 43, PivCl, pyridine; (ii) I2, H2O; iii. Sc(OTf)3, MeCN, CHCl3, 74% (three steps); (j) (i) 7, PivCl, pyridine; (ii) I2, H2O; (iii) H2NNH2, AcOH, 82% (three steps); (k) H2, Pd/C, CHCl3–MeOH–AcOH–H2O, 85%.](/image/article/2013/SC/c2sc21515b/c2sc21515b-s5.gif) | ||
| Scheme 5 Synthesis of the GPI anchor of T. congolense VSG. Reagents and conditions: (a) (i) 44, NIS, TMSOTf, 4 Å MS, CH2Cl2, −30 °C to 0 °C; (ii) NEt3, 95% (two steps); (b) 45, TMSOTf, EtCN, −78 °C, 81%; (c) HF·pyridine, THF, 89%; (d) 4, TBSOTf, 4 Å MS, thiophene, Et2O, 0 °C, 84%; (e) Zn, AcOH, 55 °C, 87%; (f) (i) [Ir(COD)(PPh2Me)2]PF6, H2, THF; (ii) HgCl2, HgO, H2O, acetone; (iii) Cl3CCN, DBU, CH2Cl2, 56% (three steps); (g) 5, TMSOTf, thiophene, PhMe, −40 °C, 87%; (h) (i) [Ir(COD)(PPh2Me)2]PF6, H2, THF (ii) HgCl2, HgO, H2O, acetone, 79% (two steps); (i) (i) 43, PivCl, pyridine; (ii) I2, H2O; iii. Sc(OTf)3, MeCN, CHCl3, 74% (three steps); (j) (i) 7, PivCl, pyridine; (ii) I2, H2O; (iii) H2NNH2, AcOH, 82% (three steps); (k) H2, Pd/C, CHCl3–MeOH–AcOH–H2O, 85%. | ||
Reduction of the trichloroacetamide to acetamide 49 proceeded smoothly to provide 50 in 87% yield. The anomeric allyl ether present in 50 was removed via isomerization with the iridium catalyst, followed by mild hydrolysis using mercury salts. The resulting hemiacetal was transformed into trichloroacetimidate 51 that was used in an α-stereoselective [5 + 2] glycosylation of the pseudodisaccharide 5 to afford pseudoheptasaccharide 52, isolated as a single diastereomer in 87% yield. Removal of the allyl ether furnished alcohol 53 in 79% yield. Phosphonylation of 53 with H-phosphonate 43 followed by oxidation and subsequent removal of the TIPS group under acidic conditions afforded glycolipid 54. The second phosphorylation with H-phosphonate 7 installed the phosphoethanolamine unit on ManIII and, after the hydrazynolysis of the levulinyl ester, gave glycolipid 55. Palladium catalyzed hydrogenolysis in a mixture of water, methanol, chloroform and acetic acid provided GPI 42 in 52% yield over seven steps starting from 53.
Synthesis of the Trypanosoma brucei VSG 117 GPI anchor
While many GPIs feature a side branch attached to the C4 position of ManI, GPI anchors of T. brucei VSGs carry oligogalactose side branches attached to the C3 position of ManI (Fig. 1). The GPI anchor of T. brucei VSG 117 (56, Fig. 7) was selected as a target to test the applicability of our general synthetic route to the synthesis of GPIs with branching at the C3 position of ManI. In addition to the challenge of introducing the branch with different connectivity within the ManI residue, the T. brucei VSG 117 GPI also requires an efficient synthesis of a challenging all-α-configured oligogalactose side branch.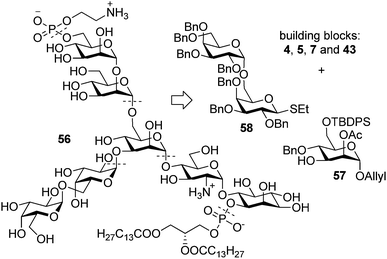 | ||
| Fig. 7 Retrosynthetic analysis of the T. brucei VSG 117 GPI anchor. | ||
The general retrosynthetic analysis breaks the target GPI down into H-phosphonates 7 and 43, and disaccharides 4 and 5, all of which have been used in previous syntheses, and the branching trisaccharide. Considering the difficulties associated with construction of α-configured oligogalactosides, the requisite α-Gal-(1 → 6)-α-Gal-(1 → 3)-α-Man trisaccharide was envisioned from the previously reported digalactose thioglycoside 58 (ref. 43) and mannoside 57 (Fig. 7). Since none of the GPIs with a C3 branch at ManI have additional phosphodiester substituents that would require Lev group for the late stage phosphorylation, for the ease of building block synthesis, the central mannose building block 2 was modified by introducing the acetate in place of the Lev ester (57, Fig. 7).44 Some GPIs isolated from T. brucei have additional galactose residues on ManII and ManIII. To facilitate easier modification of the final GPI by exchange of the oligomannoside building blocks, the order of glycosylations to append the top oligomannoside and the bottom pseudodisaccharide was reversed.
The synthesis began with construction of the oligogalactoside branch starting from mannoside 57 (Scheme 6). Glycosylation with digalactoside 58 (ref. 43) in the presence of DMF furnished the trisaccharide 59 in high yield and good selectivity.34 Exchange of the allyl ether in 59 for a tricholoacetimidate provided the glycosylating agent 60 for the [3 + 2] glycosylation of pseudodisaccharide 5 to provide pseudopentasaccharide 61. To avoid a late stage removal of acetyl in the presence of fatty acid esters, the acetate in 61 was exchanged for benzyl ether. Removal of TBDPS ether provided alcohol 62 in 77% yield over three steps. Further glycosylation of the pseudopentasaccharide 62 with dimmanosyl imidate 4 afforded the fully protected GPI glycan in good overall yield. This demonstrates the flexibility of our general synthetic route and its ability to accommodate minor changes in the structures of building blocks or in the order of glycosylations that create glycosidic bonds around ManI. Removal of the orthogonal protecting groups and installation of phosphodiesters employing the well-established protocols furnished the fully phosphorylated precursor of GPI anchor of T. brucei VSG 117 66. Hydrogenolysis of 66 provided the target GPI 56 in 85% yield.
![Synthesis of T. brucei VSG 117 GPI 56. Reagents and conditions: (a) 58, NIS, TMSOTf, DMF, 4 Å MS, PhMe–thiophene (3 : 1), 0 °C, sonication, 83%; (b) (i) [IrCOD(PPh2Me)2]PF6, H2, THF; (ii) HgCl2, HgO, H2O, acetone; (iii) Cl3CCN, DBU, DCM, 93%; (c) 5, TBSOTf, 4 Å MS, Et2O, 0 °C, 97%; (d) (i) NaOMe, MeOH, THF, 88%; (ii) NaH, BnBr, DMF, 92%; (iii) HF-pyridine, THF, 95% (e) 4, TBSOTf, 4 Å MS, Et2O, 0 °C, 70%; (f) (i) [IrCOD(PPh2Me)2]PF6, H2, THF (ii) HgCl2, HgO, H2O, acetone, 72%; (g) (i) 43, PivCl, pyridine; (ii) I2, H2O; (iii) Sc(OTf)3, MeCN, CHCl3, 72%; (h) (i) 7, PivCl, pyridine; (ii) I2, H2O, 78%; (i) H2, Pd/C, CHCl3–MeOH–H2O (3 : 3 : 1), 85%.](/image/article/2013/SC/c2sc21515b/c2sc21515b-s6.gif) | ||
| Scheme 6 Synthesis of T. brucei VSG 117 GPI 56. Reagents and conditions: (a) 58, NIS, TMSOTf, DMF, 4 Å MS, PhMe–thiophene (3:1), 0 °C, sonication, 83%; (b) (i) [IrCOD(PPh2Me)2]PF6, H2, THF; (ii) HgCl2, HgO, H2O, acetone; (iii) Cl3CCN, DBU, DCM, 93%; (c) 5, TBSOTf, 4 Å MS, Et2O, 0 °C, 97%; (d) (i) NaOMe, MeOH, THF, 88%; (ii) NaH, BnBr, DMF, 92%; (iii) HF-pyridine, THF, 95% (e) 4, TBSOTf, 4 Å MS, Et2O, 0 °C, 70%; (f) (i) [IrCOD(PPh2Me)2]PF6, H2, THF (ii) HgCl2, HgO, H2O, acetone, 72%; (g) (i) 43, PivCl, pyridine; (ii) I2, H2O; (iii) Sc(OTf)3, MeCN, CHCl3, 72%; (h) (i) 7, PivCl, pyridine; (ii) I2, H2O, 78%; (i) H2, Pd/C, CHCl3–MeOH–H2O (3:3:1), 85%. | ||
A long standing interest in glycobiology and immunology of parasitic infections directed the choice of our synthetic targets to parasitic GPIs. One of the structural implications of this is the lack of additional phosphoethanolamine residues at C2 of ManI in the targets we have reported thus far. We have demonstrated that the Lev group can be efficiently removed and that the phosphoethanolamine unit can be installed in the presence of other phosphodiesters, both suggesting that this route can also afford triphosphorylated mammalian GPIs. To test this, we have intercepted a penultimate intermediate to the GPI of T. gondii in which the levulinic ester has already been cleaved (22, Scheme 2) and we have phosphorylated the C2 of ManI to produce, in 86% yield, the fully protected triphosphorylated GPI with an additional phosphoethanolamine.45
Reflecting on the development and optimization of the described synthetic route, it is important to underline several features of the described strategy. The GPIs prepared using this synthetic route feature all major modifications of the conserved GPI core including mono and oligosaccharide branches, branches at both C3 and C4 positions of ManI, and both di- and triphosphorylated structures. The conserved core in all synthesized GPIs is constructed from only 4 interchangeable building blocks. While each side branch requires a unique approach, some flexibility is allowed in respect to the order of assembly of the branch itself and the identity of the building blocks.
In addition to the three fully orthogonal protecting groups, an oxidatively cleavable NAP group was used as well. A comprehensive study that would show expansion of this set to four groups (TIPS, Lev, Allyl and NAP) has not been performed yet but the syntheses of side branches, and the work with the fully orthogonally protected central mannose building blocks that incorporate all five protecting groups, strongly suggests that this is feasible. It is evident that the described synthetic route can easily accommodate modifications such as other monosaccharide units in the side branch and can produce unnatural GPI analogues. Should the orthogonal set of protecting groups become expanded, the ability to produce additional analogues that could include various chemical tags unrelated to carbohydrates would be significantly improved.
The inability to introduce the side branch at a late stage of the synthesis, as intended in the [1 + 5] glycosylation strategy for the GPI anchor of T. gondii, was initially seen as a shortcoming of this route. From the standpoint of the synthesis of diverse GPI structures, the construction of the side branch, which requires a unique synthetic approach for each GPI target, may require less optimization effort if executed with substrates of lower complexity early on in the synthesis. This is especially important considering that benzyl ethers are used as permanent protecting groups in the described route. Due to their prominence in oligosaccharide protection, a vast amount of data is available about the feasibility and stereochemical outcome of glycosylation reactions with benzyl protected building blocks. Utilizing this database of knowledge, concise routes to the required GPI branches can be designed efficiently as demonstrated in the synthesis of the T. brucei VSG 117 GPI that features a challenging oligogalactose branch. The use of benzyl groups, on the other hand, requires hydrogenolysis for global deprotection and thereby prevents incorporation of unsaturated lipids into the synthetic GPIs prepared by this route. Complementary strategies that successfully complete this task have been described but are, at this point, not applicable to this synthetic route due to protecting group incompatibilities.9l,n,p
One goal of our efforts directed towards the synthesis of a diverse set of GPIs was to enable biological studies. In addition to native GPIs, such studies often call for analogues with suitable linkers for immobilization on microarray slides or conjugation to other molecules. While phosphoethanolamines of GPIs have been used for this purpose,10k,14 immobilization via a linker placed in the position of the lipid may be preferred as it mimics the presentation of GPIs on the cell membrane. Instead of diacylglycerol, this synthetic strategy allows for the installation of a 6-hydroxy-1-hexanethiol linker that has been used for immobilization of synthetic GPIs and related molecules on microarrays.7a,10m It also allows for the introduction of modified phosphoethanolamines at the C6 position of ManIII that can be utilized for native chemical ligation of synthetic GPIs with proteins and peptides.18 The C2 position of ManI may also serve as a site for introduction of chemical tags or fluorescent probes owing to the presence of Lev ester.9o,46
Conclusions
A general strategy for the synthesis of GPI glycolipids by modular assembly of common building blocks was developed and applied to the syntheses of structurally distinct GPIs: the GPI of T. gondii, the low molecular weight antigen of T. gondii, the GPI anchor of T. congolense VSG and the GPI of T. brucei VSG 117.One of the key features of this strategy is the use of a fully orthogonal set of protecting groups that enables regioselective introduction of the required phosphodiesters and the efficient assembly of the building blocks into the GPI glycans. The assembly of the glycan is dictated by the positioning of the temporary protecting groups which is kept constant across the set of common building blocks. The glycosylations required for the assembly of different GPI glycans, therefore, evoke similar coupling partners making the reactions conditions broadly transferable between different GPI syntheses. Both the late stage phosphorylations and glycosylations that form glycosidic bonds around the central mannose have been optimized with respect to yield and stereoselectivity, and have been shown to be good for the syntheses of a diverse set of targets.
This report constitutes the first general approach to the synthesis of diverse GPI structures and the first synthetic route capable of producing GPIs with various substitution patterns including: simple monosaccharide branches and complex oligosaccharide branches with synthetically challenging glycosidic bonds, branches at both C3 or C4 position of ManI, di- and triphosphorylated structures, and generally diverse GPIs isolated from different organisms. With the ability to produce homogeneous native GPI structures and the flexibility that can accommodate further changes and produce unnatural analogues, this convergent route for the synthesis of diverse GPI structures is poised to enable extensive investigation into the biological roles of these glycolipids.
Acknowledgements
This work was supported by the Max Planck Society, the Körber foundation (Körber European Science Award to P.H.S.), the German Academic Exchange Service (doctoral scholarship to Y.-H. T.). The research leading to these results has received funding from the European Commission's Seventh Framework Programme FP7/2007-2013 (postdoctoral fellowship to I.V.). We thank Ms Martha Kozakowski and Mr Heung Sik Hahm for assistance with synthesis of building blocks.Notes and references
- M. Fujita and T. Kinoshita, FEBS Lett., 2010, 584, 1670–1677 CrossRef CAS.
- Y. Maeda and T. Kinoshita, Prog. Lipid Res., 2011, 50, 411–424 CrossRef CAS.
- M. G. Paulick and C. R. Bertozzi, Biochemistry, 2008, 47, 6991–7000 CrossRef CAS.
- (a) G. A. M. Cross, Annu. Rev. Cell Biol., 1990, 6, 1–39 CrossRef CAS; (b) H. Ikezawa, Biol. Pharm. Bull., 2002, 25, 409–417 CrossRef CAS.
- (a) M. A. J. Ferguson, J. Cell Sci., 1999, 112, 2799–2809 CAS; (b) M. Fujita and Y. Jigami, Biochim. Biophys. Acta, Gen. Subj., 2008, 1780, 410–420 CrossRef CAS; (c) M. Fujita, Y. Maeda, M. Ra, Y. Yamaguchi, R. Taguchi and T. Kinoshita, Cell, 2009, 139, 352–365 CrossRef CAS.
- M.-L. Hecht, Y.-H. Tsai, X. Liu, C. Wolfrum and P. H. Seeberger, ACS Chem. Biol., 2010, 5, 1075–1086 CrossRef CAS.
- (a) F. Kamena, M. Tamborrini, X. Liu, Y.-U. Kwon, F. Thompson, G. Pluschke and P. H. Seeberger, Nat. Chem. Biol., 2008, 4, 238–240 CrossRef CAS; (b) P. H. Seeberger and T. Horlacher, Chem. Soc. Rev., 2008, 37, 1414–1422 RSC; (c) N. Azzouz, F. Kamena and P. H. Seeberger, OMICS, 2010, 14, 445–454 CrossRef CAS.
- Y.-H. Tsai, X. Liu and P. H. Seeberger, Angew. Chem., Int. Ed., 2012, 51 DOI:10.1002/anie.201203912.
- (a) C. Murakata and T. Ogawa, Tetrahedron Lett., 1991, 32, 671–674 CrossRef CAS; (b) T. G. Mayer, B. Kratzer and R. R. Schmidt, Angew. Chem., Int. Ed. Engl., 1994, 33, 2177–2181 CrossRef; (c) A. S. Campbell and B. Fraser-Reid, J. Am. Chem. Soc., 1995, 117, 10387–10388 CrossRef CAS; (d) D. K. Baeschlin, A. R. Chaperon, V. Charbonneau, L. G. Green, S. V. Ley, U. Lucking and E. Walther, Angew. Chem., Int. Ed., 1998, 37, 3423–3428 CrossRef CAS; (e) D. K. Baeschlin, A. R. Chaperon, L. G. Green, M. G. Hahn, S. J. Ince and S. V. Ley, Chem.–Eur. J., 2000, 6, 172–186 CrossRef CAS; (f) J. Xue and Z. Guo, J. Am. Chem. Soc., 2003, 125, 16334–16339 CrossRef CAS; (g) N. Shao, B. Xue and Z. Guo, Angew. Chem., Int. Ed., 2004, 43, 1569–1573 CrossRef CAS; (h) J. Lu, K. N. Jayaprakash, U. Schlueter and B. Fraser-Reid, J. Am. Chem. Soc., 2004, 126, 7540–7547 CrossRef CAS; (i) Z. Guo and L. Bishop, Eur. J. Org. Chem., 2004, 3585–3596 CrossRef CAS; (j) Y.-U. Kwon, X. Liu and P. H. Seeberger, Chem. Commun., 2005, 2280–2282 RSC; (k) X. Liu, Y.-U. Kwon and P. H. Seeberger, J. Am. Chem. Soc., 2005, 127, 5004–5005 CrossRef CAS; (l) D. V. Yashunsky, V. S. Borodkin, M. A. J. Ferguson and A. V. Nikolaev, Angew. Chem., Int. Ed., 2006, 45, 468–474 CrossRef CAS; (m) X. Wu and Z. Guo, Org. Lett., 2007, 9, 4311–4313 CrossRef CAS; (n) B. M. Swarts and Z. Guo, J. Am. Chem. Soc., 2010, 132, 6648–6650 CAS; (o) B. M. Swarts and Z. Guo, Chem. Sci., 2011, 2, 2342–2352 RSC; (p) A. Ali and R. A. Vishwakarma, Tetrahedron, 2010, 66, 4357–4369 CrossRef CAS.
- (a) X. M. Wu, Z. H. Shen, X. Q. Zeng, S. H. Lang, M. Palmer and Z. Guo, Carbohydr. Res., 2008, 343, 1718–1729 CrossRef CAS; (b) H. Dietrich, J. F. Espinosa, J. L. Chiara, J. Jimenez-Barbero, Y. Leon, I. Varela-Nieto, J. M. Mato, F. H. Cano, C. Foces-Foces and M. Martin-Lomas, Chem.–Eur. J., 1999, 5, 320–336 CrossRef CAS; (c) M. Martin-Lomas, N. Khiar, S. Garcia, J. L. Koessler, P. M. Nieto and T. W. Rademacher, Chem.–Eur. J., 2000, 6, 3608–3621 CrossRef CAS; (d) J. Lopez-Prados, F. Cuevas, N. C. Reichardt, J. L. de Paz, E. Q. Morales and M. Martin-Lomas, Org. Biomol. Chem., 2005, 3, 764–786 RSC; (e) P. J. Garegg, P. Konradsson, S. Oscarson and K. Ruda, Tetrahedron, 1997, 53, 17727–17734 CrossRef CAS; (f) M. Lahmann, P. J. Garegg, P. Konradsson and S. Oscarson, Can. J. Chem., 2002, 80, 1105–1111 CrossRef CAS; (g) A. Crossman, M. J. Paterson, M. A. J. Ferguson, T. K. Smith and J. S. Brimacombe, Carbohydr. Res., 2002, 337, 2049–2059 CrossRef CAS; (h) A. P. Dix, C. N. Borissow, M. A. J. Ferguson and J. S. Brimacombe, Carbohydr. Res., 2004, 339, 1263–1277 CrossRef CAS; (i) A. Crossman, T. K. Smith, M. A. J. Ferguson and J. S. Brimacombe, Tetrahedron Lett., 2005, 46, 7419–7421 CrossRef CAS; (j) M. C. Hewitt, D. A. Snyder and P. H. Seeberger, J. Am. Chem. Soc., 2002, 124, 13434–13436 CrossRef CAS; (k) L. Schofield, M. C. Hewitt, K. Evans, M. A. Siomos and P. H. Seeberger, Nature, 2002, 418, 785–789 CrossRef CAS; (l) P. H. Seeberger, R. L. Soucy, Y.-U. Kwon, D. A. Snyder and T. Kanemitsu, Chem. Commun., 2004, 1706–1707 RSC; (m) Y.-U. Kwon, R. L. Soucy, D. A. Snyder and P. H. Seeberger, Chem.–Eur. J., 2005, 11, 2493–2504 CrossRef CAS; (n) U. E. Udodong, R. Madsen, C. Roberts and B. Fraser-Reid, J. Am. Chem. Soc., 1993, 115, 7886–7887 CrossRef CAS.
- For general reviews of methods for oligosaccharide synthesis see: (a) C.-Y. Wu and C.-H. Wong, Top. Curr. Chem., 2011, 301, 223–252 CrossRef CAS; (b) C.-H. Hsu, S.-C. Hung, C.-Y. Wu and C.-H. Wong, Angew. Chem., Int. Ed., 2011, 50, 11872–11923 CrossRef CAS; (c) J. Seibel and K. Buchholz, Adv. Carbohydr. Chem. Biochem., 2010, 63, 101–138 CrossRef CAS; (d) S. Eller, M. Weishaupt and P. H. Seeberger, Carbohydr. Chem., 2010, 36, 127–141 CAS; (e) X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS; (f) S. Muthana, H. Cao and X. Chen, Curr. Opin. Chem. Biol., 2009, 13, 573–581 CrossRef CAS; (g) A. Ishiwata and Y. Ito, Trends Glycosci. Glycotechnol., 2009, 21, 266–289 CrossRef CAS; (h) P. H. Seeberger and D. B. Werz, Nat. Rev. Drug Discovery, 2005, 4, 751–763 CrossRef CAS.
- A notable exception is the GlcN-Ino pseudodisaccharide that has often been used as a building block due to the difficulties associated with glycosylations that produce α-glucosamine.
- A. V. Nikolaev and N. Al-Maharik, Nat. Prod. Rep., 2011, 28, 970–1020 RSC.
- Y.-H. Tsai, S. Götze, N. Azzouz, H.-S. Hahm, P. H. Seeberger and D. Varon Silva, Angew. Chem., Int. Ed., 2011, 50, 9961–9964 CrossRef CAS.
- (a) B. Striepen, C. F. Zinecker, J. B. L. Damm, P. A. T. Melgers, G. J. Gerwig, M. Koolen, J. F. G. Vliegenthart, J. F. Dubremetz and R. T. Schwarz, J. Mol. Biol., 1997, 266, 797–813 CrossRef CAS; (b) P. Gerold, L. Schofield, M. J. Blackman, A. A. Holder and R. T. Schwarz, Mol. Biochem. Parasitol., 1996, 75, 131–143 CrossRef CAS; (c) I. R. E. Nett, A. Mehlert, D. Lamont and M. A. J. Ferguson, Glycobiology, 2010, 20, 576–585 CrossRef CAS; (d) D. Oxley and A. Bacic, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14246–14251 CrossRef CAS; (e) R. S. Naik, O. H. Branch, A. S. Woods, M. Vijaykumar, D. J. Perkins, B. L. Nahlen, A. A. Lal, R. J. Cotter, C. E. Costello, C. F. Ockenhouse, E. A. Davidson and D. C. Gowda, J. Exp. Med., 2000, 192, 1563–1575 CrossRef CAS; (f) N. Stahl, M. A. Baldwin, R. Hecker, K. M. Pan, A. L. Burlingame and S. B. Prusiner, Biochemistry, 1992, 31, 5043–5053 CrossRef CAS; (g) A. Treumann, M. R. Lifely, P. Schneider and M. A. J. Ferguson, J. Biol. Chem., 1995, 270, 6088–6099 CrossRef CAS; (h) S. W. Homans, M. A. J. Ferguson, R. A. Dwek, T. W. Rademacher, R. Anand and A. F. Williams, Nature, 1988, 333, 269–272 CrossRef CAS; (i) J. I. MacRae, A. Acosta-Serrano, N. A. Morrice, A. Mehlert and M. A. J. Ferguson, J. Biol. Chem., 2005, 280, 12201–12211 CrossRef CAS; (j) P. Schneider, M. A. J. Ferguson, M. J. Mcconville, A. Mehlert, S. W. Homans and C. Bordier, J. Biol. Chem., 1990, 265, 16955–16964 CAS; (k) M. A. J. Ferguson, S. W. Homans, R. A. Dwek and T. W. Rademacher, Science, 1988, 239, 753–759 CAS; (l) Y. Nakano, K. Noda, T. Endo, A. Kobata and M. Tomita, Arch. Biochem. Biophys., 1994, 311, 117–126 CrossRef CAS; (m) I. A. Brewis, M. A. J. Ferguson, A. Mehlert, A. J. Turner and N. M. Hooper, J. Biol. Chem., 1995, 270, 22946–22956 CrossRef CAS.
- K. Pekari and R. R. Schmidt, J. Org. Chem., 2003, 68, 1295–1308 CrossRef CAS.
- Two requisite phosphates extend from the GPI glycan towards the protein and the lipid (Fig. 1). An additional phosphoethanolamine unit at the C2 position of ManI (R6, Fig. 1) is ubiquitous among mammalian GPIs, some of which carry another phosphoethanolamine at the C6 position of ManII (R5, Fig. 1). The lone examples of additional GPI phosphorylation among the non-mammalian species are the GPIs of Trypanosoma cruzi that feature a (2-aminoethyl)phosphonate unit at the C6 position of glucosamine (R7, Fig. 1).
- C. F. Becker, X. Liu, D. Olschewski, R. Castelli, R. Seidel and P. H. Seeberger, Angew. Chem., Int. Ed., 2008, 47, 8215–8219 CrossRef CAS.
- The protocols for cleavage of benzyl ethers from advanced intermediates normally require minimal effort in work up and product purification making them particularly suitable for the synthesis of materials that are difficult to handle such as GPIs.
- P. G. M. Wuts and T. W. Greene, in Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., 2006, pp. 16–366 Search PubMed.
- P. Gerold, A. Dieckmannschuppert and R. T. Schwarz, J. Biol. Chem., 1994, 269, 2597–2606 CAS.
- D. S. Tan, Nat. Chem. Biol., 2005, 1, 74–84 CrossRef CAS.
- Synthesis of building blocks is described in ESI.† Discussion is limited only to the building block used in the forward synthetic routes described in this manuscript.
- J. Park, S. Kawatkar, J.-H. Kim and G.-J. Boons, Org. Lett., 2007, 9, 1959–1962 CrossRef CAS.
- J. J. Oltvoort, C. A. A. Vanboeckel, J. H. Dekoning and J. H. Vanboom, Synthesis, 1981, 305–308 CrossRef.
- R. R. Schmidt, M. Stumpp and J. Michel, Tetrahedron Lett., 1982, 23, 405–408 CrossRef CAS.
- Z. Li and J. C. Gildersleeve, J. Am. Chem. Soc., 2006, 128, 11612–11619 CrossRef CAS.
- C.-T. Ren, Y.-H. Tsai, Y.-L. Yang, W. Zou and S.-H. Wu, J. Org. Chem., 2007, 72, 5427–5430 CrossRef CAS.
- R. Kakarla, R. G. Dulina, N. T. Hatzenbuhler, Y. W. Hui and M. J. Sofia, J. Org. Chem., 1996, 61, 8347–8349 CrossRef CAS.
- A. K. Misra and R. Panchadhayee, J. Carbohydr. Chem., 2010, 29, 76–83 CrossRef.
- H. Lönn, Carbohydr. Res., 1985, 139, 105–113 Search PubMed.
- S. G. Duron, T. Polat and C. H. Wong, Org. Lett., 2004, 6, 839–841 CrossRef CAS.
- S. Mehta and B. M. Pinto, Tetrahedron Lett., 1991, 32, 4435–4438 CrossRef CAS.
- S.-R. Lu, Y.-H. Lai, J.-H. Chen, C.-Y. Liu and K.-K. T. Mong, Angew. Chem., Int. Ed., 2011, 50, 7315–7320 CrossRef CAS.
- F. Weygand and H. Ziemann, Liebigs Ann. Chem., 1962, 179–198 CrossRef CAS.
- Revisiting the mannoside 15 and performing the glycosylation with selenoglycoside 25 in the presence of NIS and TMSOTf followed by oxidative removal of the NAP ether provided disaccharide 33 in 40% yield over two steps. This further supports the use of glycosyl phosphate over the selenoglycoside despite the additional step needed for its preparation.
- L. Kröck, D. Esposito, B. Castagner, C.-C. Wang, P. Bindschädler and P. H. Seeberger, Chem. Sci., 2012, 3, 1617–1622 RSC.
- (a) R. R. Schmidt and M. Stumpp, Liebigs Ann. Chem., 1984, 680–691 CrossRef CAS; (b) H. Isobe, K. Cho, N. Solin, D. B. Werz, P. H. Seeberger and E. Nakamura, Org. Lett., 2007, 9, 4611–4614 CrossRef CAS.
- G. Soldaini, F. Cardona and A. Goti, Org. Lett., 2005, 7, 725–728 CrossRef CAS.
- J. Yang, C. Cooper-Vanosdell, E. A. Mensah and H. M. Nguyen, J. Org. Chem., 2008, 73, 794–800 CrossRef CAS.
- D. J. Cox, M. D. Smith and A. J. Fairbanks, Org. Lett., 2010, 12, 1452–1455 CrossRef CAS.
- C.-S. Chao, Y.-F. Yen, W.-C. Hung and K.-K. T. Mong, Adv. Synth. Catal., 2011, 353, 879–884 CrossRef CAS.
- K. Ruda, J. Lindberg, P. J. Garegg, S. Oscarson and P. Konradsson, J. Am. Chem. Soc., 2000, 122, 11067–11072 CrossRef CAS.
- Compounds 2 and 57 are prepared from a common intermediate but synthesis of 2 requires three additional steps.
- For details on the synthesis of triphosphorylated GPI precursor see ESI.†.
- (a) M. G. Paulick, A. R. Wise, M. B. Forstner, J. T. Groves and C. R. Bertozzi, J. Am. Chem. Soc., 2007, 129, 11543–11550 CrossRef CAS; (b) M. G. Paulick, M. B. Forstner, J. T. Groves and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20332–20337 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental details and spectroscopic data for new compounds. See DOI: 10.1039/c2sc21515b |
| This journal is © The Royal Society of Chemistry 2013 |