Structure and kinetics in the freezing of nearly hard spheres
Jade
Taffs
a,
Stephen R.
Williams
b,
Hajime
Tanaka
c and
C. Patrick
Royall
*a
aSchool of Chemistry, University of Bristol, Bristol, BS8 1TS, UK. E-mail: Paddy.Royall@bristol.ac.uk
bResearch School of Chemistry, Australian National University, Canberra, ACT 0200, Australia
cInstitute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8505, Japan
First published on 18th October 2012
Abstract
We consider homogeneous crystallisation rates in confocal microscopy experiments on colloidal nearly hard spheres at the single particle level. These we compare with Brownian dynamics simulations by carefully modelling the softness in the colloid interactions with a Yukawa potential, which takes account of the electrostatic charges present in the experimental system. Both structure and dynamics of the colloidal fluid are very well matched between experiment and simulation, so we have confidence that the system simulated is close to that in the experiment. In the regimes we can access, we find reasonable agreement in crystallisation rates between experiment and simulations, noting that the larger system size in experiments enables the formation of critical nuclei and hence crystallisation at lower supersaturations than in the simulations. We further examine the metastable fluid with a novel structural analysis, the topological cluster classification. We find that at densities where the hard sphere fluid becomes metastable, the dominant structure is a cluster of m = 10 particles with five-fold symmetry. Analysing histories of the local environment of single particles, we find fluctuations into crystalline configurations in the metastable fluid, and that the crystalline state a very often preceeded by a transition region of frequent hopping between crystal-like environments and other (m ≠ 10) structures.
1 Introduction
Crystallisation is a long-standing challenge, due not least to its local nature, where rare events on microscopic time- and length-scales initiate the phase transition.1 This lack of understanding of crystallisation can have very significant practical consequences, for example in control of drug production.2 It appears challenging to make much progress with conventional materials, due to difficulties in accessing local nucleation events which lead to crystallisation. However, particle-resolved studies of model systems such as colloidal dispersions, which capture the essential thermodynamics, provide the necessary detail required.3Colloidal ‘hard’ spheres are important in the understanding of crystallisation. Few systems have received so much attention, not least because both simulations and experiments can access relevant timescales and particle-level structural lengthscales.4–8 The general phenomenology of hard sphere crystallisation has been well established for a decade:9 at low supersaturations, close to the hard sphere freezing transition at a volume fraction of ϕf = 0.494,10 crystallisation is dominated by rare events leading to the formation of large nuclei. Higher supersaturation results in a very strong rise in nucleation rate, and upon increasing the volume fraction, approaching the hard sphere glass transition, crystallisation has been observed at times less than the structural relaxation time.11,12 At higher volume fractions still (ϕ = 0.62), crystallisation is not seen on the experimental timescale. Despite this phenomenology, very large discrepancies have been found in nucleation rates predicted by simulation using biased ensemble averaging and experiment,4,5,13 which remain unexplained.14,15 Neither the inclusion of polydispersity5,13 nor electrostatic charge16 in the simulations has resolved this situation, although the former has been shown to have profound and complex consequences for nucleation.17,18
The advent of particle-resolved studies3 enables the investigation of local mechanisms of crystallisation in experiment as well as simulation, which could shed light on the discrepancy between simulation and experiment. Charles Frank originally suggested that five-fold symmetric 13-membered icosahedra might suppress crystallisation in the Lennard-Jones system,19 and the structure of various simple liquids has recently been shown to exhibit a high degree of five-fold symmetry,20 which would suppress the process of crystallisation, implying a competition between local order and the global energy minimum (a crystal).21 Recently there has been a resurgence of interest in the role of such local structure in crystallisation. In simulations of hard spheres, five-fold symmetry has been identified both with the suppression of crystallisation22,23 and found at the centre of crystal nuclei.24 Locally dense amorphous crystal precursors have been identified in the metastable hard sphere fluid25 and have also been found in softened systems.26,27 One of us identified a mechanism for crystallisation through increased crystal-like ordering in the fluid prior to the formation of a nucleus, thereby lowering the free energy barrier,28–30 and that this entails no change in local density.31 It was also shown that in weakly size-asymmetric binary hard sphere systems, crystallites can form quickly, but apparently become ‘poisoned’.32As intriguing as these results are, relatively little attention has focussed on the mechanism by which five-fold symmetric structures transform into crystal nuclei.
Pioneering particle-resolved experiments6 identified local structure, and more recent experiments on ‘hard’ spheres too polydisperse to crystallise have shown a degree of fivefold symmetry which, along with local crystalline order, has been related to slow dynamics.33 Here we consider local structure in crystallisation in a particle-resolved colloidal model system. While such experimental studies can in principle resolve mechanisms of crystallisation, quantitative comparison to simulation and theory is very challenging, due to the limited accuracy with which colloidal volume fractions can be measured,34 combined with the lack of control over (and often knowledge of) interparticle interactions upon which crystallisation rates critically depend.16,35,36 Quantitative agreement between experiment and simulation has been obtained in the case of heterogeneous crystallisation of nearly hard spheres, initiated by a wall, where the crystallisation rate is less sensitive to the volume fraction compared to homogenous crystallisation.37
Here we present a careful comparison of experiment and simulation in a system of nearly hard spheres which undergo homogenous nucleation. We interpret our results with a novel structural method, the topological cluster classification (TCC),38–40 which directly identifies a number of local structures. Our mapping between experiment and simulation reveals good agreement in crystallisation rates at the range of supersaturation we accessed. We find that the metastable fluid is dominated by 10-membered fivefold symmetric structures reminiscent of the 13-membered icosahedra proposed long ago as a mechanism for the suppression of crystallisation.19
2 Methods
2.1 Experimental
We used polymethyl methacrylate (PMMA) particles of diameter 2.00 μm with a polydispersity of 4.0% as determined by static light scattering. This degree of polydispersity is insufficient to have much impact on phase behaviour.41 Swelling of the colloids cannot be ruled out in the density and refractive index matching solvent mixture of cis-decalin and cyclohexyl bromide used.34 However, as far as crystallisation is concerned, electrostatic charge, which is not entirely screened by the tetrabutyl ammonium salt added, contributes a further degree of uncertainty in determining the effective colloid volume fraction. If ignored, the effects of electrostatics are quite sufficient to leave measures of crystallisation rates quantitatively meaningless.16,36 For this reason we map simulations carefully to the experiments. We use confocal microscopy (Leica SP5) to track the particle coordinates. Heterogeneous nucleation is prevented by weakly sintering larger (3.5 μm) polydisperse colloids onto the wall of the sample cell. We imaged at least 50 μm from the wall and saw no sign of heterogeneous crystallisation.2.2 Mapping simulation to experiment
Crystallisation experiments are compared with standard Brownian dynamics (BD) simulations, with a system size of N = 2048 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 976 particles and a timestep of 0.1 simulation time units. Fitting of the experimental radial distribution function is carried out using Monte Carlo (MC) simulations with N = 2048 particles. Both the BD and MC simulations are carried out in the canonical (NVT) ensemble. Our particle-resolved experiments enable an innovation: simulations take as their starting point experimental coordinates sampled from a fluid at a time small compared to that required for crystallisation. These are treated with 160 MC sweeps to remove small overlaps resulting from coordinate tracking errors.
976 particles and a timestep of 0.1 simulation time units. Fitting of the experimental radial distribution function is carried out using Monte Carlo (MC) simulations with N = 2048 particles. Both the BD and MC simulations are carried out in the canonical (NVT) ensemble. Our particle-resolved experiments enable an innovation: simulations take as their starting point experimental coordinates sampled from a fluid at a time small compared to that required for crystallisation. These are treated with 160 MC sweeps to remove small overlaps resulting from coordinate tracking errors.
Particle interactions are modelled using a truncated Morse potential with a Yukawa component, which approximate the hard core and electrostatic charging of the colloidal particles respectively.
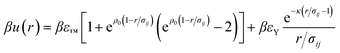 | (1) |
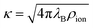 where ρion is the number density of (monovalent) ions.
where ρion is the number density of (monovalent) ions.
We fix the Yukawa parameters to the experimental data. Our approach follows ref. 42 and 43 where the Yukawa interaction parameters βεY and κσ are adjusted such that the experimental radial distribution function is well reproduced by the simulation (Fig. 1). In this case we find κσ = 30.0 ± 5.0 and βεY = 1.0 ± 0.25, which corresponds to a Debye length of 67 nm (or an ionic strength of 1.4 μM) and colloid charge of Z = 200. These are comparable to previous work on similar systems.42–46
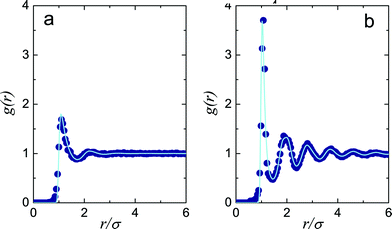 | ||
| Fig. 1 Radial distribution functions in experiment and simulation. (a) Low density, ϕ = 0.27, (b) higher density ϕ = 0.53. In both cases, simulations were carefully fitted to experiment by adjusting charge and volume fraction (see 2.2). Line, simulation data, circles, experimental data. | ||
We treat polydispersity with a Gaussian distribution in σ with 4% standard deviation (the same value as the size polydispersity in the experimental system). The radial distribution function of each experimental state point was fitted for a (metastable) fluid with MC simulation. We then quote the state point in units of ϕ = Vpart/Vbox where the volume of the particles is taken as 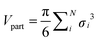 .
.
2.3 Estimating phase boundaries
There are a variety of ways to estimate the freezing transition for a system of weakly repulsive spheres. Among the more accurate36 appears to be to interpolate exact simulation results for hard-core Yukawa systems47 with the hard sphere values. This yields volume fractions for freezing and melting for a weakly charged system. Since we use a slightly softened core here, we estimate the impact of this softening on the phase behaviour by calculating the Barker–Henderson effective hard sphere diameter σeff:48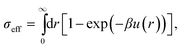 | (2) |
2.4 Structural analysis – topological cluster classification
To analyse the structure, we identify the bond network using a maximum bond length of 1.4σ and a modified Voronoi construction.38 For bond lengths greater than 1.4σ, the network in condensed systems is insensitive to the bond length. Having identified the bond network, we use the Topological Cluster Classification (TCC) to determine the nature of the local environment of each particle.38,40 This analysis identifies all the shortest path three, four and five membered rings in the bond network. We use the TCC to find structures topologically identical to clusters which are global energy minima of the Morse potential for the range we consider (ρ0 = 25.0), as listed in ref. 49 and illustrated in Fig. 2.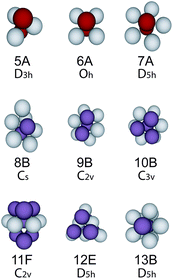 | ||
| Fig. 2 Clusters detected by the topological cluster classification. These structures are minimum energy clusters of the Morse potential with ρ0 = 25.0. We follow the nomenclature of Doye et al.49 where the number represents the number of particles within the cluster. | ||
Now the system we consider interacts not via a full Morse potential, rather our truncation takes the repulsive component only, in a similar spirit to the approach Weeks, Chandler and Andersen used for the Lennard-Jones model.50 While that approach is well-known to reproduce accurately the fluid structure at the pair level, one might expect deviations for higher-order structure such as that probed by the TCC. In fact we found that for short-ranged systems, clustering is enhanced in the case that the attractive part of the potential is removed by truncation.20 Unlike many analyses, for example those which use bond-orientational order parameters,51 our emphasis on bond topology distinguishes between icosahedra and the 13-membered D5h structure illustrated in Fig. 2 which is the minimum energy cluster for the Morse potential with (ρ0 = 25.0). We have also checked for the icosahedron and found only small quantities (≲1%).20 In addition we identify the thirteen particle structures which correspond to FCC and HCP in terms of a central particle and its twelve nearest neighbours. For more details see ref. 38 and 40. If a particle is a member of more than one cluster, we take it to reside in the larger cluster.
3 Results
Our analysis of results is divided into five sections. The first two deal with matching the structure and dynamics of the experimental and simulated systems respectively. We then proceed to discuss our comparison of experimental and simulated data. This is followed by a detailed analysis of structure in crystallisation, which leads us to our final results section, a structural mechanism for crystallisation.3.1 Fluid structure: matching state point
We begin our presentation of results by considering the stable and metastable fluids. For metastable fluids, the cluster populations are sampled from the period prior to crystallisation (see Section 3.3). The topological cluster classification (TCC) analysis shows a considerable increase in cluster populations as a function of volume fraction, as shown in Fig. 3. Some smaller clusters are subsumed into larger clusters for ϕ ≳ 0.55. We see a sharp rise in the fivefold symmetric 10-membered C2v cluster we term ‘10B’ following ref. 49 such that by ϕ ≳ 0.54, it is the dominant cluster in the fluid. We estimate freezing in our nearly hard sphere system at ϕ = 0.487 as noted in Section 2.4. We note that the experimental and simulation data are very well-matched in Fig. 3. This gives us confidence that the fluid structure of the experiments is accurately reproduced in simulation, in other words that the state point is well matched and that we are therefore in a strong position to investigate any possible discrepancies between the experimental and simulated system.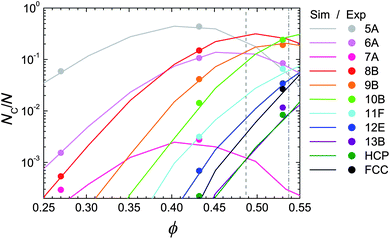 | ||
| Fig. 3 Structural changes upon increasing density in the nearly hard sphere fluid. Lines are simulation, according to eqn (1), circles are experiment. Data for metastable fluids (some of which subsequently crystallise) are taken at times ≪τx. Dashed lines are estimated freezing and melting volume fractions for our system, as described in Section 2.3.47 | ||
3.2 Matching timescales
To account for the different timescales of simulation and experiment, all times are reported in terms of the structural relaxation time, τα, as determined from the intermediate scattering function (ISF), F(k, t) = 〈cos(k·(r(t + t′) − r(t′))2)〉 where the wavevector k is taken at 2π/σ, close to the main peak in the static structure factor. r is the location of each particle at time t and the angle brackets denote a statistical average. An ISF for our experimental system is shown in Fig. 4. We fit the tail of the ISF with a stretched exponential of the form F(k, t) = Cexp[(−t/τα)b] to obtain the structural relaxation time τα. For the experimental system at ϕ = 0.43, we obtain C = 0.99, τα = 31 s, and b = 0.81. We repeat the ISF fitting for simulations across a range of densities. τα from simulation is then plotted as a function of ϕ in Fig. 5. The data are well described by a Vogel–Fulcher–Tammann (VFT) relation τα = τ0exp[D/(ϕ0 − ϕ)] where τ0 is a relaxation time in the normal fluid, D is the ‘fragility index’, and ϕ0 ≈ 0.62 is the ideal glass transition volume fraction.30 From fitting, we obtain a value of τ0 = 416 and D = 0.275. We assume the scaling of the dynamics at this volume fraction is the same for both experiment and simulation, and equate the experimental and simulation structural relaxation times at ϕ = 0.43. Thus both experiment and simulation τα are taken from the VFT fit (Fig. 5).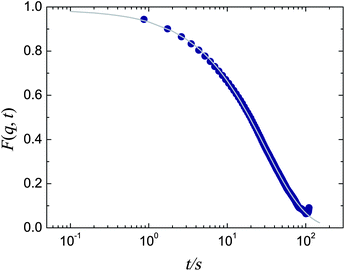 | ||
| Fig. 4 Intermediate scattering function for experimental data at ϕ = 0.43. The wavevector is taken at 2π/σ, close to the first peak in the static structure factor. Grey line is a stretched exponential fit (see text for details). | ||
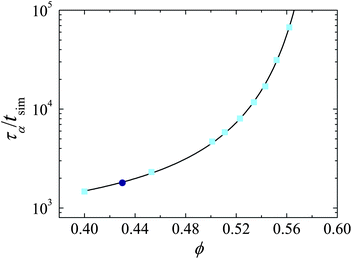 | ||
| Fig. 5 Structural relaxation time in terms of simulation time steps (tsim) as a function of ϕ. Light blue squares are simulation, dark blue circle scaled experiment. Solid line is a Vogel–Fulcher–Tammann fit to simulation data (see text for details). | ||
3.3 Comparision between experiment and simulation
The process of crystallisation is shown in Fig. 6. Here ϕ = 0.54. Note that some small regions of high local order are present at t = 0. We have previously demonstrated that even stable fluids have populations of particles in crystalline environments.20 As shown in Fig. 3, this rises markedly around the freezing transition. These images suggest that the metastable fluid shows relatively little change for 28τα ≤ t, but crystallisation subsequently occurs at 28τα ≤ τx ≤ 317τα. Henceforth we define τx as the time at which 40% of the particles are identified in crystalline environments. Moderate changes to this threshold have no impact upon our conclusions. | ||
| Fig. 6 Confocal microscopy images of crystallisation in nearly hard spheres for ϕ = 0.54. (a) 600 s (2.3 τα), (b) 4500 s (17.4 τα), (c) 7200 s (27.9 τα) and (d) 81900 s (316.9 τα), bar = 10 μm. | ||
We now compare crystallisation times in simulation and experiment. Recall that nucleation of ‘hard’ spheres is found to exhibit strong deviations between experiment and simulation.4,14 We compare crystallisation times as shown in Fig. 7. We see a reasonable agreement for moderate values of ϕ ≳ 0.56, but at lower supersaturation ϕ ≲ 0.55 or ϕ − ϕm ≲ 0.01, we find an emergent discrepancy between experiment and simulation. While no mapping between experiment and simulation is perfect,34,36 our careful analysis of state point and timescale leads us to believe that this discrepancy is not accounted for by a shift of ϕ.
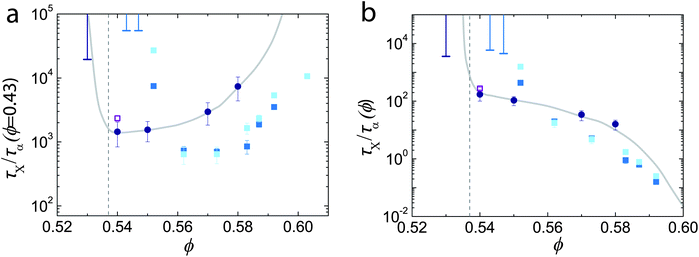 | ||
| Fig. 7 Crystallisation times in terms of τα (ϕ = 0.43) (a) and τα (ϕ) (b). Circles are experimental data, light and dark squares are simulation data for polydisperse systems of N = 2048 and N = 10976 respectively. Unfilled square is for a monodisperse system with N = 10976. Dashed lines are melting estimated as described in Section 2.3. Solid lines are to guide the eye. Error bars extending upwards are lower bounds for crystallisation times determined from experiments (light lines) and simulations (dark lines) which did not crystallise. | ||
Now neither the simulations we employ here, nor the confocal microscopy experiments access the regime of low supersaturation where the formation of a large nucleus is a rare event. In simulation, biasing techniques can be employed, and while similar methodologies are in principle possible in experiment52 the kind of precision required to determine nucleation rates quantitatively remains some way off. An important point then, is that unbiased simulation and confocal microscopy experiment access similar regimes of supersaturation. However the experiment has a rather larger system size than does the simulation. The simulation box size is typically 2000 and 10000σ3 for the N = 2048 and 10976 system sizes respectively, while the experiments are confined in capillaries of size 250σ × 250σ × 2500σ ≈ 1.6 × 108σ3. The imaging volume (50σ × 50σ × 25σ ≈ 6.3 × 105σ3) is rather smaller than the whole system and crystals can nucleate outside this region (Fig. 6). The rate of crystal growth has recently been determined in a very similar system,37 and the associated timescales are of order 100–1000τα, suggesting that for long times, crystals can spread throughout the sample, so the relevant volume is that of the entire system, rather than just the imaging volume. Thus, decreasing the supersaturation to ϕ − ϕm ≈ 0.005, we see that experiments continue to crystallise, but for simulations, the time for crystallisation moves outside the accessible timescale. Note that, at higher supersaturation, nucleation rates increase strongly, so the crystallisation time τx is somewhat independent of system size.
While the argument presented above is physically attractive, we seek a more quantitative validation, and for this we turn to the results of Filion et al.14 Clearly, for the system to crystallise, at least one nucleation event must occur. We take the nucleation rate for the highest volume fraction in ref. 14, for a monodisperse system J = 1.4 × 10−5σ3τB where τB is the time to diffuse a diameter in the dilute limit and is equal to 0.63τα (ϕ = 0.43). The corresponding volume fraction relative to melting is ϕ − ϕm = 0.0051. For our system, this corresponds to one nucleation event every 4.0 and 21.6 τα (ϕ = 0.43) for N = 10976 and N = 2048 respectively. In other words, in our simulation timescales, we expect crystallisation.
However, the rate of nucleation is highly sensitive to polydispersity,5,13 which is 4% here. Recall this is expected to have little effect on the equilibrium phase diagram.41 To the best of our knowledge, no precise predictions for nucleation rates in polydisperse systems have been made for the regime we access (ϕ ≳ ϕm). Previous work shows that the impact of a small (≳ 5%) polydispersity on nucleation rate is equivalent to a reduction in volume fraction of ∼0.015 for a monodisperse system.5,13
We note that the shape of the particle size distribution, rather than just its second moment, can be important,17,18 however here we shall assume that the effect of polydispersity is to effect a shift in ϕ of 0.015 in the nucleation rate. In other words, we take the rate for ϕ − ϕm = −0.01 for a monodisperse system to apply for ϕ − ϕm = 0.005 for our system. The nucleation rate at ϕ − ϕm = −0.01 is four orders of magnitude lower than that at ϕ − ϕm = 0.005.14 At such low rates, we expect no nucleation in simulation, but the larger system size in experiment (coincidentally four orders of magnitude larger than the simulation) is sufficient for nucleation to occur on our timescales, as is consistent with the crystallisation that we see.
If this analysis is correct, for a monodisperse system at ϕ − ϕm = 0.005 we should expect a much higher nucleation rate, and crystallisation on the simulation timescale. To verify this point, we carried out some simulations with a monodisperse system. These are shown in Fig. 7a and b, and indeed crystallise in the regime of interest. We thus conclude that, in the regime we access, the discrepancy between experiment and simulation is likely due to the larger system size in the case of the experiments.
3.4 Structure in crystallisation
We now turn to a TCC analysis of the crystallisation process. Fig. 8 shows the population in each TCC cluster as a function of time for experiment (a) and simulation (b). In both experiment and simulation, we identify three regimes. For t ≲ 10τα in the experiment and t ≲ 40τα in the simulation there is little change in cluster populations. At intermediate times approaching τx (here τx = 107 and 64τα for this experiment and simulation respectively), we see a steady growth in particles identified in crystalline environments (predominantly FCC) at the expense of particles in fluid environments. Most notable of the non-crystalline clusters is 10B, whose population drops continuously throughout this period. At the bulk level, crystallisation is thus interpreted as the conversion of 10B clusters into FCC environments. However, crystallisation is a local phenomenon, which we discuss in the next section. Note that at times larger than τx, there is a further decrease in non-crystalline clusters. On the timescale of these experiments and simulations, a reasonable population (a few percent) of non-crystalline clusters remain at all times.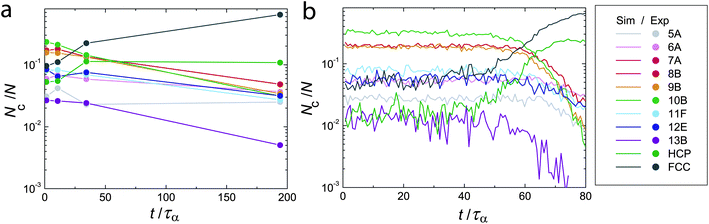 | ||
| Fig. 8 Topological cluster classification analysis of crystallisation, experimental (a) and simulation data (b). Here ϕ = 0.55. | ||
3.5 Particle-level crystallisation mechanism
We have observed that the metastable fluid is dominated by the fivefold symmetric 10B cluster. Since 10B cannot tile space, there must be some local structural transformation associated with crystallisation. We can gain insight by considering the history of a single particle. In Fig. 9, we show the history of four particles, throughout a simulation for ϕ = 0.55. We see that each particle fluctuates between different structures and is identified in a number of different structures, including local crystalline environments. Three regimes emerge: the metastable fluid, dominated by the 10B cluster, the final crystal, and a transition regime between the two. These three regimes are defined as follows: metastable fluid refers to a regime where the rate of visitation to a locally crystalline environment is less than τα−1. We sample a snapshot every 0.1 τα,† and find a rate of visitation to a crystalline environment to be 0.7 ± 0.2 τα−1. The crystalline regime is defined where particles do not leave a crystalline environment for more than τα, and the transition regime lies between these two as indicated in Fig. 9. Note that situations where a particle transitions directly from the metastable fluid to the crystal with no intermediate regime do occur, although infrequently (with a probability of 0.14). In the intermediate regime, the rate of visitation to a crystalline environment is 7 ± 2 τα−1. The duration of this intermediate transition regime is ∼2 τα. This shows there are multiple pathways to crystallisation at the particle level.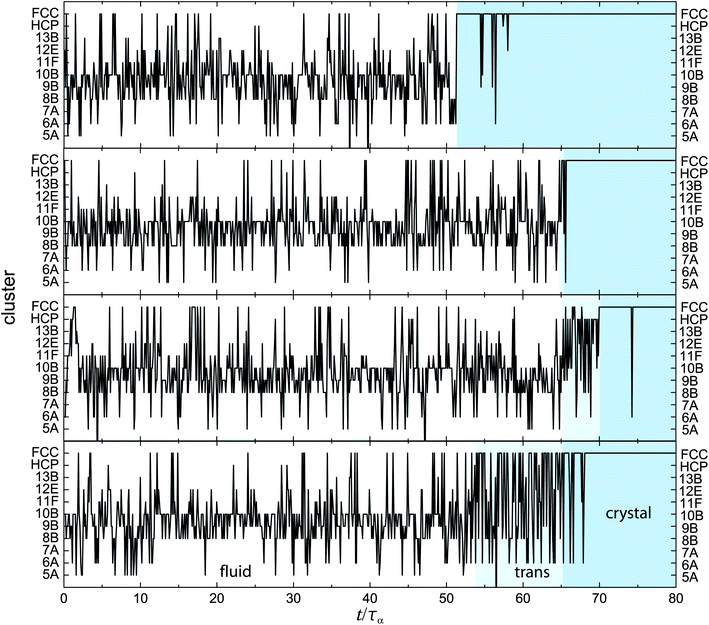 | ||
| Fig. 9 Histories of four particles at ϕ = 0.55. Shaded areas mark the different regimes of fluid, transition and crystal, as described in the text. Data are shown from Brownian dynamics simulations. Here the timespan of the transition is 0, 0.50, 5.19, 11.1 τα from top to bottom. | ||
Note that in the transition regime, although the particle is often found in amorphous structures, these are rarely 10B. This suggests that 10B-crystal transitions may be somehow suppressed. This is consistent with long-standing ideas that locally favoured five-fold symmetry can suppress crystallisation19 and very recent experimental33 work which suggests frustration between five-fold symmetry and local crystalline order.21 We would thus expect that 10B clusters are rather stable (see below). During growth, however, it is possible that a crystalline surface may disrupt the five-fold symmetry in the fluid, leading to more rapid transformation between 10B and crystalline structures. At a coarse-grained level, it was shown that crystal nucleation occurs in regions of high crystal-like bond orientational order.28 Thus, the presence of the stable transition regime for a particle may reflect that it is involved in a critical crystal nucleus.
We close by considering the stability of the fivefold symmetric 10B cluster. This seems to dominate the metastable fluid at densities where crystallisation occurs. In Fig. 10 we show transition probabilities from the 10B cluster to various geometries. We see there is a tendency to remain in the 10B cluster. That is, the 10B cluster shows a higher degree of stability than other clusters. In other words, for the nearly hard sphere fluid, the 10B is a locally favoured structure, similar to icosahedra and related polyhedra in glass-forming systems.51,53–57 This is consistent with previous reports that five-fold symmetry is favoured in ‘hard’ spheres.22,23,33 The fact that this ordering tendency to structures of five-fold character such as 10B appears to be enhanced at such high ϕ is intriguing: as the volume fraction is increased further, ‘spinodal’ crystallisation takes place in a small fraction of τα as found previously.11,12 In such unstable fluids, however, we cannot measure the lifetime of 10B clusters: the stability of 10B is exceeded by the thermodynamic driving force of crystal nucleation.
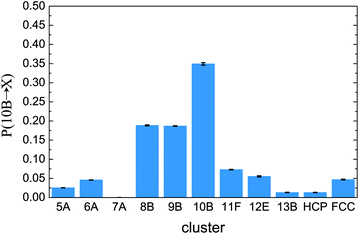 | ||
| Fig. 10 Transitions from the 10B cluster for ϕ = 0.55 in the metastable fluid. These are the probability for a particle to be found in a cluster at time t + τt, given that it was in a 10B cluster at t. Here τt = 0.08τα. Data are shown from Brownian dynamics simulations. | ||
4 Conclusions
We have carefully matched simulation to experiment for a nearly hard sphere system. For the regimes in which we can access crystallisation, the kinetics are similar in both simulation and experiment with the exception that, at lower volume fraction, experiments crystallise faster than simulations. We believe this is associated with the onset of low nucleation rates as the supersaturation is decreased. Under these conditions, the larger experimental system size means nucleation events occur on accessible timescales, enabling crystallisation to be observed in experiments but not in simulations. While accurate simulation data has been obtained for monodisperse systems across a wide range of ϕ, the effect of polydispersity characteristic of colloidal experiments has only been considered for supersaturations where nucleation rates were too low to be accessed by confocal microscopy experiments. In other words, for the regime of supersaturation we access, no evidence is found of a discrepancy in nucleation rate between experiment and simulation. In order to be confident that no discrepancy exists, predictions for polydisperse systems in the regime accessible to experiments such as ours would be helpful.Our topological cluster classification reveals insight into the mechanism of crystallisation. In particular, around the freezing transition, both experiment and simulation show that nearly hard sphere fluids become dominated by a five-fold symmetric ten-membered cluster which we term 10B. By considering particle histories, we find that transitions between this 10B cluster and crystalline environments are suppressed. Instead, after some time in a metastable fluid state, with occasional excursions to a crystalline environment, which usually occur through an intermediate structure, the particle often finds itself in a transition state, presumeably due to the proximity of a crystalline region which stabilises local crystalline environments. In the transition state, the particle spends large amounts of time in a crystalline environment, and little time in a 10B cluster, instead it is found in other amorphous clusters. Eventually, the particle spends all its time in a local crystalline environment and is said to be crystalline. This behaviour may be related to the ‘cloud’ identified in the case of softened particles.26 We note that the rather stable transition regime may also be related to the presence of long-lived crystal-like bond orientational order.28,30,33 Some particles which do not experience the transition regime may be those suddenly involved in a crystal when the crystal growth front passes through them. These indicate multiple pathways to crystallisation at the particle level.
At high supersaturations, the structural relaxation time exceeds the crystallisation time, “spinodal crystallisation”. Under such conditions, the crystallisation time may be shorter than the 10B lifetime (or, its formation time) and the system may be no longer in the regime of competition between fivefold symmetry and crystallisation.
Finally, we emphasise that, since absolutely hard spheres are not found in nature,36 it is essential to take account of the inherent softness in any experimental system. However, comparison with true hard spheres suggests that the main effect of the softness we have considered is to shift the state point such that we must consider effective volume fractions. This is consistent with previous observations that mapping the effective packing fraction to hard spheres results in a practically identical fluid structure.20
Acknowledgements
It is a pleasure to thank Kurt Binder, Bob Evans, Daan Frenkel, Rob Jack, Mathieu Leocmach, Alex Malins, Thomas Palberg, John Russo, Richard Sear, and Frank Schrieber for stimulating discussions and Monica Moreno for preliminary simulations and experiments. C.P.R. gratefully acknowledges the Royal Society for financial support and EPSRC grant code EP/H022333/1. H.T. acknowledges a grant-in-aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan and Aihara Project, the FIRST program from JSPS, initiated by CSTP.References
- R. P. Sear, J. Phys.: Condens. Matter, 2007, 19, 033101 CrossRef.
- S. L. Morissette, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 2180 CrossRef CAS.
- A. Ivlev, H. Loewen, G. E. Morfill and C. P. Royall, Complex Plasmas and Colloidal Dispersions: Particle-Resolved Studies of Classical Liquids and Solids, World Scientific, 2012 Search PubMed.
- S. Auer and D. Frenkel, Nature, 2001, 409, 1020–1023 CrossRef CAS.
- S. Auer and D. Frenkel, Nature, 2001, 413, 711–713 CrossRef CAS.
- U. Gasser, E. R. Weeks, A. Schofield, P. N. Pusey and D. A. Weitz, Science, 2001, 292, 258–262 CrossRef CAS.
- D. W. Aastuen, N. A. Clark, L. K. Cotter and B. J. Ackerson, Phys. Rev. Lett., 1986, 57, 1733–1736 CrossRef CAS.
- J. L. Harland and W. van Megen, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1997, 55, 3054–3067 CrossRef CAS.
- T. Palberg, J. Phys.: Condens. Matter, 1999, 11, R323–R360 CrossRef CAS.
- W. G. Hoover and F. Ree, J. Chem. Phys., 1968, 49, 3609–3617 CrossRef CAS.
- E. Zaccarelli, C. Valeriani, E. Sanz, W. C. K. Poon, M. E. Cates and P. N. Pusey, Phys. Rev. Lett., 2009, 103, 135704 CrossRef CAS.
- E. Sanz, C. Valeriani, W. C. K. Zaccarelli, E. Poon, P. N. Pusey and M. E. Cates, Phys. Rev. Lett., 2011, 106, 215701 CrossRef.
- S. Auer and D. Frenkel, Annu. Rev. Phys. Chem., 2004, 55, 333–361 CrossRef CAS.
- L. Filion, R. Ni, D. Frenkel and M. Dijkstra, J. Chem. Phys., 2011, 134, 134901 CrossRef CAS.
- T. Schilling, S. Dorosz, H. J. Schoepe and G. Opletal, J. Phys.: Condens. Matter, 2011, 23, 194120 CrossRef CAS.
- S. Auer and D. Frenkel, J. Phys.: Condens. Matter, 2002, 14, 7667–7680 CrossRef CAS.
- H. J. Schope, G. Bryant and W. van Megen, Phys. Rev. Lett., 2006, 96, 175701 CrossRef.
- H. J. Schope, G. Bryant and W. van Megen, J. Chem. Phys., 2007, 127, 084505 CrossRef.
- F. C. Frank, Proc. R. Soc. London, Ser. A, 1952, 215, 43–46 CrossRef CAS.
- J. Taffs, A. Malins, S. R. Williams and C. P. Royall, J. Chem. Phys., 2010, 133, 244901 CrossRef.
- H. Tanaka, J. Phys.: Condens. Matter, 1998, 10, L207–L214 Search PubMed.
- N. C. Karayiannis, R. Malshe, J. J. de Pablo and M. Laso, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 061505 CrossRef.
- N. C. Karayiannis, R. Malshe, M. Kröger, J. J. de Pablo and M. Laso, Soft Matter, 2012, 8, 844–858 RSC.
- B. O'Malley and I. Snook, Phys. Rev. Lett., 2003, 90, 085702 CrossRef.
- T. Schilling, H. J. Schöpe, M. Oettel, G. Opletal and I. Snook, Phys. Rev. Lett., 2010, 105, 025701 CrossRef CAS.
- W. Lechner, C. Dellago and P. G. Bolhuis, Phys. Rev. Lett., 2011, 106, 085701 CrossRef.
- J. Russo and H. Tanaka, Soft Matter, 2012, 8, 4206–4215 RSC.
- T. Kawasaki and H. Tanaka, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14036–14041 CrossRef CAS.
- T. Kawasaki and H. Tanaka, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 156335 Search PubMed.
- K. Kawasaki and H. Tanaka, J. Phys.: Condens. Matter, 2010, 22, 232102 CrossRef.
- J. Russo and H. Tanaka, Sci. Rep., 2012, 2, 505, DOI:10.1038/srep00505.
- S. R. Williams, C. P. Royall and G. Bryant, Phys. Rev. Lett., 2008, 100, 225502 CrossRef.
- M. Leocmach and H. Tanaka, Nat. Commun., 2012, 3, 974, DOI:10.1038/ncomms1974.
- W. C. K. Poon, E. R. Weeks and C. P. Royall, Soft Matter, 2012, 8, 21–30 RSC.
- S. Auer, W. C.-K. Poon and D. Frenkel, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 67, 020401(R) Search PubMed.
- C. P. Royall, W. C. K. Poon and E. R. Weeks, Soft Matter, 2012 Search PubMed , arxiv preprint arXiv: 1205.6665.
- K. Sandomirski, H. Allahyarov, E. Loewen and S. U. Egelhaaf, Soft Matter, 2011, 7, 8050 RSC.
- S. R. Williams, 2007, arxiv preprint arXiv: 0705.0203v1.
- C. P. Royall, S. R. Williams, T. Ohtsuka and H. Tanaka, Nat. Mater., 2008, 7, 556–561 CrossRef CAS.
- A. Malins, PhD Thesis, A Structural Approach to Glassy Systems, University of Bristol, 2012.
- P. Sollich and N. B. Wilding, Phys. Rev. Lett., 2010, 104, 118302 CrossRef.
- C. P. Royall, M. E. Leunissen and A. van Blaaderen, J. Phys.: Condens. Matter, 2003, 15, S3581–S3596 CrossRef CAS.
- C. P. Royall, M. E. Leunissen, A.-P. Hyninnen, M. Dijkstra and A. van Blaaderen, J. Chem. Phys., 2006, 124, 244706 CrossRef.
- A. Yetiraj and A. van Blaaderen, Nature, 2003, 421, 513–517 CrossRef CAS.
- C. P. Royall, A. A. Louis and H. Tanaka, J. Chem. Phys., 2007, 127, 044507 CrossRef.
- A. I. Campbell, V. J. Anderson, J. S. van Duijneveldt and P. Bartlett, Phys. Rev. Lett., 2005, 94, 208301 CrossRef.
- A. P. Hynninen and M. Dijkstra, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 68, 021407 CrossRef.
- J. A. Barker and D. Henderson, Rev. Mod. Phys., 1976, 48, 587–671 CrossRef CAS.
- J. P. K. Doye, D. J. Wales and R. S. Berry, J. Chem. Phys., 1995, 103, 4234–4249 CrossRef CAS.
- J. D. Weeks, D. Chandler and H. C. Andersen, J. Chem. Phys., 1971, 54, 5237–5247 CrossRef CAS.
- P. J. Steinhardt, D. R. Nelson and M. Ronchetti, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 784–805 CrossRef CAS.
- M. Hermes, E. C. M. Vermolen, M. E. Leunissen, D. L. J. Vossen, P. D. J. van Oostrum, M. Dijkstram and A. van Blaaderen, Soft Matter, 2011, 7, 4623 RSC.
- H. Jonsson and H. C. Andersen, Phys. Rev. Lett., 1988, 60, 2295–2298 CrossRef CAS.
- T. Tomida and T. Egami, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 3290–3308 Search PubMed.
- M. Dzugutov, S. I. Simdyankin and F. H. M. Zetterling, Phys. Rev. Lett., 2002, 89, 195701 CrossRef.
- D. Coslovich and G. Pastore, J. Chem. Phys., 2007, 127, 124504 Search PubMed.
- A. Malins, J. Eggers, C. P. Royall, S. R. Williams and H. Tanaka, 2012, arxiv preprint arXiv: 1203.1732.
Footnote |
| † The absolute values for these transitions are sensitive to the sampling rate. However that local crystalline environments are sampled very much more frequently in the transition regime than in the metastable fluid holds for reasonable sampling rates. Likewise the time the particle spends in each regime does not vary with sampling rate. |
| This journal is © The Royal Society of Chemistry 2013 |