Sensitive and selective electrochemical detection of chromium(VI) based on gold nanoparticle-decorated titania nanotube arrays
Wei
Jin
,
Guosheng
Wu
and
Aicheng
Chen
*
Department of Chemistry, Lakehead University, 955 Oliver Road, Thunder Bay, ON P7B 5E1, Canada. E-mail: aicheng.chen@lakeheadu.ca; Fax: +1 807 346 7775; Tel: +1 807 343 8318
First published on 16th October 2013
Abstract
Owing to the severe toxicity and mobility of Cr(VI) in biological and environmental systems, it is of great importance to develop convenient and reliable methods for its detection. Here we report on a facile and effective electrochemical technique for monitoring Cr(VI) concentrations based on the utilization of Au nanoparticle-decorated titania nanotubes (TiO2NTs) grown on a titanium substrate. It was found that the electrochemical reduction of Cr(VI) at the Ti/TiO2NT/Au electrode exhibited an almost 23 fold improvement in activity as compared to a polycrystalline gold electrode, due to its nanoparticle/nanotubular heterojunction infrastructure. As a result, the Ti/TiO2NT/Au electrode demonstrated a wide linear concentration range from 0.10 μM to 105 μM, a low detection limit of 0.03 μM, and a high sensitivity of 6.91 μA μM−1 Cr(VI) via amperometry, satisfying the detection requirements of the World Health Organization (WHO). Moreover, the Ti/TiO2NT/Au electrode exhibited good resistance against interference from coexisting Cr(III) and other metal ions, and excellent recovery for Cr(VI) detection in both tap and lake water samples. These attributes suggest that this hybrid sensor has strong potential in applications for the selective detection of Cr(VI).
1. Introduction
Due to the large volume of Cr compounds that are discharged into the environment from various industrial processes such as mineral processing,1 pigments2 and electroplating,3 the Cr speciation in terrestrial and aquatic systems has attracted considerable attention over the last several years. The two stable Cr oxidation states are Cr(III) and Cr(VI), which have significantly different physicochemical properties and toxicities.4 Cr(III) compounds are considered to be harmless; indeed trace amounts of this element are required for proper metabolism in living organisms,5 whereas Cr(VI) compounds exert extreme adverse effects on biochemical systems.6 Cr(VI) may be easily reduced to Cr(III) with the formation of reactive free radicals within the cell, leading to the inhibition of the metallo-enzyme system,7 therefore Cr(VI) has been classified as human carcinogen. Furthermore, Cr(VI) compounds are highly soluble and mobile in both biological and natural ambient systems.8 In the groundwater system of USA, up to 220 μg L−1 (4.2 μM) naturally occurring Cr(VI) have been reported.9 In order to protect the human life and environment from Cr(VI) toxicity, a provisional limit of 50 μg L−1 (0.96 μM) in groundwater was set by World Health Organization (WHO).10Considerable efforts have been devoted to the selective detection of Cr(VI) for environmental applications, encompassing spectroscopic, chromatographic and electrochemical methods. Normally, coexisting Cr(III) concentrations are from 10 to 1000 times higher than Cr(VI) concentrations in test samples,11 which results in serious interference for the detection of Cr(VI). Therefore, prior separation, complexing or reaction is required for certain analytical techniques, such as inductively coupled plasma atomic emission (ICP-AES),12,13 UV-vis spectroscopy,14,15 high-performance liquid chromatography (HPLC),16 atomic absorption spectroscopy,17,18 fluorescence19,20 and voltammetry,21,22 leading to the disadvantages of complicated and time-consuming procedures.
In contrast, because the electrochemical reduction potentials of Cr(III) and Cr(VI) are distinct,10 they offer a facile and selective pathway for Cr(VI) detection. The electrochemical detection of Cr(VI) has been studied at the pyridine modified glass carbon electrode,23 poly-L-histidine film modified screen-printed carbon electrode24 and 4-(mercapto-n-alkyl)pyridinium monolayer modified gold electrode.25 However, there is a preconcentration or accumulation step that is required prior to electrochemical analysis. Consequently, the Ag nanoparticle based electrode,26,27 boron diamond electrode (BDD),10,28 graphite screen printed electrode29 and Prussian blue modified glass carbon electrode30 have been successfully employed to directly detect the Cr(VI) concentration, while the Cr(VI) linear concentration ranges at these electrodes are relatively narrow. Welch and Compton10 demonstrated that Cr(VI) can be electrochemically detected directly at a polycrystalline Au electrode by cyclic voltammetry with a LoD (limit of detection) of 4.3 μM Cr(VI) in the presence of 5 mM Cr(III), and a Cr(VI) linear concentration range of 100–1500 μM was obtained as compared to 20–200 μM Cr(VI) at BDD electrode. Most recently, Metters and Banks31 achieved a wider linear range (10–1600 μM) for the detection of Cr(VI) with a LoD of 4.4 μM using a gold screen printed macro-electrode. Subsequently, due to the attractive physicochemical and catalytic properties of nanometric noble metal particles, gold nanoparticle (Au NP) modified electrodes have been developed in an attempt to detect Cr(VI) concentrations. Dominguez-Renedo32 developed the Au NP modified carbon screen-printed electrode with a LoD of 0.4 μM and a linearity of 0.4–30 μM. Jena33 developed an Au NP-modified silicate network electrode with a LoD of 0.002 μM and a linearity of 0.004–0.057 μM. Ouyang34 identified a LoD of 5.6 × 10−5 μM and a linearity of 2 × 10−4 to 2 × 10−2 μM was obtained at flower-like Au NP on a glass carbon electrode. Clearly, as compared to the polycrystalline Au electrode, the detection limit of the Au/NP electrode for Cr(VI) was significantly improved owing to the superior conduction capability and high surface-to-volume ratio of the gold nanoparticles.35 However, the linear concentration range was substantially decreased, inhibiting the practical applications. Therefore, it is highly desirable to develop an electrode system with satisfactory Cr(VI) detection limit and wide linear concentration range.
It has been demonstrated that the nanoparticle support also plays a critical role in the performance of electroanalysis.36–38 Since Zwilling39 developed a fabrication method for the synthesis of highly ordered titania nanotubes (TiO2NTs) utilizing the anodic oxidation of Ti in 1999, there has been a widespread application of TiO2 nanotubes in electrochemistry and photocatalysis, due to their semiconducting nature, insertion host capacity and superior chemical stability.40 For the same loading of Au NPs, a TiO2 nanotubular support provides a significant enhancement in electrochemical activity towards oxygen reduction in comparison with a flat TiO2 substrate or a pure Au electrode.41 Although the photocatalytic activity of Au/TiO2 nanotube arrays for the reduction of Cr(VI) has been reported,42 no investigation of their electrochemical performance for the detection or reduction of Cr(VI) has yet been carried out. In this regard, the aim of this study is to develop the Au nanoparticle decorated titania nanotube electrode for the electrochemical detection of Cr(VI), which may offer a facile and reliable Cr(VI) quantification technique with wide linear range, high sensitivity and good LoD.
2. Experimental
2.1 Reagents and apparatus
The reagents used in this study included dimethyl sulfoxide (≥99.5 wt%), hydrofluoric acid (48 wt% in H2O), gold(III) chloride hydrate (99.8 wt%), methanol (99.8 wt%), hydrochloric acid (37 wt% in H2O), chromium(III) nitrate (99 wt%) and iron(III) nitrate (≥98 wt%), which were of analytic purity grade and used as received, from Sigma-Aldrich. Pure water (18 MΩ cm) was obtained from a NANO pure® Diamond™ UV ultrapure water purification system. The electrolytes used in this investigation were prepared by dissolving the corresponding chemicals in ultrapure water.The anodic oxidation was carried out with a DC power supply using a dual-electrode system, while other electrochemical experiments were performed with a CHI (660B) workstation using a three-electrode system. The photo-assisted deposition was performed using a BlueWave TM 50 AS UV spot lamp (365 nm emission) with an intensity of ca. 20 mW cm−2. The surface morphology and composition of the prepared electrodes were characterized using a Hitachi SU-70 Field emission scanning electron microscope (FE-SEM) with an X-ray energy dispersive spectrometer (EDS). X-ray diffraction (XRD) was recorded using an X'pert PANalytical Diffractometer with Cu Kα radiation.
2.2 Preparation of the Ti/TiO2NT/Au electrode
The Ti/TiO2NT/Au electrodes were fabricated via a two-step process involving anodic oxidation followed by photo-assisted chemical deposition.43–45 The Ti substrates (1 cm2) were cleaned by sonication in acetone for 10 min followed by 10 min in distilled water, and then etched in 18% HCl at approximately 85 °C for 20 min. After being rinsed with pure water and dried in a N2 stream, the etched Ti substrate was anodized in a dimethyl sulfoxide (DMSO) + 2% (wt) HF solution, whereas the counter electrode was a platinum foil. Anodization was carried out at 40 V for 7 h, after which the sample was annealed at 450 °C for 3 h to obtain an anatase structure. For the deposition of Au NPs, the TiO2 nanotube electrode was placed in deaerated 5 mL of 50% (v/v) methanol solution containing 0.2 mM HAuCl4, after which photo-assisted deposition was performed under UV light illumination for 10 min. Finally, the electrode was rinsed with pure water and dried in a vacuum oven at 40 °C.2.3 Electrochemical reduction and detection of Cr(VI)
The fabricated Ti/TiO2NT/Au electrode and polycrystalline gold were employed as the working electrodes, while the counter and reference electrodes were a Pt foil and an Ag/AgCl (KCl saturated) electrode, respectively. For the cyclic voltammetry, the working electrode was cycled in stationary solution with the corresponding scan rate and voltage range. Amperometric measurements were performed in a stirred solution with an applied potential of 0.28 V.All electrolytes were deaerated with Ar for 30 min prior to each measurement and then maintained under an Ar atmosphere for the duration of the measurements at room temperature (25 °C). For the measurement of actual samples, tap water was obtained directly from our laboratory without treatment, while the lake water was collected from a local lake located in Lakehead University and filtered through a 0.45 μm filter prior to measurement. Both the tap and lake water samples were acidified by the dropwise addition of a 1 M HCl solution with rapid stirring, until a pH of 1 was attained.
3. Results and discussion
3.1 Characterization of the Ti/TiO2NT/Au electrode
The electrochemical performance of the electrode was expected to depend upon the morphology and structure of the substrate. As shown in Fig. 1, a self-organized array of TiO2 nanotubes was grown directly on a Ti substrate via anodic anodization. The uniform diameter of these nanotubes was ∼120 nm, with a wall thickness of ∼60 nm and a length of ∼3–4 μm, which were estimated from the cross-section of the SEM image (data not shown). The EDS of Fig. 2a displayed strong Ti and O peaks, and the corresponding XRD peaks as shown in Fig. 2b were attributed to tetragonal anatase, which confirmed that the formed nanotubes were anatase TiO2. As shown in Fig. 1, the gold nanoparticles (∼10 nm in diameter) were well dispersed on the as prepared TiO2NTs, which was further confirmed by the corresponding EDS and XRD peaks in Fig. 2a and b, respectively. It should be noted that a small population of ∼30 nm diameter Au nanoparticles were presented on the slightly longer nanotubes due to the aggregate nature of the Au NPs.46 Clearly, the formed TiO2NTs were highly ordered with open top architectures and high surface areas; and the well-dispersed Au nanoparticles on the TiO2NTs would result in a metal–semiconductor heterojunction with potentially high electrochemical activity. | ||
| Fig. 1 SEM image of the as prepared Au nanoparticle decorated TiO2 nanotube electrode. | ||
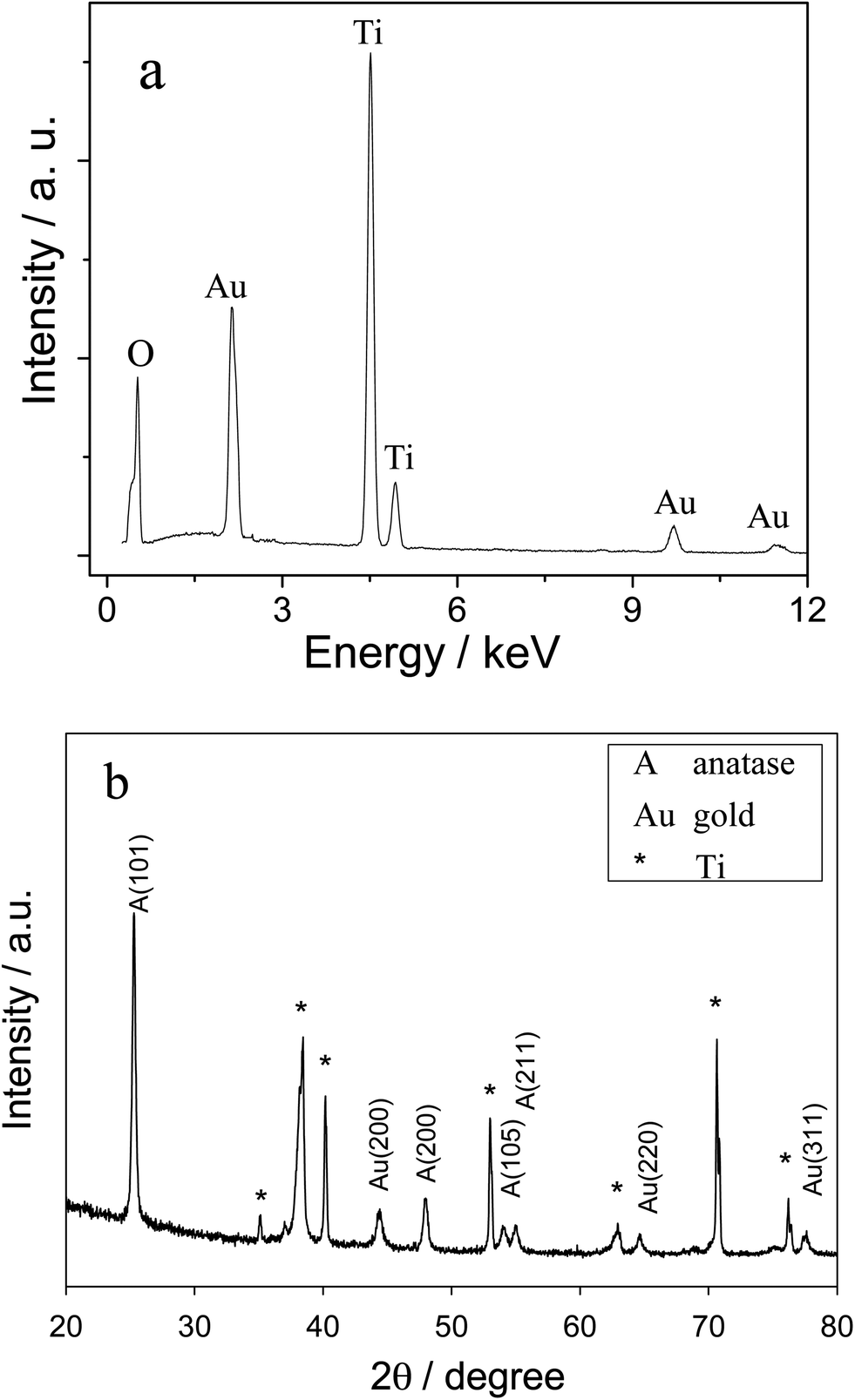 | ||
| Fig. 2 (a) EDX and (b) XRD spectra of the Ti/TiO2NT/Au electrode. | ||
3.2 Electrochemical reduction of Cr(VI)
As shown by Banks and co-workers,29,31 the choice of the acidic electrolytes plays some roles in the accurate detection of Cr(VI). Due to the fact that chloride ions are commonly present in real samples, the HCl electrolyte was employed in this study. In order to characterize the performance of the Ti/TiO2NT/Au electrode toward the electrochemical reduction of Cr(VI), cyclic voltammetric (CV) measurements were performed in a 0.1 M HCl solution. As shown in Fig. 3a, a well-defined reduction peak occurred at 0.28 V with a current density of 545 μA cm−2 in the presence of 0.1 mM Cr(VI). No reduction peak was observed in the same potential region of its corresponding blank CV curve in the absence of Cr(VI), showing that this peak was attributed to the Cr(VI) reduction. In addition, there was no corresponding oxidation peak in the reverse scan of the CV curve with Cr(VI), suggesting an electrochemically irreversible process. As a comparison, the CV curves at a polycrystalline gold electrode were also recorded, as shown in Fig. 3b, presenting the irreversible Cr(VI) reduction peak at 0.27 V with a current density of 23 μA cm−2. Clearly, the Cr(VI) reduction at the Ti/TiO2NT/Au electrodes had a more positive peak potential and a nearly 23 fold improvement of peak current, indicating the significant beneficial effect of the TiO2NT/Au NP heterojunction structure on the electrochemical reduction of Cr(VI). This considerable enhancement might be attributed to the high surface area of the nanotubular support,47 and the augmented diffusion of electroactive species to the Au nanoparticles.48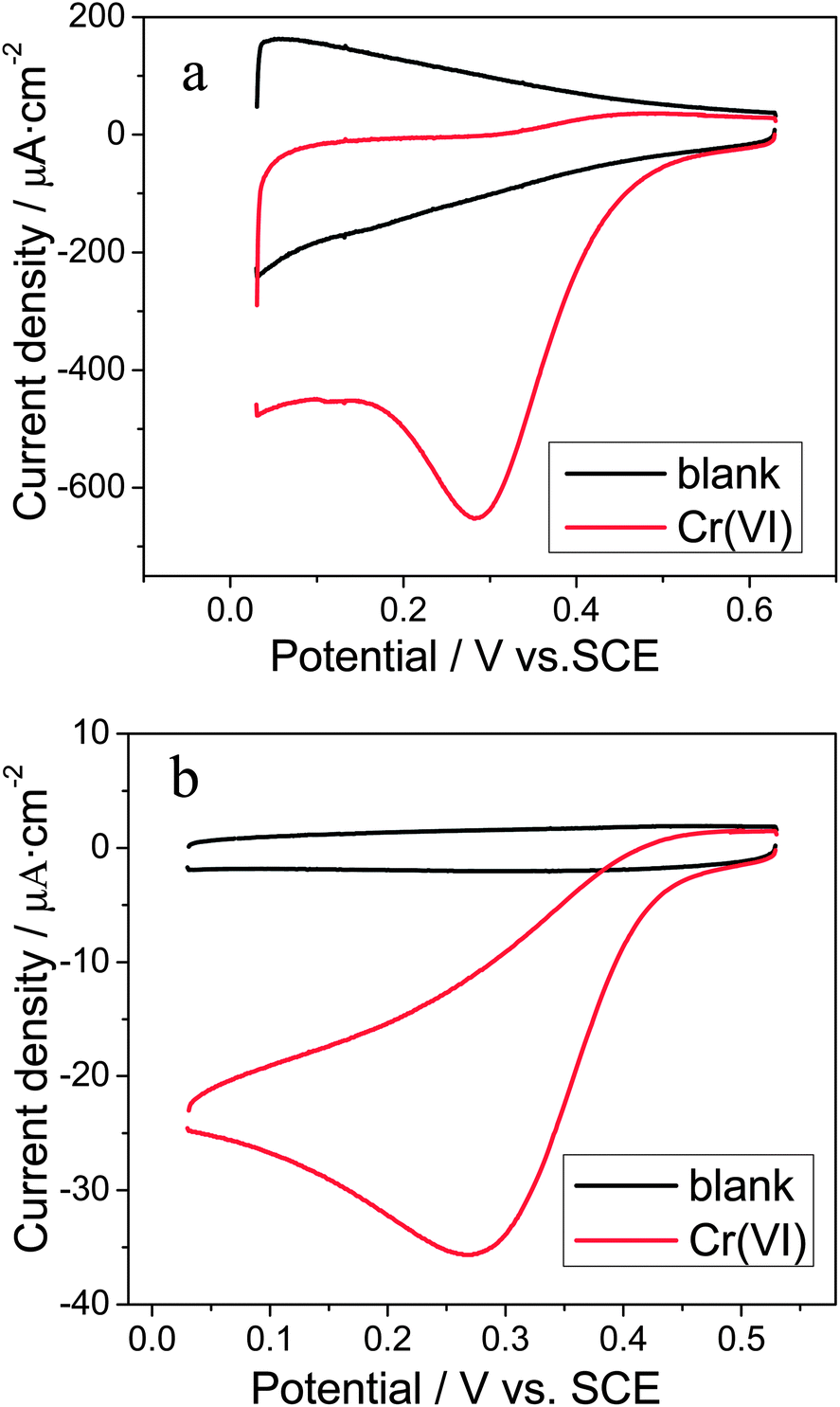 | ||
| Fig. 3 Cyclic voltammograms recorded at (a) Ti/TiO2NT/Au, (b) polycrystalline gold in a 0.1 M HCl solution in the absence and presence of 0.1 mM Cr(VI) at a scan rate of 50 mV s−1. | ||
Moreover, the CV curves at different scan rates (from 10 to 150 mV s−1) were recorded in a 0.1 mM Cr(VI) + 0.1 M HCl solution to determine the reaction kinetics. As shown in Fig. 4a, the Cr(VI) reduction peak potential (Epc) negatively shifted from 0.295 V at 10 mV s−1 to 0.211 V at 150 mV s−1, which is consistent with the characteristics of an irreversible reaction.49 Additionally, a linear relationship can be seen between the Cr(VI) reduction peak current (ipc) and the square root of the scan rate, as shown in Fig. 4b, indicating diffusion controlled reaction kinetics.50 Therefore, the Cr(VI) reduction activity is expected to be determined primarily by the mass transport of electroactive species. The Au/TiO2NT heterojunction infrastructure presented a porous surface and linear channels51 decorated with electroactive gold nanoparticles, which allowed for the free exchange of reaction intermediates, and resulted in significantly enhanced Cr(VI) reduction activity.
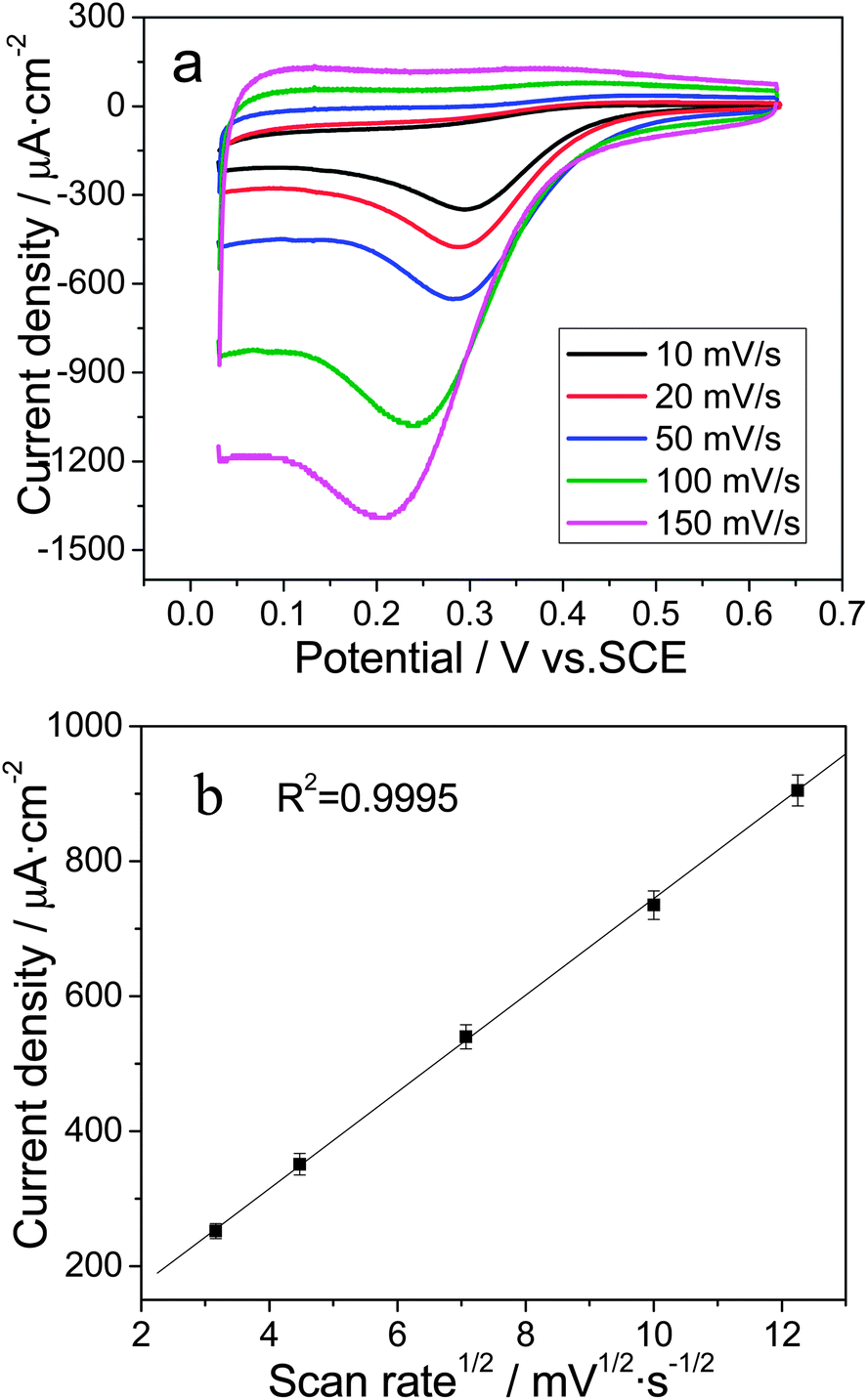 | ||
| Fig. 4 (a) Cyclic voltammograms of 0.1 mM Cr(VI) at the Ti/TiO2NT/Au electrode with scan rates of 10, 20, 50, 100 and 150 mV s−1 in a 0.1 M HCl solution; (b) the corresponding plot of the Cr(VI) reduction peak current vs. the square root of the scan rate (n = 3). | ||
For further mechanistic insight, the influence of pH on the reduction of Cr(VI) was investigated in HCl solutions of different concentrations, while the Cr(VI) concentration remained constant at 0.1 mM. As can be seen in Fig. 5, with the decreased concentration of HCl, the Cr(VI) reduction peak current was significantly reduced from 545 μA cm−2 in a 0.1 M HCl solution to 230 μA cm−2 in a 0.01 M HCl solution, and the peak potential negatively shifted from 0.28 V in a 0.1 M HCl solution to 0.15 V in a 0.01 M HCl solution. Thereafter, the Cr(VI) reduction peak disappeared in the 0.001 M HCl solution, indicating that this reaction was largely dependent on the H+, as illustrated in eqn (1):
| HCrO4− + 7H+ + 3e− → Cr3+ + 4H2O | (1) |
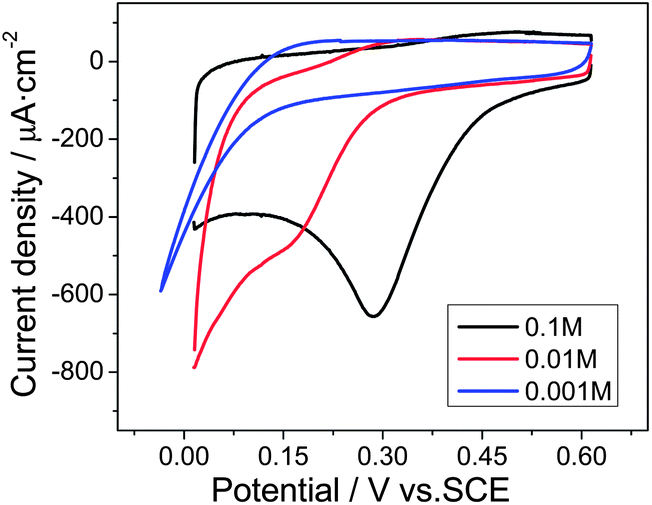 | ||
| Fig. 5 Cyclic voltammograms of 0.1 mM Cr(VI) at the Ti/TiO2NT/Au electrode in 0.001, 0.01 and 0.1 M HCl solutions at a scan rate of 50 mV s−1. | ||
This suggested that the pH of the supporting electrolyte played an important role in the electrochemical detection of Cr(VI). Therefore, the 0.1 M HCl solution was utilized for the following analytical investigations.
3.3 Amperometric detection of Cr(VI)
The analytical performance of the Ti/TiO2NT/Au electrode towards Cr(VI) was evaluated using an amperometry technique, while the potential was set at a constant value of 0.28 V, which is the peak potential observed in Fig. 3a. As shown in Fig. 6a, a rapid response was exhibited as a result of the successive addition of 0.1, 2.5 and 10 μM Cr(VI) in a 0.1 M HCl solution. More importantly, there was an excellent linear relationship range, from 0.10 to 105 μM Cr(VI), with a correlation coefficient of R2 = 0.9997, as illustrated in Fig. 6b. However, the current response deviated below the correlated slope when the Cr(VI) was further increased to above 105 μM, and became saturated at 160 μM Cr(VI), which was likely due to the accumulation of reduction intermediates on the electrode surface. The detection limit of the Ti/TiO2NT/Au electrode toward Cr(VI) was estimated to be 0.03 μM (S/N = 3) and its corresponding % Relative Standard Deviation (RSD) was found to be 3.2% (n = 3), which satisfied the detection requirement of the WHO standard.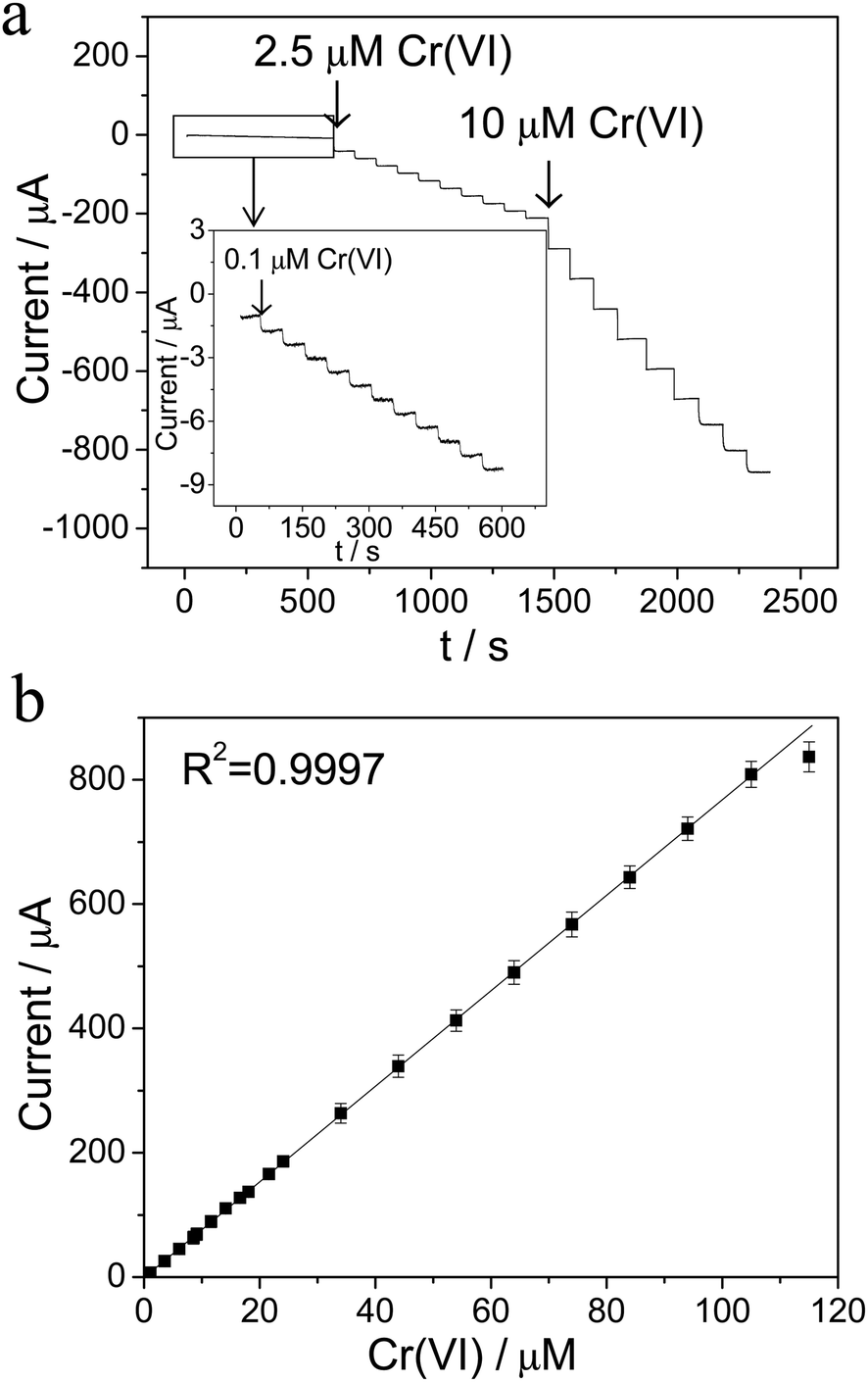 | ||
| Fig. 6 (a) Amperometric current responses of the Ti/TiO2NT/Au electrode as the result of the successive addition of Cr(VI) at the electrode potential of 0.28 V in a 0.1 M HCl solution. The inset is the enlarged amperometric responses of the low Cr(VI) concentration area (marked by a black rectangle); (b) the calibration plot of the response current against the Cr(VI) concentration (n = 3). | ||
As compared to previous results of gold-based electrodes10,32–34,52,53 for the detection of Cr(VI) in Table 1, the Ti/TiO2NT/Au electrode exhibited an improved performance, and in particular, the highest sensitivity (6.91 μA μM−1) which may be attributed to the enhanced catalytic activity of the diminutive gold nanoparticles in combination with the high surface area of the Ti/TiO2NT/Au heterojunction infrastructure. Moreover, our proposed method possesses the advantages of a wide linear range, a satisfied detection limit, convenient detection procedures and simple electrode preparation, which fulfill the requirements for potential practical applications.
| Electrode | Method | Linear range (μM) | Detection limit (μM) | Sensitivity (μA μM−1) | Ref. |
|---|---|---|---|---|---|
| Gold screen printed macro-electrode | Linear sweep voltammetry | 10–1600 | 4.4 | 0.05 | 31 |
| Polycrystalline gold electrode | Cyclic voltammetry | 100–1500 | 4.3 | N/A | 10 |
| Au NP on indium tin oxide | Cyclic voltammetry | 5–100 | 2 | 0.30 | 53 |
| Au NP modified carbon screen printed electrode | Differential pulse voltammetry | 0.4–30 | 0.4 | N/A | 32 |
| Flower-like Au NP on glass carbon electrode | Accumulation followed by stripping square voltammetry | 2 × 10−4 to 2 × 10−2 | 5.6 × 10−5 | N/A | 34 |
| Au NP modified glassy carbon | Accumulation followed by square wave voltammetry | 2.5 × 10−3 to 0.86 | 2 × 10−4 | N/A | 52 |
| Au NP on indium tin oxide | Amperometry | 0.5–50 | 0.1 | 0.35 | 53 |
| Au NP on thiol functionalized silicate network | Amperometry | 4 × 10−3 to 5.7 × 10−2 | 2 × 10−3 | 1.56 | 33 |
| Au NP on TiO2NTs | Amperometry | 0.1–105 | 0.03 | 6.91 | This study |
3.4 Investigation of the possible interference of Cr(III) and other ions
It is of great importance for an electrochemical sensor to discriminate between target analytes, i.e. Cr(VI), from other potential interfering species. As mentioned above, Cr(III) concentrations are typically from 10 to 1000 times higher than Cr(VI) concentrations in actual samples, which may result in serious interference for the detection of Cr(VI). To facilitate the evaluation of the selective detection of Cr(VI) using the fabricated Ti/TiO2NT/Au electrode in the presence of interfering Cr(III), amperometric measurements were carried out at 0.28 V. Initially, 10 μM Cr(VI) was injected into a stirred 0.1 M HCl supportive electrolyte, generating a 70 μA current response as shown in Fig. 7, which is consistent with the corresponding value seen in Fig. 6a. Thereafter, a 30-fold excess of Cr(III) was added, with no obvious change in the amperometric response observed, showing a high resistance of the Ti/TiO2NT/Au electrode against the Cr(III) interference. As Fe(III) is the common ion that is present in groundwater, 10 μM Fe(III) was also added to the test solution, whereupon the amperometric current did not change. Moreover, the subsequent addition of 10 μM Cr(VI) generated an almost identical amperometric response as the initial 10 μM Cr(VI) injection, further confirming the high interference resistance of the Ti/TiO2NT/Au electrode for the detection of Cr(VI).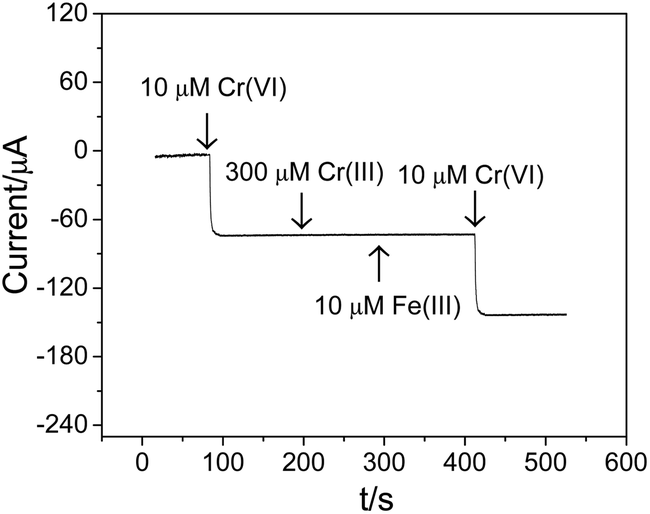 | ||
| Fig. 7 Interference of Cr(III) and Fe(III) on the detection of Cr(VI) at the Ti/TiO2NT/Au electrode in a 0.1 M HCl solution. | ||
To evaluate its practical applications, the Ti/TiO2NT/Au electrode was employed to detect Cr(VI) concentrations in tap and lake water samples. The trace Cr content of the acidified tap and lake water samples was 1.1 ppb (∼0.02 μM) and 0.5 ppb (∼0.01 μM) based on ICP-AES analysis, respectively, which are below the electrochemical detection limit. Consequently, a certain amount of Cr(VI) was spiked into each actual sample to determine the recovery using the proposed electrochemical detection method. As shown in Table 2, the recovery was from 96% to 99% for the tap water samples and from 94% to 97.8% for the lake water samples, respectively. Furthermore, the Cr(VI) was also accurately determined using ICP-AES, as Cr(VI) was the only Cr species present in the samples. Clearly, the results obtained via the electrochemical method are in good agreement with those derived from the ICP-AES analysis, indicating that the developed electrochemical Ti/TiO2NT/Au sensor shows promise for practical implications for the detection of Cr(VI) in environmental systems.
| Sample | Added (μM) | Using the method developed in this study | By ICP-AES (μM) | ||
|---|---|---|---|---|---|
| Found | Recovery (%) | RSD (%) | |||
| Tap water | 0.5 | 0.48 ± 0.02 | 96.0 ± 4.0 | 3.2 | 0.50 |
| 1.0 | 0.99 ± 0.03 | 99.0 ± 3.0 | 2.7 | 0.99 | |
| 5.0 | 4.90 ± 0.23 | 98.0 ± 4.6 | 4.2 | 4.97 | |
| Lake water | 0.5 | 0.47 ± 0.03 | 94.0 ± 6.0 | 5.1 | 0.49 |
| 1.0 | 0.96 ± 0.04 | 96.0 ± 4.0 | 3.4 | 0.98 | |
| 5.0 | 4.89 ± 0.21 | 97.8 ± 4.2 | 3.7 | 4.93 | |
4. Conclusions
A novel Ti/TiO2NT/Au electrode was fabricated via the anodic oxidation of a Ti substrate and photo-assisted chemical deposition of Au nanoparticles on the formed TiO2 nanotube arrays, and its application for the sensitive detection of Cr(VI) was investigated by cyclic voltammetry and amperometry. The electrochemical reduction of Cr(VI) at the Ti/TiO2NT/Au electrode demonstrated a nearly 23 fold improvement of peak current as compared to a polycrystalline gold electrode, due to the nanoparticle/nanotubular heterojunction infrastructure. As a result, a wide linear concentration range from 0.10 to 105 μM, a low detection limit of 0.03 μM, and a high sensitivity of 6.91 μA μM−1 Cr(VI) were achieved, which satisfy the detection requirements of the WHO. Moreover, the Ti/TiO2NT/Au electrode exhibited high resistance against interference from coexisting Cr(III) and other ions, and good recovery was obtained for both the tap and lake water samples. These results suggest that the electrochemical sensor developed in this study has strong potential for the detection of Cr(VI) in environmental applications.Acknowledgements
This work was supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (NSERC). A. Chen acknowledges NSERC and the Canada Foundation of Innovation (CFI) for the Canada Research Chair Award in Material and Environmental Chemistry.References
- C. Arslan and D. F. Duby, Hydrometallurgy, 1997, 46, 46337–46348 Search PubMed.
- M. Kendig, S. Jeanjaquet, R. Addison and J. Waldrop, Surf. Coat. Technol., 2001, 140, 58–66 CrossRef CAS.
- W. Zhang, L. Zhuang, L. Tong, I. M. C. Lo and R. Qiu, Chemosphere, 2012, 86, 809–816 CrossRef CAS PubMed.
- L. E. Eary and D. Rai, Environ. Sci. Technol., 1987, 21, 1187–1193 CrossRef.
- D. Rai, B. M. Sass and D. A. Moore, Inorg. Chem., 1987, 26, 345–349 CrossRef CAS.
- M. Cespon-Romero, M. C. Yebru-Biurru and M. P. Bermejo-Barrera, Anal. Chim. Acta, 1996, 327, 37–45 CrossRef.
- K. P. Lee, C. E. Ulrich, R. G. Geil and H. J. Trochimowicz, Sci. Total Environ., 1989, 86, 83–108 CrossRef CAS.
- J. Kotas and Z. Stasicka, Environ. Pollut., 2000, 107, 263–283 CrossRef CAS.
- State of the Science of Hexavalent Chromium in Drinking Water, Water Research Foundation, 2012.
- C. M. Welch, O. Nekrassova and R. G. Compton, Talanta, 2005, 65, 74–80 CAS.
- K. Ndungu, N.-K. Djane, F. Malcus and L. Mathiasson, Analyst, 1999, 124, 1367–1372 RSC.
- Y.-J. Lai and W.-L. Tseng, Analyst, 2011, 136, 2712–2717 RSC.
- A. G. Cox, I. G. Cook and C. W. McLeod, Analyst, 1985, 110, 331–333 RSC.
- G. Y. Jung, Y. S. Kim and H. B. Lim, Anal. Sci., 1997, 13, 463–467 CrossRef CAS.
- A. Krushevska, A. Waheed, J. Nobrega, D. Amarisiriwardena and R. M. Barnes, Appl. Spectrosc., 1998, 52, 205–211 CrossRef CAS.
- J. Posta, A. Alimonti, F. Petrucci and S. Caroli, Anal. Chim. Acta, 1996, 325, 185–193 CrossRef CAS.
- R. Millacic and J. Stupar, Analyst, 1994, 119, 627–632 RSC.
- M. Gardner and S. Comber, Analyst, 2002, 127, 153–156 RSC.
- H. Zhang, Q. Liu, T. Wang, Z. Yun, G. Li, J. Liu and G. Jiang, Anal. Chim. Acta, 2013, 770, 140–146 CrossRef CAS PubMed.
- Y. Xiang, L. Mei, N. Li and A. Tong, Anal. Chim. Acta, 2007, 581, 132–136 CrossRef CAS PubMed.
- Y. Li and H. Xue, Anal. Chim. Acta, 2001, 448, 121–134 CrossRef CAS.
- J. Li, J. Zhang, H. Wei and E. Wang, Analyst, 2009, 134, 273–277 RSC.
- N. A. Carrington, L. Yong and Z.-L. Xue, Anal. Chim. Acta, 2006, 572, 17–24 CrossRef CAS PubMed.
- M. F. Bergamini, D. P. dos Santos and M. V. B. Zanoni, Sens. Actuators, B, 2007, 123, 902–908 CrossRef CAS PubMed.
- I. Turyan and D. Mandler, Anal. Chem., 1997, 69, 894–897 CrossRef CAS PubMed.
- S. Xing, H. Xu, J. Chen, G. Shi and L. Jin, J. Electroanal. Chem., 2011, 652, 60–65 CrossRef CAS PubMed.
- H. Xu, Q.-L. Zheng, P. Yang, J.-S. Liu, S.-J. Xing and L.-T. Jin, Sci. China: Chem., 2011, 54, 1001–1010 Search PubMed.
- S. Fierro, T. Watanabe, K. Akai and Y. Einaga, Electrochim. Acta, 2012, 82, 9–11 CrossRef CAS PubMed.
- P. M. Hallam, D. K. Kampouris, R. O. Kadara and C. E. Banks, Analyst, 2010, 135, 1947–1952 RSC.
- S. Xing, H. Xu, G. Shi, J. Chen, L. Zeng and L. Jin, Electroanalysis, 2009, 21, 1678–1684 CrossRef CAS.
- J. P. Metters, R. O. Kadara and C. E. Banks, Analyst, 2012, 137, 896–902 RSC.
- O. Dominguez-Renedo, L. Rauiz-Espelt, N. Garcia-Astorgano and M. J. Arcos-Martinez, Talanta, 2008, 76, 854–858 CrossRef CAS PubMed.
- B. K. Jena and C. R. Raj, Talanta, 2008, 76, 161–165 CrossRef CAS PubMed.
- R. Ouyang, S. A. Bragg, J. Q. Chambers and Z.-L. Xue, Anal. Chim. Acta, 2012, 722, 1–7 CrossRef CAS PubMed.
- J. Zhao, X. Zhu, T. Li and G. Li, Analyst, 2008, 133, 1242–1245 RSC.
- S. Ranganathan and R. L. McCreery, Anal. Chem., 2001, 73, 893–900 CrossRef CAS.
- A. Chen and S. Chatterjee, Chem. Soc. Rev., 2013, 42, 5425–5438 RSC.
- A. Chen and B. Shah, Anal. Methods, 2013, 5, 2158–2173 RSC.
- V. Zwilling, E. Darque-Ceretti and A. Boutry-Forveille, Electrochim. Acta, 1999, 45, 921–929 CrossRef CAS.
- S. Chen, M. Malig, M. Tian and A. Chen, J. Phys. Chem. C, 2012, 116, 3298–3304 CAS.
- J. M. Macak, F. Schmidt-Stein and P. Schmuki, Electrochem. Commun., 2007, 9, 1783–1787 CrossRef CAS PubMed.
- S. Luo, Y. Xiao, L. Yang, C. Liu, F. Su, Y. Li, Q. Cai and G. Zeng, Sep. Purif. Technol., 2011, 79, 85–91 CrossRef CAS PubMed.
- M. Tian, S. S. Thind, S. Chen, N. Matyasovzsky and A. Chen, Electrochem. Commun., 2011, 13, 1186–1189 CrossRef CAS PubMed.
- M. Tian, M. Malig, S. Chen and A. Chen, Electrochem. Commun., 2011, 13, 370–373 CrossRef CAS PubMed.
- T. Hezard, K. Fajerwerg, D. Evrard, V. Colliere, P. Behra and P. Gros, Electrochim. Acta, 2012, 73, 15–22 CrossRef CAS PubMed.
- J. Nam, N. Won, H. Jin, H. Chung and S. Kim, J. Am. Chem. Soc., 2009, 131, 13639–13645 CrossRef CAS PubMed.
- A. K. M. Kafi, G. Wu and A. Chen, Biosens. Bioelectron., 2008, 24, 566–571 CrossRef CAS PubMed.
- M. Oyama, Anal. Sci., 2010, 26, 1–12 CrossRef CAS.
- W. Jin, M. S. Moats, S. Zheng, H. Du, Y. Zhang and J. D. Miller, Electrochim. Acta, 2011, 56, 8311–8318 CrossRef CAS PubMed.
- W. Jin, H. Du, S. Zheng, H. Xu and Y. Zhang, J. Phys. Chem. B, 2010, 114, 6542–6548 CrossRef CAS PubMed.
- Z. Liu, X. Zhang, S. Nishimoto, T. Murakami and A. Fujishima, Environ. Sci. Technol., 2008, 42, 8547–8551 CrossRef CAS.
- M.-C. Tsai and P.-Y. Chen, Talanta, 2008, 76, 533–539 CrossRef CAS PubMed.
- B. Liu, L. Lu, M. Wang and Y. Zi, J. Chem. Sci., 2008, 120, 493–498 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |