 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAltered selectivity of a dipicolylamine based metal ion receptor†
Joydev
Hatai
and
Subhajit
Bandyopadhyay
*
Indian Institute of Science Education and Research (IISER) Kolkata, BCKV Main Campus PO, Mohanpur, Nadia WB 741252, India. E-mail: sb1@iiserkol.ac.in
First published on 6th November 2013
Abstract
2,2-Dipicolylamine (DPA) based receptors detect Zn2+ with high fidelity. A fluorescent sensor with thiocarbamate-linked DPA turns it into a highly selective module for Ag+ with up to ∼7-fold enhancement of fluorescence. The sensor exhibits a double ratiometric behavior. It also displays an interesting [Ag+] dependent aggregation induced emission.
The 2,2-dipicolylamine (DPA) unit has been the most widely used receptor for Zn2+ ions since the discovery of the Zynpyr family of fluorescent probes by the Lippard group.1 The high selectivity of the receptor was extensively studied in conjugation with various fluorophores in order to use it in aqueous media both for cell imaging and detection of Zn2+.2,3 Yoon and Spring's group has discovered that the DPA-amide system binds to Zn2+ in a unique imidic acid tautomeric form, whereas all the other metal ions bind in the amide form.4 We have frequently made use of the thiocarbamate linker to prepare a repertoire of fluorophore–spacer–receptor triads for the detection and imaging of various heavy metal ions.5 Intrigued by the unparallel fidelity and high sensitivity of DPA-amide based sensors for the Zn2+ ion, we have linked a DPA unit to a simple fluorophore through a thiocarbamate linker (Fig. 1) and studied its sensing behaviour. Here we show that the selectivity of the DPA-amide receptor is altered as a consequence of modifying the functionality: in contrast to Zn2+ selectivity of the DPA-amide unit, the DPA-thiocarbamate acts as a Ag+ selective receptor. Despite its simple structure, the response of the sensor molecule to a varying amount of Ag+ was rather curious.
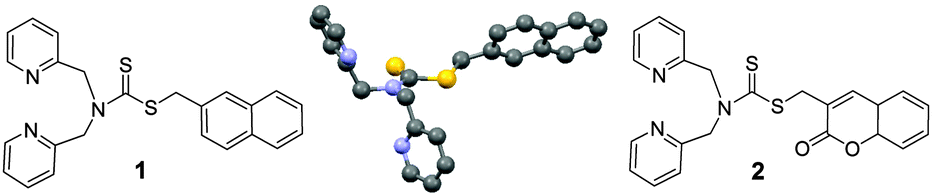 | ||
| Fig. 1 Structures of 1, 2 and X-ray crystal structure of 1. | ||
Combining results from steady-state fluorescence, UV, NMR, dynamic light scattering, lifetime, AFM and SEM experiments, the reason for this behavior was attributed to Ag+ mediated aggregation and its depletion upon addition of higher equivalents of Ag+.
Chemosensor 1 was obtained in 82% yield following our method for the synthesis of thiocarbamates starting from dipicolylamine, carbon disulphide and 2-bromomethyl naphthalene in a water–dioxane (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solvent mixture.5 Details of the synthesis and characterization data, including a summary of crystallographic parameters, are provided in the ESI† (Fig. S15).
:1) solvent mixture.5 Details of the synthesis and characterization data, including a summary of crystallographic parameters, are provided in the ESI† (Fig. S15).
The emission spectra of sensor 1 (λex = 350 nm, Fig. S1, ESI†) were recorded systematically under various conditions (Table 1). In methanol with 1% DMSO as a cosolvent, it exhibited a weak emission band (quantum yield, Φ = 0.04) at 495 nm (Fig. S2, ESI†).
In water (with 1% DMSO as the cosolvent, 1 mM HEPES buffered at pH = 7.0), the band shifted to 500 nm (Fig. 2D). In water–methanol (H2O–MeOH, 3:2, v/v) (1 mM, pH = 7.0, HEPES) the intensity of the band at 495 nm was found to be the highest among the varying ratios of H2O and MeOH (Fig. S7, ESI†).
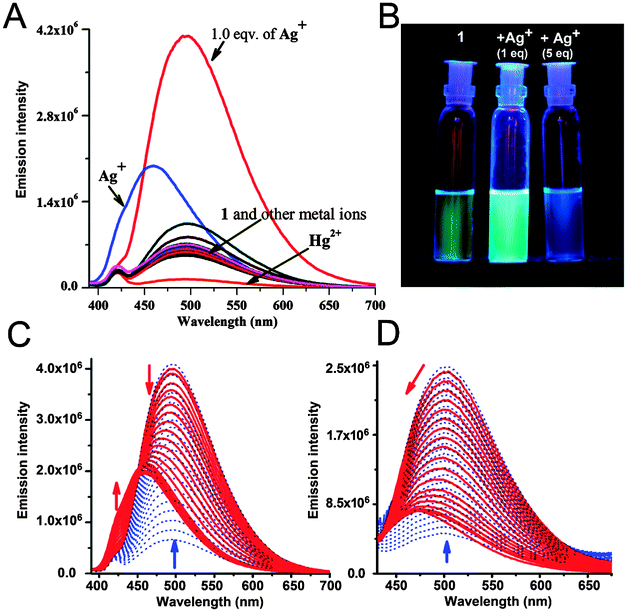 | ||
| Fig. 2 (A) Change in fluorescence intensity of 1 (10 μM) upon addition of various metal ions (10 μM). The spectrum with Ag+ in blue refers to the change upon addition of 50 μM Ag+ in H2O–MeOH, 3:2, v/v; (B) visual change under 366 nm UV light. Change in fluorescence emission of 1 (10 μM) in (C) H2O–MeOH (3:2, v/v) and (D) water (99%) upon addition of 0–1 equivalents (blue) and 1–5 equivalents of Ag+ (red). | ||
The metal-sensing ability of chemosensor 1 (10 μM) was investigated based on its fluorescence properties in the presence of Na+, K+, Li+, Mg2+, Co2+, Ni2+, Cu2+, Mn2+, Cr3+, Fe3+, Pb2+, Ag+, Al3+, Hg2+, Ba2+ and Zn2+ (10 μM) salts. Initial screening with MeOH as the solvent gave a response with a 2.5 fold enhancement of the fluorescence signal (Φ = 0.11, Fig. S2, ESI†) upon addition of Ag+. In a mixed aqueous–organic solvent (H2O–MeOH, 3:2, v/v with 1% DMSO) (Table 1) the enhancement was maximum. Upon addition of the metal ions (10 μM) to the receptor 1 (10 μM), a 6.5 fold enhancement of intensity was observed (Φ = 0.52) at 495 nm only with Ag+ (Fig. 2A). This selectivity for Ag+ remained unaltered in water (99%). Among the other metal ions, Hg2+ quenched the emission intensity whereas Pb2+ showed a small enhancement of fluorescence with 1 (Fig. 2A). A discernible change was also visible with the naked eye under 366 nm UV light (Fig. 2B).
To gain a better insight into the fluorescence behavior, a series of experiments were carried out with a fixed concentration of chemosensor 1 (10 μM) and a varying amount of Ag+ in H2O–MeOH (3:2, v/v) for which the fluorescence response was maximum. The systematic variation of the amount of Ag+ displayed a sharp enhancement of fluorescence intensity at 495 nm upon addition of up to 1.0 equivalent of Ag+ ions, whereas at higher concentrations of Ag+ the emission intensity of 1 at 495 nm decreased significantly and gradually underwent a blue shift to 460 nm (Fig. 2C and Fig. S3A and S3B, ESI†). Also, a new emission band centered at 424 nm was observed with a new isoemission point at 450 nm, indicating a dual-ratiometric fluorescence behavior of the sensor with Ag+ (Fig. S3C, ESI†). Upon addition of 5.0 equivalents (50 μM) of the metal ions, only Ag+ displayed a 3 fold (Φ = 0.25) enhancement of fluorescence (Fig. 2A and C and Fig. S3A, ESI†).
This interesting behavior of the sensor at higher concentration of Ag+ prompted us to explore the mechanism of the fluorescence enhancement. The time dependence of the fluorescence intensity of chemosensor 1 (10 μM) was recorded. It was found that even in the absence of any metal ions the plot of fluorescence intensity recorded at regular intervals slowly increased with time and reached a saturation level in both mixed aqueous–organic (Fig. S5, ESI†) and aqueous media (Fig. S6, ESI†). The maximum enhancement, found in 60% water (Fig. 3D and Fig. S7, ESI†), was similar to the behavior observed for a number of rotor-like fluorescent probes studied by Tang and prompted us to hypothesize that aggregation might play a role in the enhancement of fluorescence intensity in our case as well.6 It should be noted that in all the systems where aggregation induced enhancement (AIE) of fluorescence is reported, the presence of a rotor-like biaryl framework is a common feature6 and the restriction of low energy molecular motions such as rotation of the aryl groups in the aggregated form is responsible for the enhanced emission.
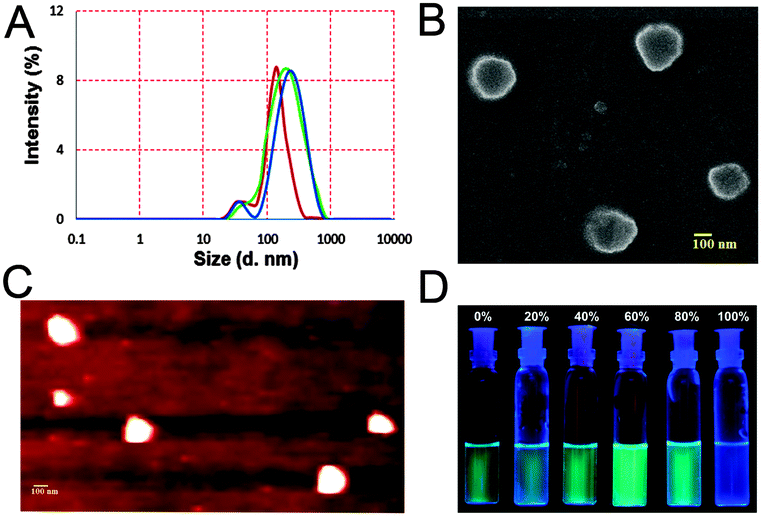 | ||
| Fig. 3 (A) Particle size analysis (DLS): sample recorded after 1.5 h (red) and 3 h (green) after preparation of the solution in mixed media, and upon addition of 1 equiv. of Ag+ (blue) to a freshly prepared sample of 1 in the same solvent system; (B) SEM image of the aggregates obtained after addition of Ag+ (1 equiv.) to a sample of 1 and (C) the corresponding AFM image; (D) visual change observed with the change of percentage of water (0–100%) to a 10 μM solution of 1 as seen under 366 nm light. | ||
The enhanced emission of 1 in highly viscous glycerol indicates that the restriction of low energy intramolecular motions plays an important role (Fig. S8, ESI†).7 However, in our case, there is no biarylic rotor-like motif. This posed a basic question: whether the presence of this motif in the probes is indeed an essential criterion for the AIE. In order to investigate this, the evidence for aggregation was systematically studied under various conditions.
Addition of only one equivalent of Ag+ to a freshly prepared solution of sensor 1 drastically changed the fluorescence intensity within a few seconds. In the absence of Ag+ this enhancement was slow (Fig. S5 and S6, ESI†). Thus, it was thought that Ag+ at this concentration range acts as a mediator for the aggregation that causes an enhancement of the emission intensity. This increase in emission intensity, however, is diminished with the addition of more Ag+ ions (Fig. 2A).
In our attempt to study the association of the metal–ligand binding, a clear break in the reciprocal plot was observed (Fig. S12A, ESI†). This prompted us to consider the whole scenario as a stepwise bi-modal process, where the first process was a Ag+ mediated aggregation of the sensor and the second one was an interaction between a second Ag+ ion and the aggregated complex which resulted in a disintegration of the aggregates. Fluorescence titration data (Fig. S3A, ESI†) indicated that the metal ligand association in the first step indeed takes place in a 1:1 stoichiometry. A peak at m/z = 522 in the ESI-MS showed the correct isotope pattern for the 1:1 association of Ag+ with 1 (Fig. S18, ESI†). At a lower concentration of Ag+ (0–10 μM) the binding constant was found to be 1.5 × 104 M−1 (Fig. S12B, ESI†) from the double reciprocal plot.
Fluorescence titration of chemosensor 1 with Ag+ was also performed in aqueous medium (99% with 1% DMSO) in the presence of HEPES (1 mM) buffered at pH 7.0 (Fig. 2D). In aqueous medium sensor 1 showed an emission maximum at 500 nm, however at higher concentrations of Ag+ the emission band at 500 nm is blue shifted to 470 nm (Fig. S4A and S4B, ESI†). The ratiometric behavior with the additional band at 424 nm that was observed in H2O–MeOH (3:2, v/v) was absent in the aqueous medium (Fig. 2D).
The leveling of the tail at a higher wavelength in the UV-vis spectra of 1 (10 μM) in the presence and absence of 1 equivalent of Ag+ ions was consistent with our hypothesis of the formation of aggregates. With the addition of more Ag+, this leveling-off of the tail diminished and coincided with the baseline indicating that the aggregates dissolved at higher concentrations of Ag+ (Fig. S9A, ESI†).
The silver assisted aggregation was further confirmed by 1H NMR titration experiments in CD3OD–D2O (1:1, v/v) (Fig. S11, ESI†). The NMR spectrum of 1 without any added metal ion and upon addition of Ag+ ions (up to 1.0 equiv.) showed a somewhat poor signal quality that is often encountered with aggregated samples. Additionally, significant up-field shifts of several aromatic protons compared to the one in pure MeOH are strongly indicative of the formation of aggregates under these conditions.8 However, further addition of Ag+ (>1.0 equiv.) to the same solution produced sharp spectra with reduced line broadening with a significant downfield shift of aromatic and methylene protons, indicating the depletion of the aggregates. Additionally, the downfield shifts of the methylene protons adjacent to the pyridyl and thiocarbamate units indicate that the binding to Ag+ involves both the moieties. Mass spectrometry of a sample of 1 with five equivalents of Ag+ showed a peak at m/z 666 corresponding to the species [1 + 2Ag + H2O + OH−] indicating the binding of two Ag+ ions (Fig. S19, ESI†). Thus, the binding of 1 to Ag+ is dependent on the concentration of the metal ion and is consistent with our suggested bimodal model.
DLS experiments (Fig. 3A and Fig. S14A, ESI†) agreed well with our hypothesis. The time dependent particle size distribution of a sample of 1 (10 μM) in mixed media increased over time (Fig. 3A). Addition of 1 equiv. Ag+ promoted fast aggregation. Thus, the Z-average diameter of 192 (±7) nm (PDI = 0.33) of the aggregates recorded after 1.5 h increased to 239 (±11) nm (0.39) (Fig. 3A) after 3 h. Upon addition of 1 equivalent of Ag+ to a freshly prepared sample, the diameter was found to be 248 (±8) nm (0.31) (Fig. 3A). Addition of 5 equivalents of Ag+ resulted in a gradual dissolution of the aggregates and the particle size became too small to determine by DLS. The AFM and the SEM (Fig. 3B and C) results obtained with 1 (10 μM) in the presence and absence of Ag+ (1 equiv.) were also in good agreement with the previous data. The particle diameter obtained with AFM and SEM was 215 (±19) nm and 220 nm respectively.
In the crystal structure, two consecutive naphthyl rings of 1 are aligned in a parallel-displaced or offset arrangement at a distance of 4.3 Å, perhaps to minimize the dispersion forces (Fig. S16, ESI†).9 It is conceivable that in the aggregates such stacking of the aromatic units may also take place. This is important since face-to-face π-stacking interactions would have disrupted the fluorescence.10
Lifetime studies described in the ESI† were consistent with the proposed model of formation and depletion of the aggregates (Fig. S14B and Table S1, ESI†). Fahrni has recently observed that the break-down of aggregates of a probe upon complexation of Cu(I) enhances fluorescence.11
The properties of probe 1 as a Ag+ sensor were tested. Competitive experiments were carried out with the chemosensor and Ag+ in the presence of various metal ions (Fig. S9B, ESI†). The results clearly indicated that the fluorescence response of the chemosensor is affected only in the presence of Hg2+ where the fluorescence intensity is quenched, whereas no significant change in the fluorescence response was observed for the other metal ions. The emission of 1 in the presence of one equivalent of Ag+ observed between pH 3 and 12 clearly indicates that the sensor can detect the Ag+ ions over a broad pH range (Fig. S10, ESI†). The detection limit determined using standard methods from the fluorescence data collected at low concentrations of Ag+ was found to be 83 nM (Fig. S13, ESI†).
In conclusion, we have shown that the selectivity of a DPA based receptor can be tuned to bind Ag+ upon modification with a thiocarbamate linkage. The probe shows a high selectivity and sensitivity for Ag+ metal ions. The presence of the aromatic ring gave rise to stacking of the molecules, thereby forming aggregates. In the aggregated state, the enhanced emission of the probe is due to the restriction of intramolecular flexibility and rotation. The formation of the aggregates was promoted by addition of up to one equivalent of Ag+, whereas the metal ion at a higher concentration led to disintegration of the aggregates. From these detailed studies, we conclude that aggregation induced enhancement can occur even in systems devoid of any rotor-like motif. These interesting results warranted the preparation of more probes with the DPA–thiocarbamate scaffold and the study of their sensing properties. In fact, on advice of a reviewer, we have synthesized compound 2 (Fig. 1) with a coumarin fluorophore (characterization: Fig. S26–S28, ESI†) that shows a similar sensing and AIE behavior (Fig. S20–S23, ESI†).
The help of Mr Arnab Maity with AFM studies and of Mr Soumik Mondal with crystallography is gratefully acknowledged. We thank the CSIR for a fellowship to JH, IISER for facilities and DST for a research grant (SR/S1/OC-26/2010).
Notes and references
- G. K. Walkup, S. C. Burdette, S. J. Lippard and R. Y. Tsien, J. Am. Chem. Soc., 2000, 122, 5644 CrossRef CAS.
- (a) S. C. Burdette, G. K. Walkup, B. Spingler, R. Y. Tsien and S. J. Lippard, J. Am. Chem. Soc., 2001, 123, 7831 CrossRef CAS PubMed; (b) C. C. Woodroofe and S. J. Lippard, J. Am. Chem. Soc., 2003, 125, 11458 CrossRef CAS PubMed; (c) C. J. Chang, E. M. Nolan, J. Jaworski, S. C. Burdette, M. Sheng and S. J. Lippard, Chem. Biol., 2004, 11, 203 CAS.
- (a) S. Atilgan, T. Ozdemir and E. U. Akkaya, Org. Lett., 2008, 10, 4065 CrossRef CAS PubMed; (b) W. Jiang, Q. Fu, H. Fan and W. Wang, Chem. Commun., 2008, 259 RSC; (c) B. Tang, H. Huang, K. Xu, L. Tong, G. Yang, X. Liu and L. An, Chem. Commun., 2006, 3609 RSC; (d) H. T. Ngo, X. Liu and K. A. Jolliffe, Chem. Soc. Rev., 2012, 41, 4928 RSC; (e) S. Maruyama, K. Kikuchi, T. Hirano, Y. Urano and T. Nagano, J. Am. Chem. Soc., 2002, 124, 10650 CrossRef CAS PubMed.
- Z. C. Xu, J. Yoon and D. R. Spring, Chem. Soc. Rev., 2010, 39, 1996 RSC.
- For example: (a) J. Hatai, S. Pal, G. P. Jose and S. Bandyopadhyay, Inorg. Chem., 2012, 51, 10129 CrossRef CAS PubMed; (b) J. Hatai, S. Pal, G. P. Jose, T. Sengupta and S. Bandyopadhyay, RSC Adv., 2012, 2, 7033 RSC.
- (a) Z. Chang, Y. Jiang, B. He, J. Chen, Z. Yang, P. Lu, H. S. Kwok, Z. Zhao, H. Qiu and B. Z. Tang, Chem. Commun., 2013, 49, 594 RSC; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC; (c) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC; (d) W. Z. Yuan, S. Chen, J. W. Y. Lam, C. Deng, P. Lu, H. H.-Y. Sung, I. D. Williams, H. S. Kwok, Y. Zhang and B. Z. Tang, Chem. Commun., 2011, 47, 112161 Search PubMed; (e) J. Liu, Q. Meng, X. Zhang, X. Lu, P. He, L. Jiang, H. Dong and W. Hu, Chem. Commun., 2013, 49, 1199 RSC.
- (a) M. A. Haidekker and E. A. Theodorakis, Org. Biomol. Chem., 2007, 5, 1669 RSC; (b) V. Bhalla, S. Kaur, V. Vij and M. Kumar, Inorg. Chem., 2013, 52, 4860 CrossRef CAS PubMed.
- (a) N. Maurer, K. F. Wong, M. J. Hope and P. R. Cullis, Biochim. Biophys. Acta, 1998, 1374, 9 CrossRef CAS; (b) I. Turcu and M. Bogdan, J. Phys. Chem. B, 2012, 116, 6488 CrossRef CAS PubMed.
- (a) T. Janowski and P. Pulay, J. Am. Chem. Soc., 2012, 134, 17520 CrossRef CAS PubMed; (b) C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191 RSC.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley, New York, 1970 Search PubMed.
- M. T. Morgan, P. Bagchi and C. J. Fahrni, J. Am. Chem. Soc., 2011, 133, 15906 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed characterization and additional spectroscopic details. CCDC 942550. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc46285d |
| This journal is © The Royal Society of Chemistry 2014 |