Chloride-catalyzed, multicomponent self-assembly of arsenic thiolates†
Matthew E.
Carnes
,
Mary S.
Collins
,
Nathan R.
Lindquist
,
Edmundo
Guzmán-Percástegui
,
Michael D.
Pluth
and
Darren W.
Johnson
*
Department of Chemistry & Biochemistry and Materials Science Institute, University of Oregon, Eugene, Oregon 97403-1253, USA. E-mail: dwj@uoregon.edu; Fax: +1 541-346-4643; Tel: +1 541-346-1695
First published on 1st November 2013
Abstract
We present the observation that chloride serves as a simple catalyst for the acceleration of a self-assembly reaction between AsCl3 and dithiolate ligands (H2L) to form As2L3 assemblies. Studies on a model monomeric arsenic complex suggest that chloride may accelerate ligand exchange dynamics in pnictogen thiolates in general.
Supramolecular self-assembly is a proven strategy for synthesizing complex molecular architectures from relatively simple components.1 Customarily, these reactions involve molecular building blocks designed with structural features to enable complementary interactions between the components. This includes idealized communications such as metal–ligand bonds,2 hydrogen bonding,3 and π-stacking effects.4 Optimally, these systems utilize strong enthalpic driving forces as well as low kinetic barriers to yield rapid, selective, and complete assembly.
In practice, the facile assembly of complex supramolecular structures is not always as simple; many self-assembly reactions fall victim to “kinetic traps” and undesirable polymerization. As such, a variety of computationally feasible self-assembled systems may be unattainable through current experimental methods. Recent progress in the field of supramolecular chemistry includes exciting examples of catalyzed self-assembly reactions where a small molecule either facilitates5 or accelerates6 the organization process. Herein, we report an example of an external species, chloride ion, accelerating the formation of a multi-component, self-assembled pnictogen complex by destabilizing oligomers and a kinetically-stable macrocyclic intermediate.
We have previously shown that a series of dinuclear pnictogen (Pn) complexes can be self-assembled from a Pn3+ source (Pn = P, As, Sb, Bi) and a variety of dithiol ligands bridged by aromatic spacers.7 The dynamic covalent chemistry of the Pn–S bond necessary for this self-assembly has been implemented in the past to explain “kinetic mistakes” and scrutinize the variety of modes through which these species are “corrected” in the self-assembly process.8 Studies incorporating 1,4-bis(mercaptomethyl)naphthalene (H2L) into these Group 15 (Pn) complexes have yielded the synthesis and solution characterization of a series of crystallographically confirmed dinuclear macrocycles (Pn2L2Cl2), larger assemblies (Pn2L3), and heterometallic Group 15 assemblies (PnPn′L3).9
The reaction of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of H2L with AsCl3 results in the spontaneous formation of a pair of syn- and anti-As2L2Cl2 macrocycles and proceeds via an As2LCl4 intermediate. These diastereomers are defined with respect to position of the chloride atom within the macrocycle and rapidly interconvert in solution at room temperature.10 Notably, all reagents are consumed within four hours, even in the absence of base. If the macrocycle As2L2Cl2 is crystallized and then redissolved in CDCl3, the 1H NMR is unchanged after heating at 50 °C for weeks. If the reaction stoichiometry of H2L to AsCl3 exceeds 1:1, macrocycle synthesis is followed by the very slow, subsequent formation of As2L3 after a lag phase of four days.11 The procedure for rapidly obtaining As2L3 in high yield is previously reported and involves the use of vacuum or an amine base.9,12
:1 ratio of H2L with AsCl3 results in the spontaneous formation of a pair of syn- and anti-As2L2Cl2 macrocycles and proceeds via an As2LCl4 intermediate. These diastereomers are defined with respect to position of the chloride atom within the macrocycle and rapidly interconvert in solution at room temperature.10 Notably, all reagents are consumed within four hours, even in the absence of base. If the macrocycle As2L2Cl2 is crystallized and then redissolved in CDCl3, the 1H NMR is unchanged after heating at 50 °C for weeks. If the reaction stoichiometry of H2L to AsCl3 exceeds 1:1, macrocycle synthesis is followed by the very slow, subsequent formation of As2L3 after a lag phase of four days.11 The procedure for rapidly obtaining As2L3 in high yield is previously reported and involves the use of vacuum or an amine base.9,12
Our preliminary investigation of the transmetalation of pnictogen dithiolate Pn2L3 assemblies,13 in particular the antimony to arsenic transmetalation, raised the question of whether a catalytic amount of the antimony assembly, Sb2L39 could template the formation of As2L3 from H2L and AsCl3 in the absence of base. This would potentially provide us with information about the mechanism of transmetalation and allow us to probe how labile pnictogen thiolate bonds interact in the process of self-assembly.
When H2L is treated with AsCl3 in a 3:2 stoichiometry in the presence of catalytic Sb2L3, the reaction rate was increased substantially. To probe the potential for a template effect of Sb2L3 in this reaction, the mononuclear antimony trithiolate (1) was synthesized and tested in the same reaction. Surprisingly, 1 is also an equally effective catalyst for forming arsenic assemblies, indicating that the observed catalysis from Sb2L3 is not due to a template effect. Notably, 1H NMR resonances characteristic of the additives Sb2L3 or 1 disappeared immediately upon addition of AsCl3 to the reaction mixture suggesting that the active catalyst is a decomposition product of both. When antimony trichloride was added as a potential pre-catalyst, it did not catalyze the reaction.
In order to gain insight into potential mechanisms of action of the catalyst and to identify the active catalyst species, a variety of potential species were screened via1H-NMR spectroscopy (Fig. 1). In all screening cases, the catalyst was loaded with 0.1 equivalents based on final As2L3 concentration. Since the reaction is known to proceed readily with stoichiometric base, tridodecylamine was also screened as a base catalyst candidate but failed to catalyze the reaction. Benzyl mercaptan, a potent thiol nucleophile, also failed to catalyze the reaction.
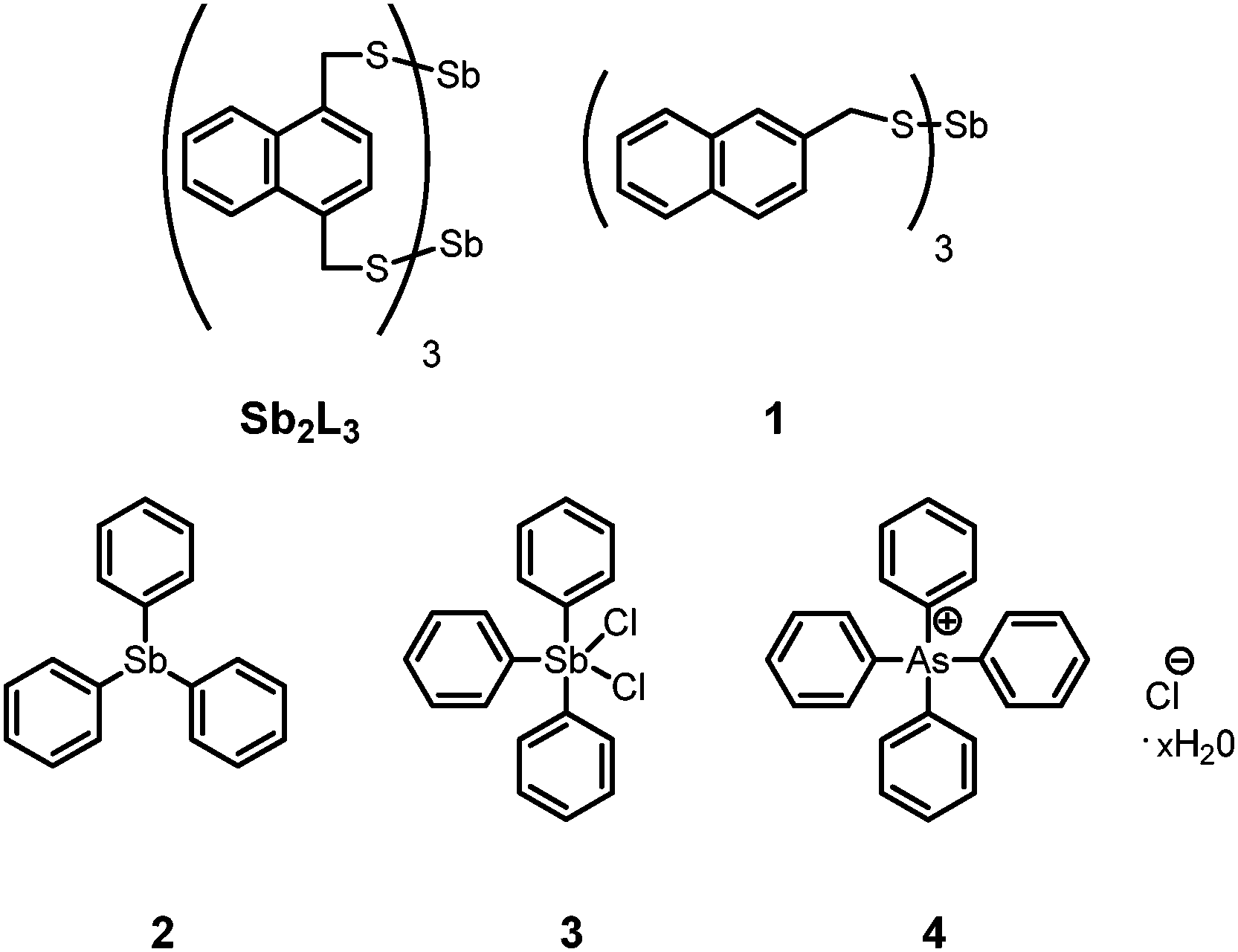 | ||
| Fig. 1 The evolution of catalysts and precatalysts screened for the self-assembly reaction of As2L2Cl2 with H2L to form As2L3. | ||
Triphenylstibine (2) was also added to the initial reaction mixture to discern if a covalently bonded, persistent antimony species could accelerate As2Cl3 formation, which, surprisingly, indeed proved to be the case. It should be noted that 2 is stable to the initial addition of AsCl3, but over time, is oxidized to triphenylstibine oxide in air over the course of the experiment. The observation that 2 is easily oxidized under such experimental conditions led to the serendipitous implementation of two “pre-oxidized” pentavalent catalysts SbCl2Ph3 (3) and AsPh4Cl (4).
Both 3 and 4 remained unchanged throughout the reaction, as monitored by 1H-NMR, but only 4 is an effective catalyst for the As2L3 formation with these conditions. Catalyst 4 is not susceptible to ligand exchange reactions with the dithiol functional groups of H2L which rules out a mechanism involving activation of the thiol. As a control, tetrabutylammonium chloride (TBACl) was also added and proved to be a competent catalyst in this system, suggesting an important role for the chloride anion in the formation of As2L3 from its precursors. Notably, the addition of tetrabutylammonium perchlorate does not result in rate acceleration, nor did the addition of catalytic amounts of amine base.
Although the methylene region in the 1H-NMR spectrum is congested, the clear resonances of the naphthalene aromatic protons allow for the reaction progress to be monitored. To investigate the dependence of the reaction rate on [Cl−], the reaction between As2L2Cl2 and H2L (Scheme 1) was run with different concentrations of TBACl while keeping the concentrations of As2L2Cl2 and H2L constant. The rate of formation for As2L3 was monitored over the course of three days for each run. The initial rate of formation of As2L3 increased linearly with increasing concentrations of TBACl (see Fig. S1, ESI†). Furthermore, log–log analysis of the rate of As2L3 formation as a function of TBACl concentration confirmed a first-order dependence on [Cl−] (Fig. S2, ESI†). All species remain in solution throughout the course of the experiment; new peaks are observed by 1H-NMR spectroscopy at low temperature and we hypothesize that these correspond to soluble oligomers (Fig. S3, ESI†).14
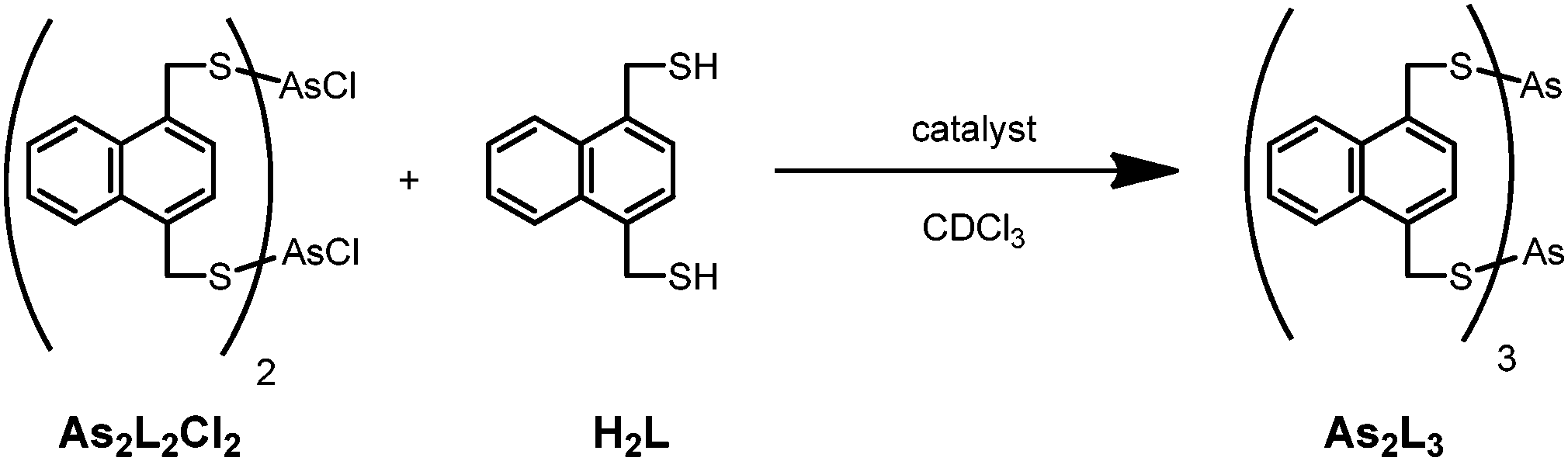 | ||
| Scheme 1 The arsenic macrocycle As2L2Cl2, which forms rapidly from H2L and AsCl3, slowly reacts with H2L in the absence of base to form As2L3 unless catalyzed. | ||
The addition of a species containing a chloride anion to the reaction mixture causes broadening of the usually distinct methylene proton resonances of As2L2Cl2 (Fig. S4, ESI†). As the amount of chloride is increased, the pair of sharp AB doublets broadens and eventually coalesces into a singlet. At these high chloride concentrations, the peaks characteristic of As2L3 begin to appear even though no additional H2L was added. Forming As2L3 can be explained by inducing rapid exchange between chloride and thiolate at the arsenic center through an associative interchange substitution mechanism (Scheme 2). This temporarily free thiolate is now able to react with neighboring As2L2Cl2 macrocycles resulting in As2L3 and free AsCl3.
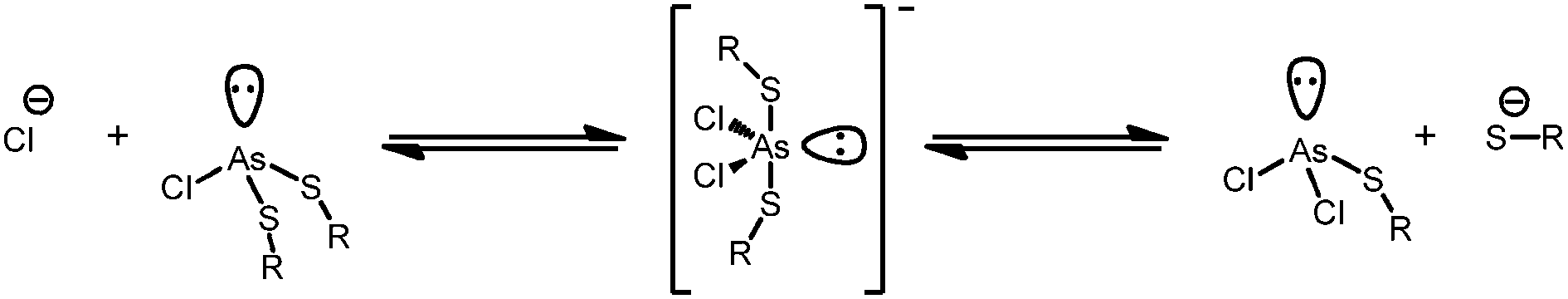 | ||
| Scheme 2 A potential associative interchange substitution mechanism for exchange of chlorides and thiolates leading to activation of As2L2Cl2 and a possible explanation for the cannibalism of As2L2Cl2 to form As2L3 in the presence of Cl−. | ||
Further support for this hypothesis can be found by analyzing the products formed initially from As2L2Cl2 and H2L, which include a disproportionate amount of an asymmetric conformer of As2L3 (As2L3-asym)9 in addition to the three-fold symmetric conformer of As2L3 (Fig. 2).15 An asymmetric conformer of As2L3 should result from a putative [As2L2Cl2·Cl]− adduct which is rapidly interconverting and may be attacked from either side of a coordinated ligand that is interconverting between symmetric (ligands eclipsed) and asymmetric (ligands staggered) conformations about the arsenic centers. The resulting asymmetric form is the dominant species observed for Bi2L3 and is observed in the 1H-NMR spectrum of redissolved crystals of As2L3 and Sb2L3 as very small peaks.9
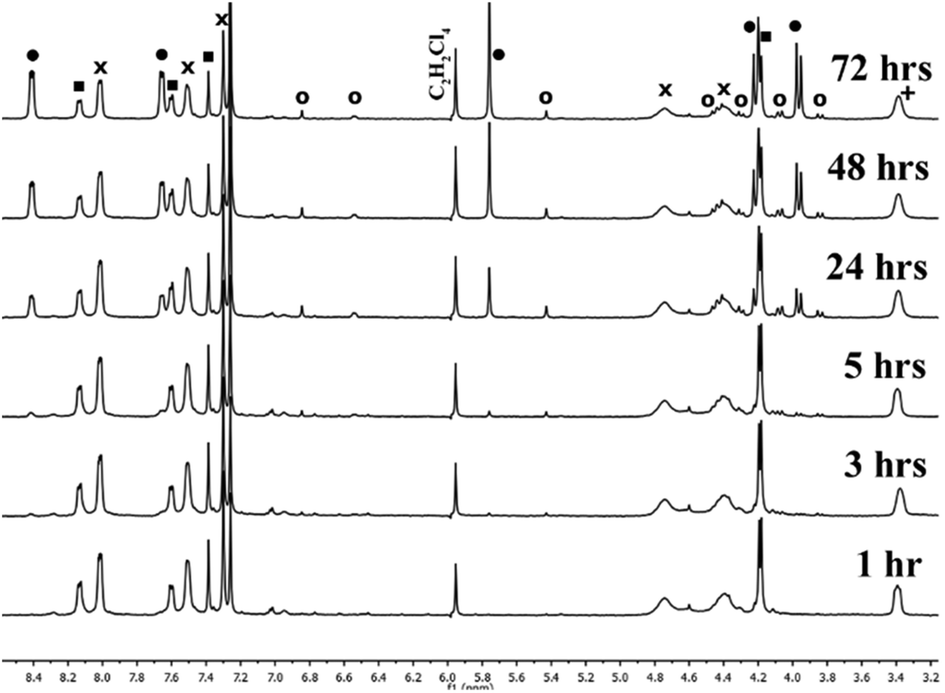 | ||
| Fig. 2 1H NMR spectra of the species distribution over the course of 3 days for a representative reaction of As2L2Cl2 and H2L in CDCl3 (20% loading of [TBACl] was used and 0.0038 mM C2H2Cl4 internal standard). Components of the reaction are labeled: As2L3 (●), H2L (■), As2L2Cl2 (x), TBACl (+), and asymmetric conformer of As2L3 (o). | ||
The effect of chloride ion on arsenic thiolate complexes is not specific to chelating macrocyclic structures such as As2L2Cl2. The simple reaction of benzyl thiol (HSBn) with arsenic trichloride leads to the spontaneous formation of As(SBn)2Cl and only a small amount of the As(SBn)3 species. The ratio of these products does not change noticeably over the course of 24 h (Fig. S5, ESI†). Addition of chloride to this system does not promote the formation of the As(SBn)3 species. However, it does cause more rapid exchange of the thiols bound to arsenic as is evident by the coalescence of the previously inequivalent methylene protons.16 As was the case with the uncatalyzed reaction, the ratio of products does not change over 24 h (Fig. S6, ESI†). This increase in exchange may be due to chloride from TBACl adding to the arsenic to form an intermediate disphenoidal, tetracoordinate arsenic anion.17
We have utilized the relatively slow kinetics of arsenic–thiolate bond to gain greater insight into the mechanism of a self assembly reaction: the formation of As2L3 using a 1,4-bis(mercaptomethyl)naphthalene ligand (H2L). Subsequently, we have shown that chloride ion is an effective accelerator of this multi-component self-assembly reaction by activating a kinetically stable As2L2Cl2 macrocycle. We have determined that the rate of complex formation has a first order dependence on chloride ion. To the best of our knowledge, this is the first reported example of the catalyzed assembly of a metal–ligand supramolecular complex. Results of a study on a model mononuclear As(thiolate)3 complex suggest that the dynamics of As–thiolates are influenced by chloride concentration, suggesting potential implications in biological/environmental arsenic coordination chemistry given the high background concentration of chloride in natural systems.
The authors thank the University of Oregon for partial support of this work. University of Oregon NMR facilities are supported by NSF CHE-0923589. D.W.J. is a Scialog Fellow of Research Corporation for Science Advancement.
Notes and references
- J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- M. D. Watson, A. Fechtenkötter and K. Müllen, Chem. Rev., 2001, 101, 1267–1300 CrossRef CAS PubMed.
- (a) T. Hasell, X. Wu, J. T. A. Jones, J. Basca, A. Steiner, T. Mitra, A. Trewin, D. J. Adams and A. I. Cooper, Nat. Chem., 2010, 2, 750–755 CrossRef CAS PubMed; (b) S. Otto, R. L. E. Furlan and J. K. M. Sanders, Science, 2002, 297, 590–593 CrossRef CAS PubMed.
- K. Patel, O. Š. Miljanić and J. F. Stoddart, Chem. Commun., 2008, 1853–1855 RSC.
- (a) S. A. Fontenot, V. M. Cangelosi, M. A. W. Pitt, A. C. Sather, L. N. Zakharov, O. B. Berryman and D. W. Johnson, Dalton Trans., 2011, 40, 12125–12131 RSC; (b) M. Watt, M. S. Collins and D. W. Johnson, Acc. Chem. Res., 2013, 46, 955–966 CrossRef CAS PubMed.
- V. M. Cangelosi, T. G. Carter, L. N. Zakharov and D. W. Johnson, Chem. Commun., 2009, 5606–5608 RSC.
- V. M. Cangelosi, T. G. Carter, J. L. Crossland, L. N. Zakharov and D. W. Johnson, Inorg. Chem., 2010, 49, 9985–9992 CrossRef CAS PubMed.
- V. M. Cangelosi, A. C. Sather, L. N. Zakharov, O. B. Berryman and D. W. Johnson, Inorg. Chem., 2007, 46, 9278–9284 CrossRef CAS PubMed.
- The dynamic nature of the arsenic thiol bonds in this assembly has been demonstrated by addition of excess arsenic trichloride to preformed As2L3 and As2L2Cl2: this extra arsenic leads to rapid scrambling. If one equivalent of AsCl3 is added to As2L3, the resulting mixture contains a statistical distribution of As2LCl4, As2L2Cl2, and As2L3.
- W. J. Vickaryous, R. Herges and D. W. Johnson, Angew. Chem., Int. Ed., 2004, 43, 5831–5833 CrossRef CAS PubMed.
- V. M. Cangelosi, L. N. Zakharov and D. W. Johnson, Angew. Chem., Int. Ed., 2010, 49, 1248–1251 CrossRef CAS PubMed.
- For more information about dynamic covalent oligomers see J. Leclaire, L. Vial, S. Otto and J. K. M. Sanders, Chem. Commun., 2005, 1959–1961 RSC.
- The asymmetric isomer of As2L3 forms in very low concentration as a result of a self-assembly reaction (with no added catalyst) and was never observed previously in higher amounts (ref. 9). The appearance of elevated amounts of this intermediate suggests a possible new mechanism in this catalyzed self-assembly.
- A. Shiotani, H. F. Klein and H. Schmidbauer, J. Am. Chem. Soc., 1971, 93, 1555–1556 CrossRef.
- S. Grewe, T. Hausler, M. Mannel, B. Rossenbeck and W. S. Sheldrick, Z. Anorg. Allg. Chem., 1998, 624, 613–619 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra, and kinetic analysis. See DOI: 10.1039/c3cc47093h |
| This journal is © The Royal Society of Chemistry 2014 |