 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and ligand-based reduction chemistry of boron difluoride complexes with redox-active formazanate ligands†
M.-C.
Chang
and
E.
Otten
*
Stratingh Institute for Chemistry, University of Groningen Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: edwin.otten@rug.nl
First published on 19th May 2014
Abstract
Mono(formazanate) boron difluoride complexes (LBF2), which show remarkably facile and reversible ligand-based redox-chemistry, were synthesized by transmetallation of bis(formazanate) zinc complexes with boron trifluoride. The one-electron reduction product [LBF2]−[Cp2Co]+ and a key intermediate for the transmetallation reaction, the six-coordinate zinc complex (L(BF3))2Zn were isolated and fully characterized.
The chemistry of coordination complexes bearing redox-active (or non-innocent) ligands has recently received increasing attention due to its potential application in small molecule activation and catalysis,1 and its relevance to biological (enzymatic) transformations.2 The most studied ligands in this class are dithiolenes/dioxolenes,3 α-diimines4 and bis(imino)pyridines.5 Formazanates (1,2,4,5-tetraazapentadienyls), which are nitrogen-rich analogues of the well-know β-diketiminates,6 have received comparatively little attention as ligands in coordination chemistry.7 Unlike β-diketiminates, which have a NCCCN backbone, formazanates feature a NNCNN backbone; the two additional nitrogen atoms provide formazanates with redox-active properties. Specifically, the reduced form of formazanates should be relatively accessible and stable due to delocalization of the SOMO over all 4 N atoms. This is in a way related to the stability of the analogous organic verdazyl radicals, which may be obtained from formazan precursors.8 In previous studies, we found that bis(formazanate) zinc complexes such as [PhNNC(p-tol)NNPh]2Zn (1a) are capable of storing one or two electrons in the ligand frameworks to form stable singly- and doubly-reduced zinc complexes.9 The crystal structures of the reduced complexes show (weak) interactions between the internal nitrogen atoms of formazanate ligands and sodium counter cations. Based on this observation we anticipated that the reduction potential of bis(formazanate) zinc compounds could be altered by coordination to neutral Lewis acids. In the course of testing a range of Lewis acids for binding to 1a, we found that BF3 reacts cleanly via salt metathesis, opening a high-yield synthetic route to obtain mono(formazanate) boron difluoride (LBF2) complexes which are difficult to access otherwise, either from the parent formazan or formazanate salts (LK or LNa). A related formazanate diacetate compound LB(OAc)2 was described by Hicks and co-workers,7b and although spectroscopic data suggested ligand-based 1-electron redox-chemistry to occur in these compounds, the ‘borataverdazyl’ radical anions obtained were too unstable to allow full characterization. Here we report the synthesis and X-ray crystallographic characterization of LBF2 and the radical anion LBF2˙−, with the formation of a relatively stable 2-electron reduction product LBF22− confirmed by cyclic voltammetry. In addition, we provide evidence for the transmetallation pathway by characterization of a likely intermediate.
Mono(formazanate) boron difluoride complexes are readily accessible by transmetallation of bis(formazanate) zinc complexes with BF3·Et2O in hot toluene. In the case of (PhNNC(p-tol)NNPh)2Zn (1a), a stoichiometric reaction does not go to completion, but full conversion is achieved by performing the reaction with 3 equivalents of BF3·Et2O at 70 °C twice. During the reaction, a colour change from deep blue to red and the precipitation of a white solid (presumably ZnF2) was observed to indicate formation of mono(formazanate) boron difluoride complex (PhNNC(p-tol)NNPh)BF2 (2a, Scheme 1), which was isolated as an air-stable, crystalline material in 86% yield. A single crystal X-ray diffraction study (Fig. S1, ESI†) revealed a distorted tetrahedral boron centre, which is displaced out of the (planar) formazanate NNCNN backbone by 0.5 Å. A similar bonding mode was observed for β-diketiminate complexes of Sc and attributed to steric interactions.10 In the absence of significant steric pressure in 2a, we ascribe the out-of-plane displacement of the B atom to packing effects; the observation of only one 19F NMR resonance for 2a even at low temperature is consistent with a low-energy C2v symmetric structure through which the two 19F environments exchange. As is the case in its parent zinc complex 1a and the related mono(formazanate) boron diacetate complexes reported by Hicks and co-workers,7b full delocalization within the formazanate framework in 2a is indicated by the similar N–N and C–N bond lengths in the NNCNN backbone.
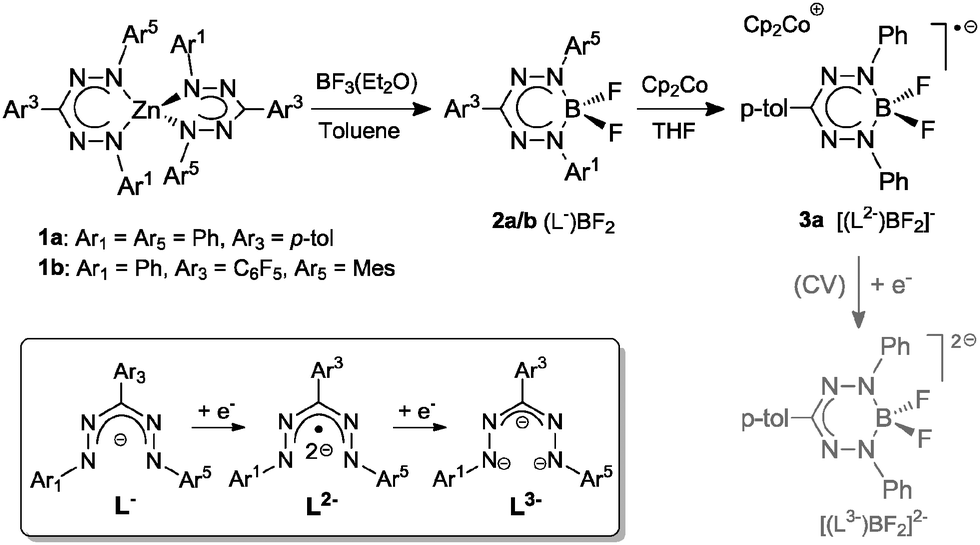 | ||
| Scheme 1 Synthesis of mono(formazanate) boron difluoride complexes and their reduction products. | ||
The redox-active nature of the formazanate ligand in 2a was established by cyclic voltammetry (CV) in THF solution with [Bu4N][PF6] electrolyte. Upon scanning to negative potentials, the CV shows two quasi-reversible one-electron redox processes (system I/I′ and II/II′, Fig. 1) that can be assigned to formazanate-based reductions. The first redox-couple of 2a occurs at more positive potential (E1/2(I/I′) = −0.98 V vs. Fc0/+) than that in 1a (−1.31 V vs. Fc0/+)9 and at similar potential as Hicks et al. reported for the analogous mono(formazanate) boron diacetate compound (−0.86 V vs. Fc0/+ in CH3CN).7b A second redox-event is observed at more negative potential (E1/2(II/II′) = −2.06 V vs. Fc0/+) but also this reduction is reversible. Based on these data, we infer that both one- and two-electron reduction products of 2a are relatively stable and accessible. Importantly, the second reduction forms the dianion LBF22−, in which two electrons have been added to a single formazanate ligand to result in a ‘L3−’ fragment coordinated to the boron centre (Scheme 1). Bard et al. recently explored the voltammetry of boron dipyrromethenes (BODIPY): in general these show one-electron reductions at more negative potentials compared to 2a.11 Related β-diketiminate compounds show irreversible reductions at much more negative potential (Ep,red ∼ −2.6 V vs. Fc0/+).12
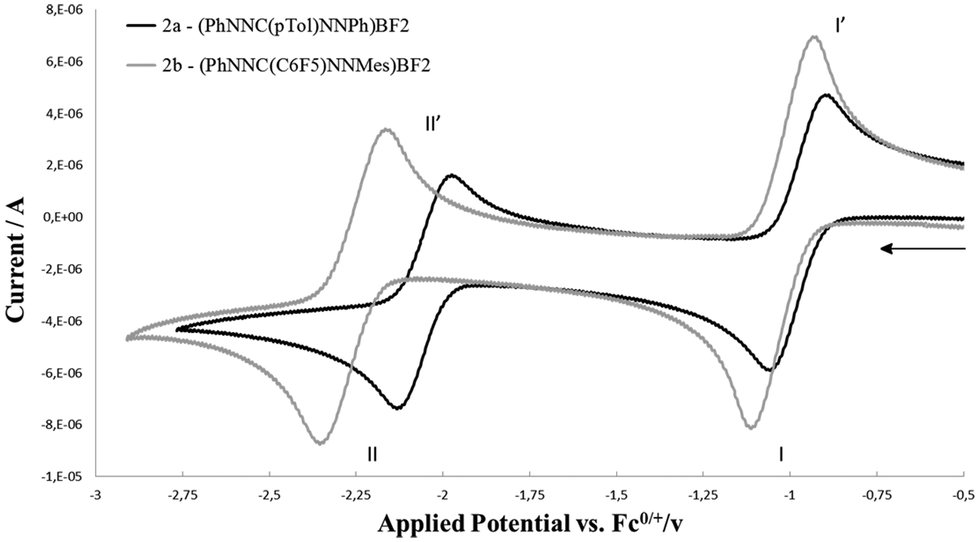 | ||
| Fig. 1 Cyclic voltammetry of 2a and 2b (1.5 mM solution in THF, 0.1 M [Bu4N][PF6]) recorded at 100 mV s−1. | ||
The first redox-couple of 2a occurs at −0.98 V vs. Fc0/+, which suggests that cobaltocene is a suitable reducing agent to selectively synthesize a singly-reduced product. Upon reaction of 2a with one equivalent of Cp2Co in THF, an immediate colour change from red to green is observed to indicate formation of the radical species [Cp2Co]+[(PhNNC(p-tol)NNPh)BF2]− (3a). In contrast to the reduction product of the diacetate analogue reported by Hicks, the difluoro complex 3a is stable in solution under an inert atmosphere for days and can even be boiled in THF–hexane without noticeable decomposition. The surprising stability of 3a is likely due to the strength of the B–F bonds in comparison to B–OAc. Crystals of 3a suitable for single-crystal X-ray diffraction analysis were obtained by diffusion of hexane into THF solution at room temperature in quantitative yield (Fig. 2, left). Compared with 2a, 3a has shortened B–N (av. 1.554 Å for 2avs. av. 1.535 Å for 3a), elongated N–N (av. 1.309 Å for 2avs. av. 1.362 Å for 3a) and elongated B–F (av. 1.376 Å for 2avs. av. 1.410 Å for 3a) bond lengths. The similar N–N bond lengths in 3a and the reduced L2Zn complexes9 suggest the ligand of 3a is a radical dianionic species (L2−) in which the unpaired electron occupies a N–N anti-bonding orbital.
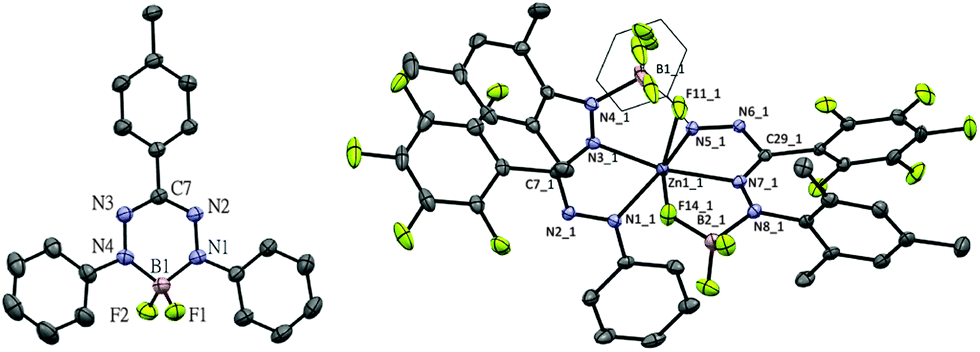 | ||
| Fig. 2 Molecular structure of 3a (left) and 4b (right) showing 50% probability ellipsoids. For 3a, [Cp2Co]+ cation and THF molecule are omitted; for 4b, one of the two independent molecules is shown; for both, hydrogen atoms are omitted for clarity. | ||
UV-Vis absorption spectroscopy of neutral and anionic mono(formazanate) difluoride compounds provides additional evidence for ligand-based reduction and formation of the dianionic ligand radical (L2−) (Fig. S3, ESI†). The neutral compound 2a shows a single broad absorption in the visible at 521 nm. In the case of 3a, absorption is at longer wavelength (716 nm) and at 454 nm with shoulders at 674 and 431 nm, respectively. These absorption bands are in agreement with the presence of reduced formazanate ligands.9,13 The EPR spectrum of 3a shows a broad EPR signal in frozen THF with g-value of ∼2 (Fig. S2, ESI†). Attempts to synthesize and characterize the two-electron reduction product have so far been unsuccessful.
In order to expand the scope of transmetallation reactions from bis(formazanate) zinc complexes and boron trifluoride, the reaction of (PhNNC(C6F5)NNMes)2Zn (1b) with BF3 was attempted. The colour of the reaction mixture fades from deep to light orange upon heating 1b in the presence of 3 eq. BF3Et2O at 70 °C overnight, but precipitation of ZnF2 was not observed. Orange crystals of the product 4b were obtained by slow diffusion of hexane into a toluene solution at −30 °C in 85% yield (Fig. 2, right). The crystal structure of 4b shows a distorted octahedral zinc centre. In the structure, there are two tridentate (PhNNC(C6F5)NNMes(BF3)) units coordinated to the Zn centre in a meridional fashion via two nitrogens and a fluorine atom to give a [NNF]2Zn compound. This unusual binding motif results from interaction of BF3 with the terminal nitrogen of the formazanate fragment (the NMes group), which gives rise to 2 five-membered chelate rings upon coordination to the Zn centre. To the best of our knowledge, the structural characterization of this BF3 binding mode has no precedent in the literature, although the ‘frustrated Lewis pair’ (tmp)MgCl/BF3 has been postulated to contain a B–F fragment appended to a Mg–N(tmp) bond.14 The 19F-NMR of 4b shows six distinct resonances with integration ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3:1:1:1 (Fig. S6, ESI†). Five resonances with the same integration (1F) suggest that all F substituents of the C6F5 ring are inequivalent due to hindered rotation around the C–C6F5 bond. The resonance integrating as 3F shows 11B and 10B coupling features and can be assigned to the BF3 unit. The appearance of the BF3 group in the 19F NMR does not change upon cooling to −55 °C, which suggests that the barrier to rotation around the N–BF3 bond is low. Conversely, heating an NMR sample of 4b to moderate temperature (65 °C for 24 h) does not result in changes in the spectroscopy, confirming that the octahedral [NNF]2Zn complex is quite stable. Upon heating the NMR tube to 130 °C overnight, full conversion to 2b is obtained. The 19F NMR spectrum of the new species shows signals characteristic for a freely rotating C6F5 group and a BF2 unit (11B NMR: −1.34 ppm, triplet with JB–F = 24 Hz). These data are consistent with formation of [PhNNC(C6F5)NNMes]BF2 (2b). Cyclic voltammetry of 2b (Fig. 1) shows two quasi-reversible redox processes similar to 2a but shifted to more negative potential. This suggests that the electron-rich N-Mes group in 2b is more important than the electron-withdrawing C–C6F5 moiety in modulating the redox-potential of the formazanate fragment.
:1:3:1:1:1 (Fig. S6, ESI†). Five resonances with the same integration (1F) suggest that all F substituents of the C6F5 ring are inequivalent due to hindered rotation around the C–C6F5 bond. The resonance integrating as 3F shows 11B and 10B coupling features and can be assigned to the BF3 unit. The appearance of the BF3 group in the 19F NMR does not change upon cooling to −55 °C, which suggests that the barrier to rotation around the N–BF3 bond is low. Conversely, heating an NMR sample of 4b to moderate temperature (65 °C for 24 h) does not result in changes in the spectroscopy, confirming that the octahedral [NNF]2Zn complex is quite stable. Upon heating the NMR tube to 130 °C overnight, full conversion to 2b is obtained. The 19F NMR spectrum of the new species shows signals characteristic for a freely rotating C6F5 group and a BF2 unit (11B NMR: −1.34 ppm, triplet with JB–F = 24 Hz). These data are consistent with formation of [PhNNC(C6F5)NNMes]BF2 (2b). Cyclic voltammetry of 2b (Fig. 1) shows two quasi-reversible redox processes similar to 2a but shifted to more negative potential. This suggests that the electron-rich N-Mes group in 2b is more important than the electron-withdrawing C–C6F5 moiety in modulating the redox-potential of the formazanate fragment.
The sequential transformation 1b → 4b → 2b suggests that a six-coordinate species related to 4b is likely also involved in the formation of 2a. Based on these observations, we propose the following mechanism for the transmetallation leading to compounds 4 (Scheme 2): (i) formazanate rearrangement from a 6- to a 5-membered chelate ring liberates the terminal N-atom, (ii) BF3 binds to this terminal N-atom and brings a B–F group in proximity of the Zn centre, and (iii) the F atom binds to the Lewis acidic Zn(II) centre to from a tridentate [NNF] ligand with two 5-membered chelate rings. Repeating this sequence for the second formazanate ligand results in formation of the [NNF]2Zn complex 4. Elimination of ZnF2 from this complex either occurs rapidly (in case of 1a → ‘4a’ → 2a), or requires heating to proceed so that the intermediate can be isolated (1b → 4b → 2b). A reason for the increased stability of 4bvs. that of putative intermediate 4a could be the favorable π-interactions between the electron-rich N-Mes and the electron-poor C–C6F5 substituents,15 which are present only when the formazanate ligands adopt a 5-membered chelate ring.
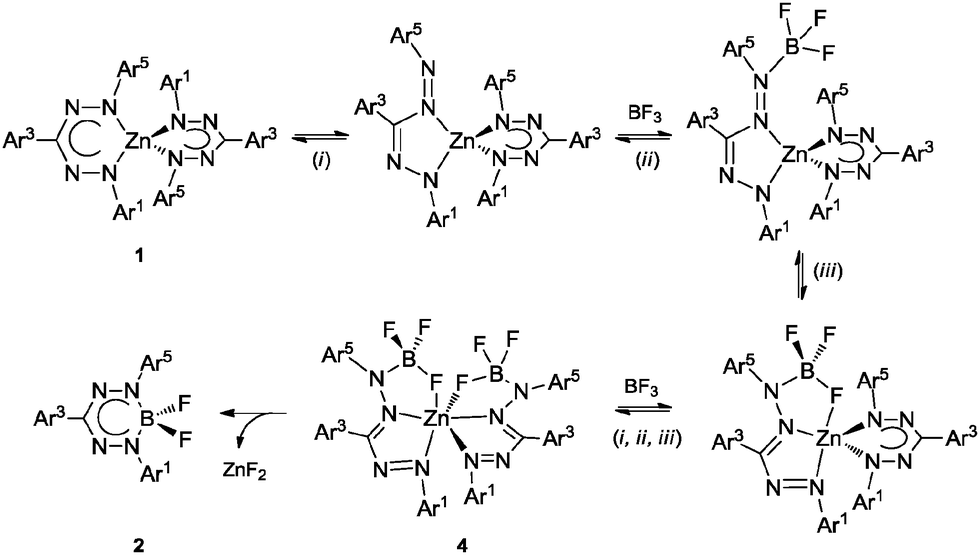 | ||
| Scheme 2 Proposed mechanism of transmetallation from bis(formazanate) zinc complex to mono(formazanate) boron difluoride complex. | ||
In conclusion, transmetallation of bis(formazanate)zinc complexes with BF3·Et2O provides a convenient entry into formazanate boron chemistry. The reaction likely occurs via initial binding of BF3 to the formazanate ligand. This pathway is possible through the flexibility of the NNCNN backbone to adopt 5-membered chelate ring isomers, which are inaccessible to their β-diketiminate congeners. One-electron reduction of LBF2 results in a fully characterized stable ligand-based radical, and cyclic voltammetry confirms a second reduction is possible, allowing access to three oxidation states (LBF20/−1/−2) that are all based the redox-chemistry of a single formazanate ligand. The further development of this unique class of stable redox-active ligands towards applications in coordination chemistry and catalysis is the focus of ongoing work in our laboratory.
Notes and references
- (a) P. J. Chirik and K. Wieghardt, Science, 2010, 327, 794 CrossRef CAS PubMed; (b) V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270 CrossRef CAS; (c) V. K. K. Praneeth, M. R. Ringenberg and T. R. Ward, Angew. Chem., Int. Ed., 2012, 51, 10228 CrossRef CAS PubMed.
- (a) L. Que and W. B. Tolman, Nature, 2008, 455, 333 CrossRef CAS PubMed; (b) J. B. Broderick, B. R. Duffus, K. S. Duschene and E. M. Shepard, Chem. Rev., 2014, 114, 4229 CrossRef CAS PubMed.
- (a) Prog. Inorg. Chem., ed. K. D. Karlin and E. I. Stiefel, John Wiley & Sons, Inc., Hoboken, New Jersey, 2004, vol. 52 Search PubMed; (b) W. Kaim and B. Schwederski, Coord. Chem. Rev., 2010, 254, 1580 CrossRef CAS PubMed; (c) R. Eisenberg and H. B. Gray, Inorg. Chem., 2011, 50, 9741 CrossRef CAS PubMed.
- (a) H. Tsurugi, T. Saito, H. Tanahashi, J. Arnold and K. Mashima, J. Am. Chem. Soc., 2011, 133, 18673 CrossRef CAS PubMed; (b) S. J. Kraft, U. J. Williams, S. R. Daly, E. J. Schelter, S. A. Kozimor, K. S. Boland, J. M. Kikkawa, W. P. Forrest, C. N. Christensen, D. E. Schwarz, P. E. Fanwick, D. L. Clark, S. D. Conradson and S. C. Bart, Inorg. Chem., 2011, 50, 9838 CrossRef CAS PubMed.
- (a) M. W. Bouwkamp, A. C. Bowman, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 13340 CrossRef CAS PubMed; (b) D. Zhu, I. Thapa, I. Korobkov, S. Gambarotta and P. H. M. Budzelaar, Inorg. Chem., 2011, 50, 9879 CrossRef CAS PubMed; (c) A. M. Tondreau, S. C. E. Stieber, C. Milsmann, E. Lobkovsky, T. Weyhermüller, S. P. Semproni and P. J. Chirik, Inorg. Chem., 2013, 52, 635 CrossRef CAS PubMed.
- (a) L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031 CrossRef CAS PubMed; (b) Y.-C. Tsai, Coord. Chem. Rev., 2012, 256, 722 CrossRef CAS PubMed.
- (a) A. R. Siedle and L. H. Pignolet, Inorg. Chem., 1980, 19, 2052 CrossRef CAS; (b) J. B. Gilroy, M. J. Ferguson, R. McDonald, B. O. Patrick and R. G. Hicks, Chem. Commun., 2007, 126 RSC; (c) J. B. Gilroy, P. O. Otieno, M. J. Ferguson, R. McDonald and R. G. Hicks, Inorg. Chem., 2008, 47, 1287 CrossRef CAS PubMed; (d) J. B. Gilroy, M. J. Ferguson, R. McDonald and R. G. Hicks, Inorg. Chim. Acta, 2008, 361, 3388 CrossRef CAS PubMed; (e) S. Hong, L. M. R. Hill, A. K. Gupta, B. D. Naab, J. B. Gilroy, R. G. Hicks, C. J. Cramer and W. B. Tolman, Inorg. Chem., 2009, 48, 4514 CrossRef CAS PubMed.
- (a) F. A. Neugebauer, Angew. Chem., Int. Ed. Engl., 1973, 12, 455 CrossRef; (b) R. G. Hicks, Verdazyls and Related Radicals Containing the Hydrazyl [R2N-NR] Groupin Stable Radicals, John Wiley & Sons, Ltd, 2010, p. 245 Search PubMed.
- M.-C. Chang, T. Dann, D. P. Day, M. Lutz, G. G. Wildgoose and E. Otten, Angew. Chem., Int. Ed., 2014, 53, 4118 CrossRef CAS PubMed.
- P. G. Hayes, W. E. Piers, L. W. Lee, L. K. Knight, M. Parvez, M. R. Elsegood and W. Clegg, Organometallics, 2001, 20, 2533 CrossRef CAS.
- A. B. Nepomnyashchii and A. J. Bard, Acc. Chem. Res., 2012, 45, 1844 CrossRef CAS PubMed.
- S. M. Barbon, V. N. Staroverov, P. D. Boyle and J. B. Gilroy, Dalton Trans., 2014, 43, 240 RSC.
- R. Kuhn and H. Trischmann, Monatsh. Chem., 1964, 95, 457 CrossRef CAS.
- M. Jaric, B. A. Haag, A. Unsinn, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2010, 49, 5451 CrossRef CAS PubMed.
- (a) L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808 CrossRef CAS PubMed; (b) C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data for compounds 2a, b, 3a and 4b. CCDC 995403–995405. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc03244f |
| This journal is © The Royal Society of Chemistry 2014 |