Sunlight, electrochemistry, and sustainable oxidation reactions†
Bichlien H.
Nguyen
,
Alison
Redden
and
Kevin D.
Moeller
*
Department of Chemistry, Washington University in St. Louis, St. Louis, MO 63130, USA. E-mail: moeller@wustl.edu
First published on 1st October 2013
Abstract
Inexpensive, readily available photovoltaic cells have been used to conduct indirect electrochemical oxidation reactions. The reactions retain the efficiency of the solar-electrochemical method while capitalizing on the unique opportunities for selectivity afforded by a chemical oxidant. The versatility of the electrochemical method allowed for the recycling of Os(VIII)-, TEMPO-, Ce(IV)-, Pd(II)-, Ru(VIII)-, and Mn(V)-oxidants all with the same very simple reaction apparatus.
Oxidation reactions are powerful synthetic tools because they allow us to selectively increase the functionality of a molecule. However, they are also inherently problematic from an environmental standpoint. Every stoichiometric oxidation reaction requires the formation of a stoichiometric reduction product. If a metal oxidant is used, then the reaction leads to one or more equivalents of a metal waste product. To circumvent this problem, efforts to conduct more benign oxidation reactions typically channel the stoichiometric reduction product to something that is non-toxic and inexpensive to dispose. For example, extensive work is being conducted to develop oxygen as a co-oxidant for recycling chemical oxidants.1,2 This work channels the required reduction reaction to the formation of water. In a similar fashion, electrochemistry can be used to conduct more benign oxidation reactions by channeling the reduction reaction to the formation of hydrogen gas.3
Recently, we have shown that electrochemical reactions can be conducted with the use of a simple photovoltaic cell as the power supply (Fig. 1).4,5
 | ||
| Fig. 1 An electrochemical reaction setup with a simple 6 V photovoltaic power supply is shown. The ammeter is spliced into the line so that the current passed through the flask can be monitored. | ||
The positive lead of the photovoltaic cell is connected to the anode of the reaction flask, and the negative lead is connected through an ammeter in series to the cathode. The photovoltaic cell is exposed to sunlight, resulting in the passage of current through the electrolysis flask. In this way, oxidation reactions can be conducted in a manner that consumes only sunlight and produces hydrogen gas as the only byproduct. The utility of the approach for conducting direct oxidation reactions is illustrated in Scheme 1 for both an oxidative cyclization and the functionalization of a proline derivative.6,7
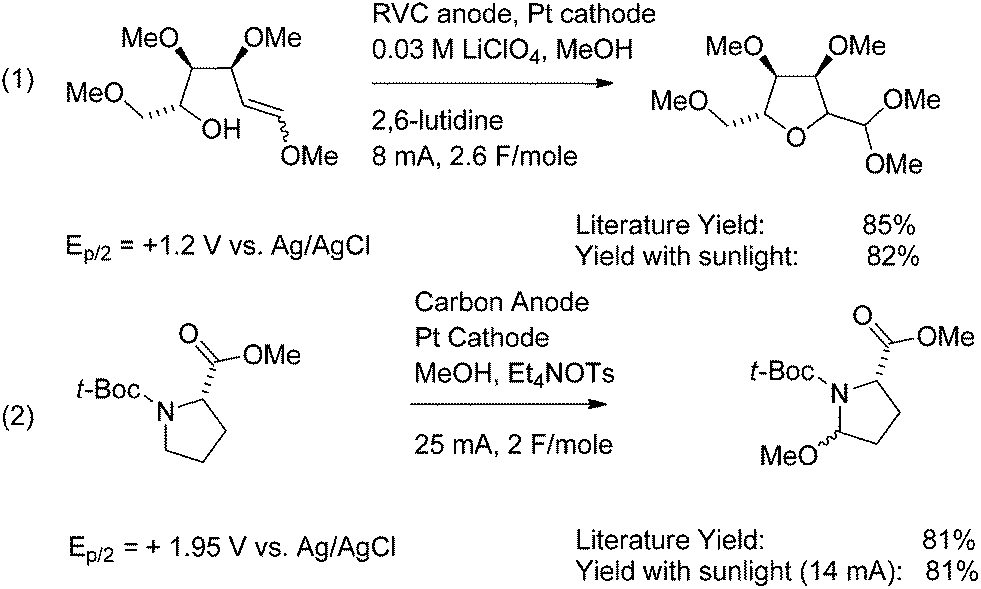 | ||
| Scheme 1 | ||
Both of the reactions illustrated in Scheme 1 were run by passing a constant current of electricity through the electrochemical cell. Reactions run in this fashion are extremely versatile. During a constant current electrolysis, the potential at the anode climbs until it matches that of the functional group with the lowest oxidation potential. The potential at the anode then holds constant until the majority of the functional group is consumed. After that point, the potential at the electrode again climbs until it matches that of the functional group with the second lowest oxidation potential in solution. By keeping the current density low, the potential at the anode can be maintained at the level of the first group until 90% or more of the original functional group is consumed. Substrates with oxidation potentials of +1.2 V vs. Ag/AgCl (Ep/2) (eqn (1)) or +1.95 V vs. Ag/AgCl (Ep/2) (eqn (2)) can both be oxidized using the exact same electrochemical apparatus without any need to manually adjust the potential.
While electrolysis reactions like those illustrated in Scheme 1 can be very powerful synthetic tools, the same feature that makes the reactions so versatile with respect to changing oxidation potentials of the substrates can severely limit the reactions. For example, if a functional group targeted for oxidation is not the group in the reaction with the lowest oxidation potential, then a direct electrolysis approach has no chance of working. A constant current electrolysis will always select for the functional group with the lowest oxidation potential.
Oxidation reactions that take advantage of chemical reagents do not have this restriction. A chemical oxidant can select based on sterics, chirality, binding properties, etc. In this way, the use of a chemical oxidant offers opportunities for selectivity in an oxidation reaction that are unavailable to a direct electrolysis. It was this observation that led to the development of indirect electrolysis reactions.8 In an indirect electrolysis, a chemical oxidant serves as a mediator for the electrolysis (Scheme 2).
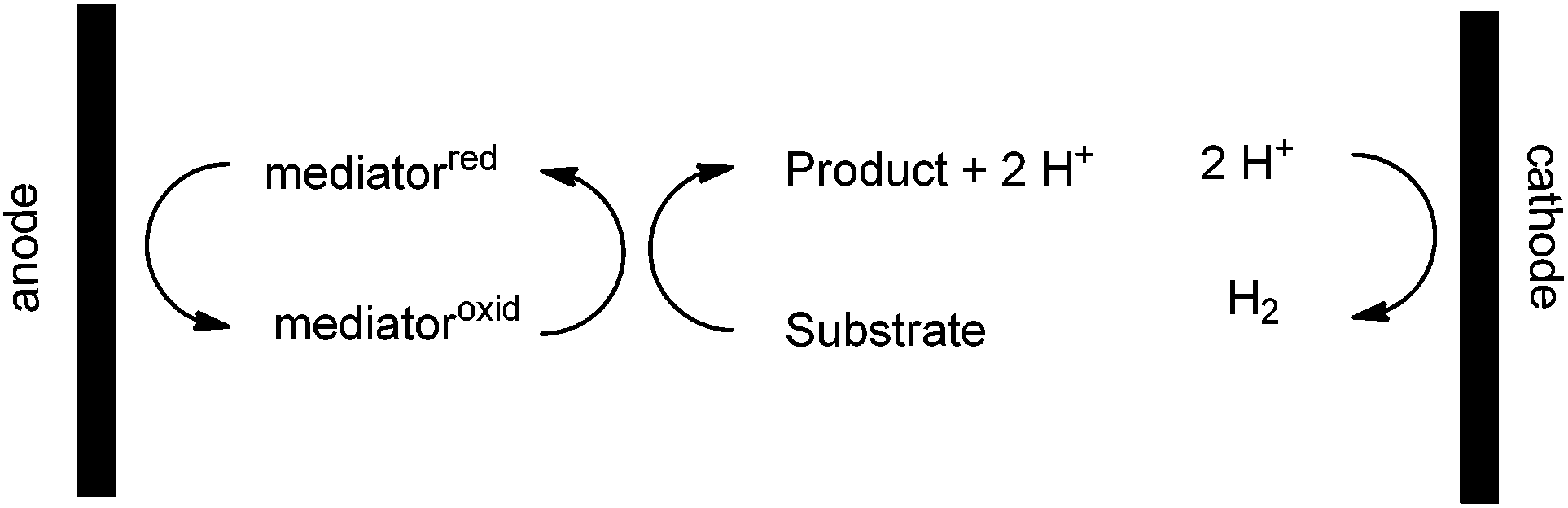 | ||
| Scheme 2 | ||
The oxidant formed at the anode oxidizes a substrate in solution and then is regenerated again at the anode. This allows the chemical oxidant to be used in a catalytic fashion while again channeling the necessary reduction reaction toward the production of hydrogen gas. Hence, the indirect electrolysis allows one to capitalize on the selectivity of the chemical reagent while retaining the advantages of the electrolysis.
Indirect electrolyses often capitalize on constant current electrolysis conditions. In an indirect oxidation, this means that the oxidation potential at the surface of the anode again climbs until it matches that of the most easily oxidized group in solution. In this case, that group is the reduced form of the mediator (chemical oxidant). The potential at the anode then holds constant for the duration of the experiment because the overall process is catalytic. Since the chemical oxidant is recycled and never consumed, the oxidation potential at the anode never climbs. The result is that the same very simple “solar-electrochemical” cell used for direct electrolysis reactions should be compatible with the recycling of a wide variety of chemical oxidants. We report here that this is the case.
Our initial focus was to demonstrate that the indirect electrolysis reactions already reported in the literature could be conducted with the use of a simple photovoltaic as the power supply. To this end, a solar-driven version of the Torii procedure for conducting electrochemical Sharpless dihydroxylations was examined (Scheme 3).9 Styrene was introduced into a vial equipped with a reticulated vitreous carbon (RVC) anode and a carbon rod cathode along with 10 mole% of potassium ferricyanide, 0.2 mole% of K2OsO2(OH)4, 1 mole% of (DHQD)2PHAL, 3 equiv. of potassium carbonate, and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 t-BuOH–H2O mixture of solvent. Current (2.5 mA, 2.5 F mole−1) was then passed through the flask using the reaction setup shown in Fig. 1 to afford a 92% yield of the diol product in 94% ee. For comparison, the literature yield for the oxidation of styrene with a more traditional electrolysis setup was 95% with a 97% ee. It was clear that the optimized electrochemical reaction could be mimicked nicely with the very simple solar-electrochemical cell, an observation that suggests that the electrochemical method can be readily adopted by synthetic chemists without any need for specialized equipment.
:1 t-BuOH–H2O mixture of solvent. Current (2.5 mA, 2.5 F mole−1) was then passed through the flask using the reaction setup shown in Fig. 1 to afford a 92% yield of the diol product in 94% ee. For comparison, the literature yield for the oxidation of styrene with a more traditional electrolysis setup was 95% with a 97% ee. It was clear that the optimized electrochemical reaction could be mimicked nicely with the very simple solar-electrochemical cell, an observation that suggests that the electrochemical method can be readily adopted by synthetic chemists without any need for specialized equipment.
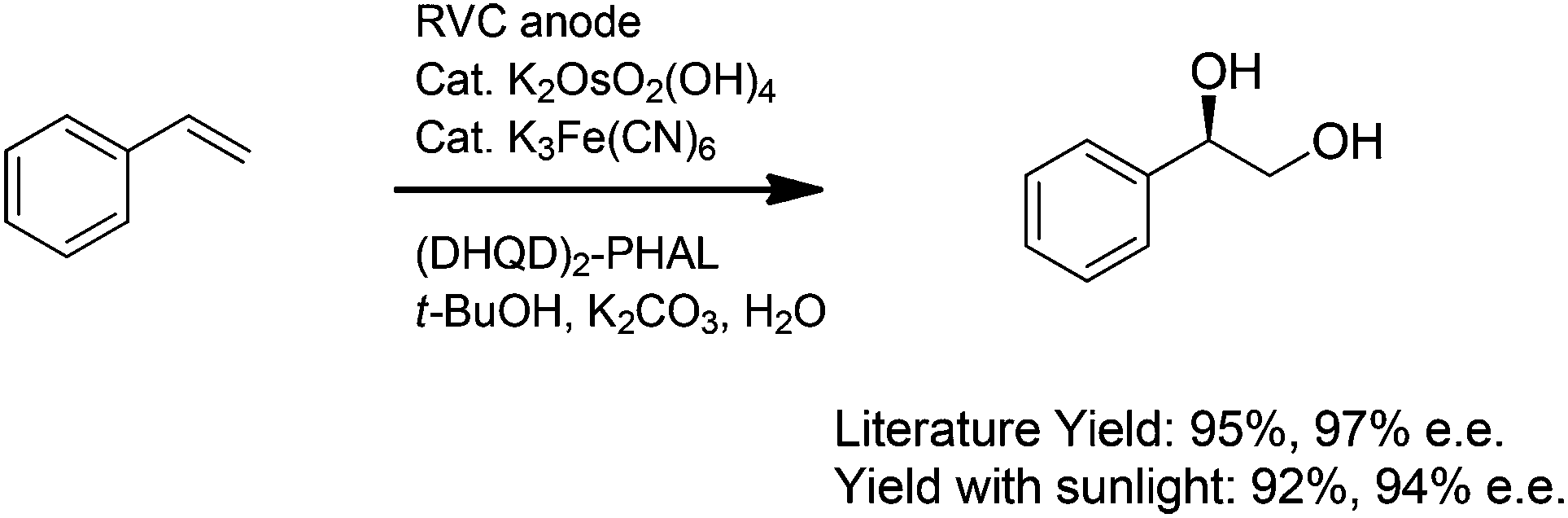 | ||
| Scheme 3 | ||
The exact same reaction setup was also used to run a mediated anodic oxidation of a sugar substrate. In this case, the less sterically-hindered primary alcohol of the sugar was selectively oxidized by using TEMPO as the mediator for the oxidation (Scheme 4).10 Once again, the yield of the sunlight-driven reaction was similar to the yield reported in the literature for an electrolysis that had utilized more traditional electrochemical equipment.
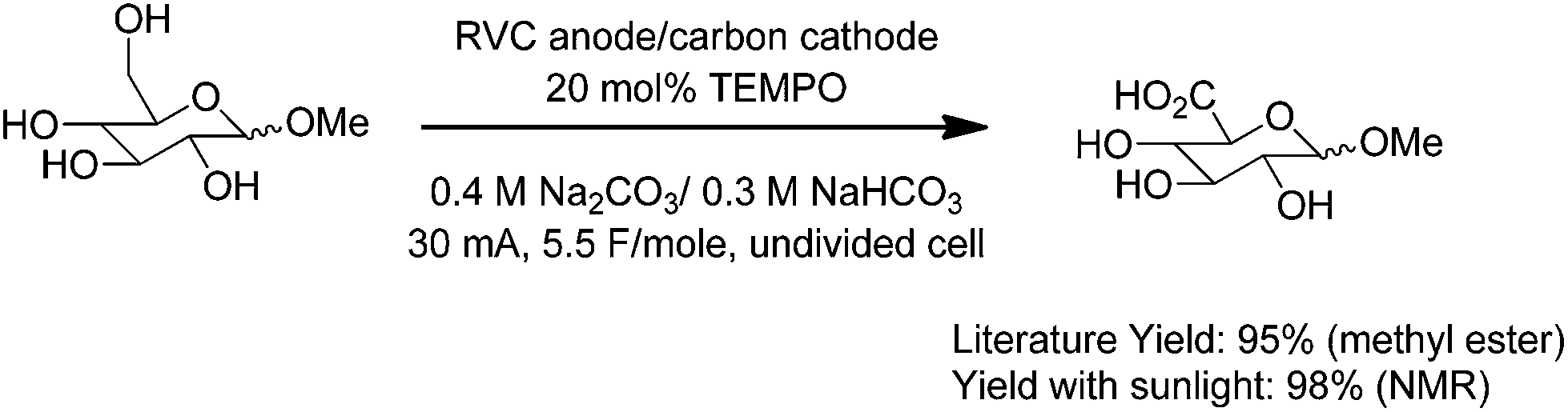 | ||
| Scheme 4 | ||
The sunlight-driven reactions can also be used to mimic more complex electrochemical reactions. In electrolyses that utilize transition metal mediators, a lack of solubility for the reduced form of the mediator can represent a significant challenge. The main issue occurs when the oxidized form of the mediator encounters the cathode. A reduction at that point leads to destruction of the oxidant. If the reduced form of the metal is soluble, then reduction at the cathode is not a major problem because the mediator will simply be reoxidized at the anode. The reaction loses current efficiency, but in a sunlight-driven process this concern is minimal. However, if the reduced form of the mediator is not soluble, then it is deposited on the cathode. This removes the mediator from solution, a situation that leads to changes in the potential at the anode and undesired direct oxidation reactions. Two methods have been employed to address this second scenario. First, the direction of the current flow in the reaction can be switched. This change converts the electrode initially used as an anode to a cathode and the electrode initially used as a cathode to an anode. For the Ru(VIII)-oxidation shown in Scheme 5,11 the optimized literature procedure calls for switching the direction of the current in the reaction every 30 seconds. Ruthenium that deposited on the original cathode during the first 30 s is oxidized when that electrode becomes an anode for the subsequent 30 s. In this way, the mediator is constantly reintroduced into the reaction medium. Mimicking the reaction with a sunlight-driven experiment requires the same switching of current direction. When the direction of the current was switched at a rate of once every 30 minutes, the yield of the reaction with the solar-electrochemical setup was only 40%. The yield was raised to 82% by switching the direction of the current every 1 to 2 min. This was done by manually switching the leads on the setup shown in Fig. 1. Clearly, the reaction could be further optimized by automating the switching process so that the 30 s time scale used in the optimized literature reaction was mimicked more accurately. However, to test whether an electrochemical reaction in the literature will work for a specific experiment of interest, this simple method is very effective.
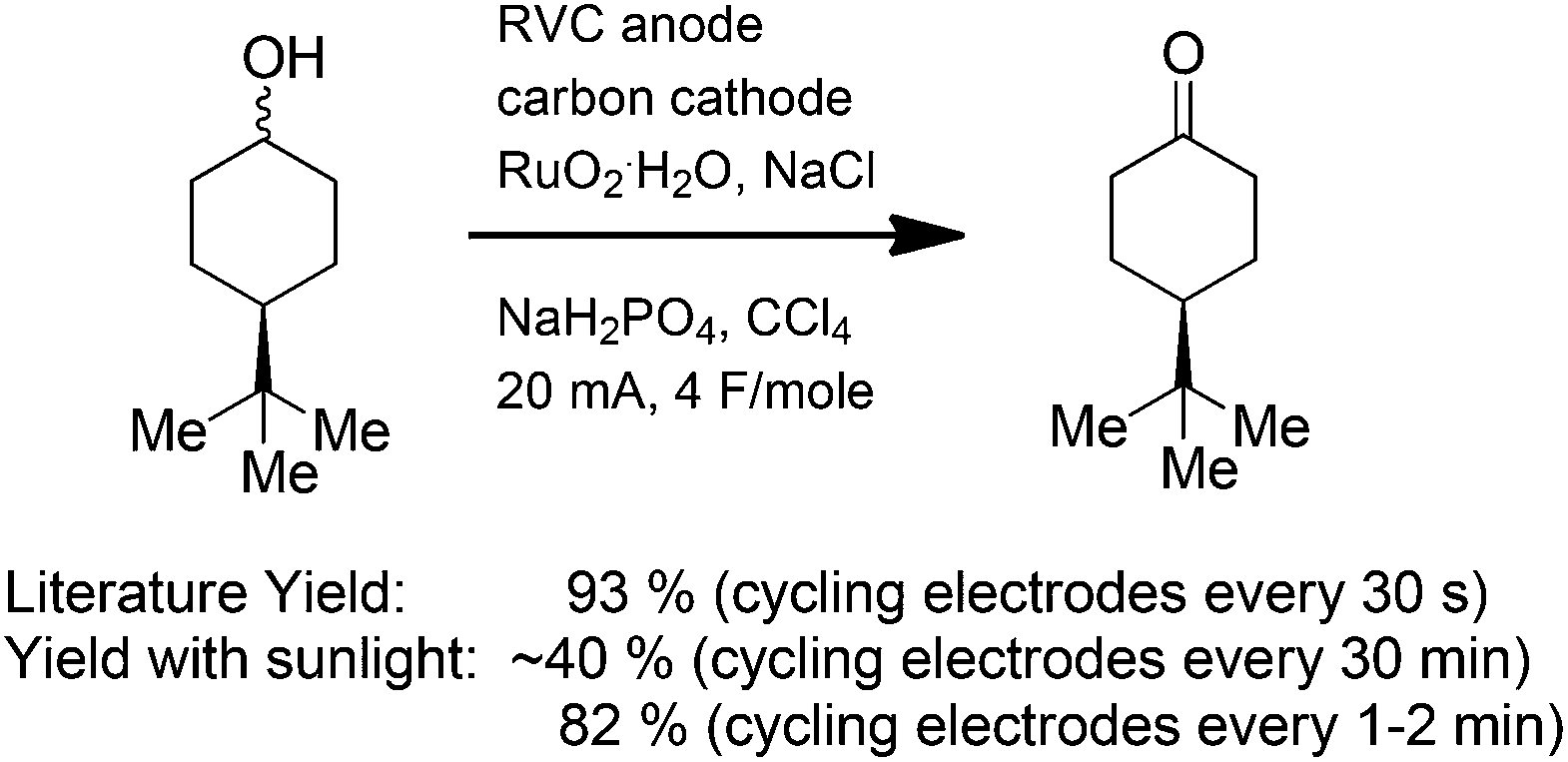 | ||
| Scheme 5 | ||
The second approach to circumvent mediator deposition on the cathode uses a divided cell for the electrolysis. This prevents the chemical oxidant generated at the anode from ever reaching the cathode. This approach is illustrated with the two Pd(II)-mediated oxidation reactions shown in Scheme 6. Both reactions were compatible with the use of the simple solar-electrochemical reaction setup shown in Fig. 1. The electrochemical Wacker oxidation (eqn (1)) proceeded in an 81% isolated yield with the use of a photovoltaic as the power supply.12 In a similar fashion, the oxidative Heck reaction (eqn (2)) proceeded in a 68% isolated yield with the use of a photovoltaic as the power supply.13
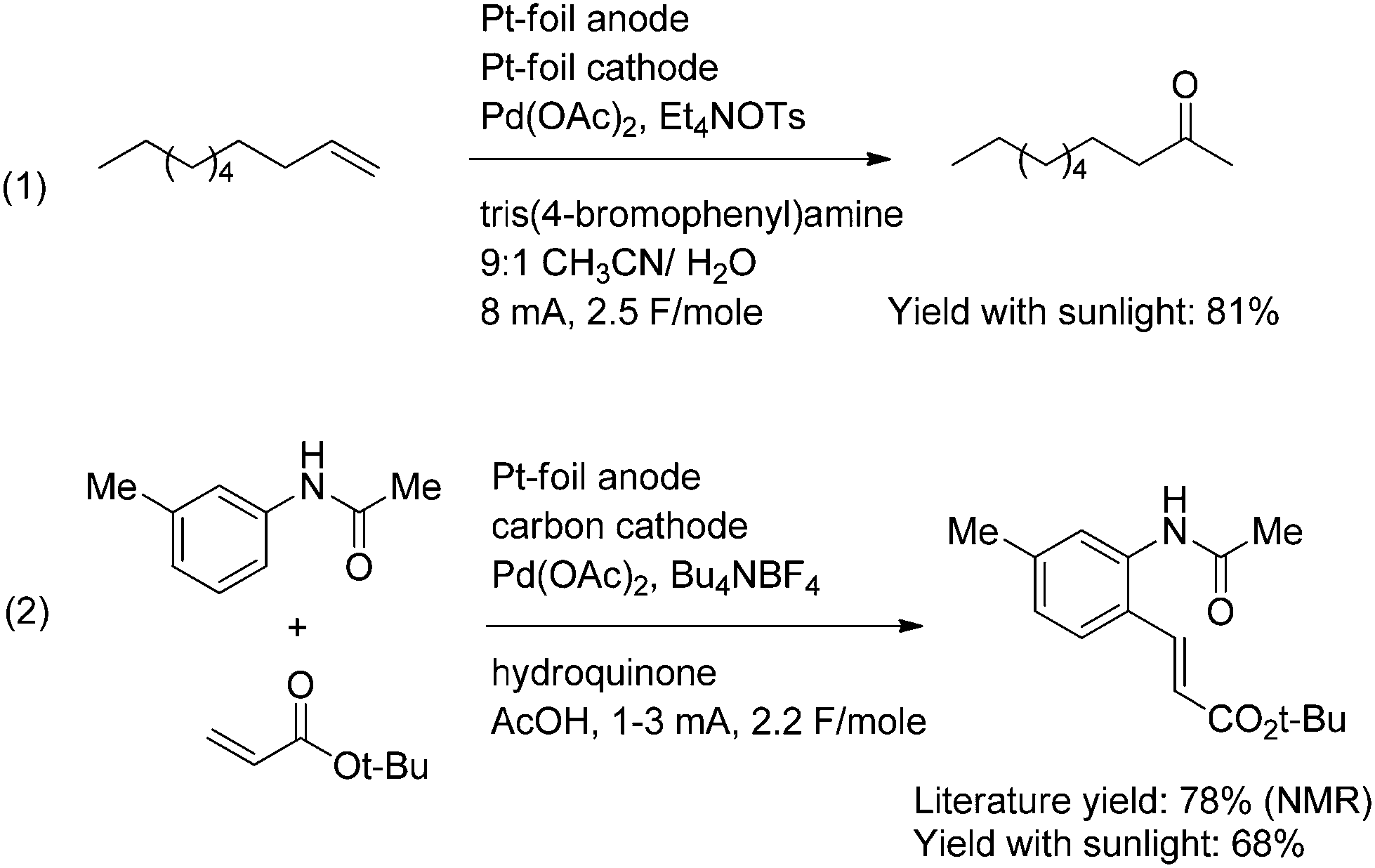 | ||
| Scheme 6 | ||
In order to further demonstrate the range of reactions that can be run in a catalytic fashion using the electrochemical apparatus shown in Fig. 1, both manganese(V)- and cerium(IV)-mediated electrolyses were conducted (Scheme 7). For Mn(V), the asymmetric epoxidation of cis-β-methylstyrene was accomplished.14 The reaction used Jacobsen's catalyst as a mediator, and it afforded a 47–71% yield (NMR) of the product in a 87% ee. In a similar fashion, the Ce(IV)-mediated deprotection of a para-methoxybenzyl alcohol proceeded in an 88% yield (NMR) of the product.15
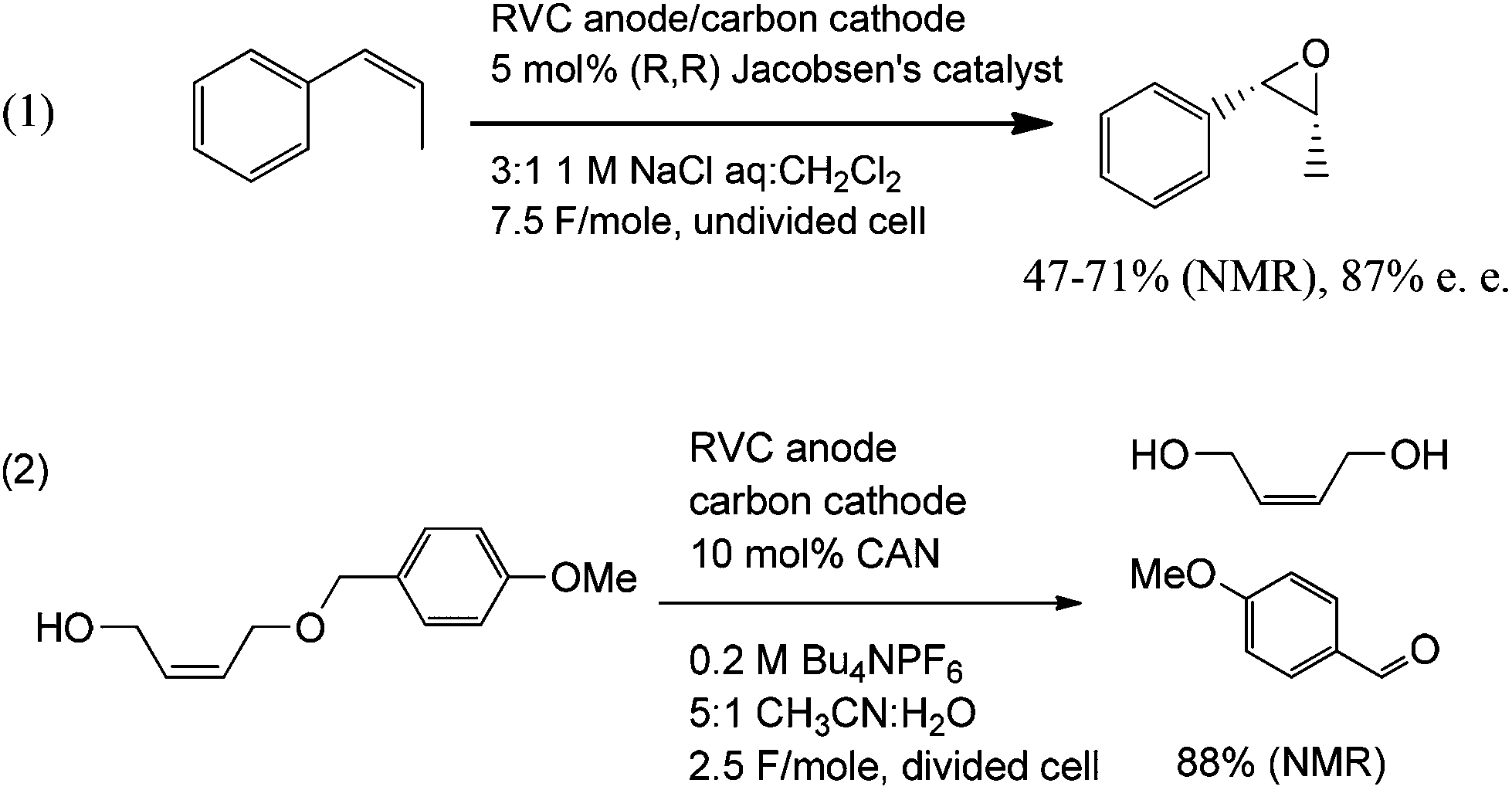 | ||
| Scheme 7 | ||
For each of the reactions illustrated in Schemes 1–7, the same very simple sunlight-driven electrochemical apparatus was used. From direct electrolyses to asymmetric oxidation reactions, the potential at the working electrode automatically adjusted to match that of the substrate or mediator. The result is a very general, readily adoptable method for running sustainable oxidation reactions.
Acknowledgements
We thank the National Science Foundation (CHE-1151121) and (CHE-1240194/CenSURF) for their generous support of this work.References
- For reviews see: (a) A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45(6), 851 CrossRef CAS PubMed; (b) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111(4), 2981 CrossRef CAS PubMed; (c) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50(47), 11062 CrossRef CAS PubMed.
- For selected recent examples see: (a) L. C. John, A. Gunay, A. J. Wood and M. H. Emmert, Tetrahedron, 2013, 69, 5758 CrossRef CAS; (b) A. M. Suess, M. Z. Ertem, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 9797 CrossRef CAS PubMed; (c) J. Kim and S. S. Stahl, ACS Catal., 2013, 3, 1652 CrossRef CAS PubMed; (d) D. Pun, T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 8213 CrossRef CAS PubMed; (e) T. Diao, D. Pun and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 8205 CrossRef CAS PubMed; (f) A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415 CrossRef CAS PubMed; (g) R. M. Leithall, V. N. Shetti, S. Maurelli, M. Chiesa, E. Gianotti and R. Raja, J. Am. Chem. Soc., 2013, 135, 2915 CrossRef CAS PubMed; (h) R. J. Angelici, Catal. Sci. Technol., 2013, 3, 279 RSC; (i) S. Kanbak-Aksu, H. M. Nahid, W. R. Hagen, R. Hollmann, D. Sordi, R. A. Sheldon and I. W. C. E. Arends, Chem. Commun., 2012, 48, 5745 RSC.
- For recent reviews of electrochemical reactions see: (a) J. B. Sperry and D. L. Wright, Chem. Soc. Rev., 2006, 35, 605 RSC; (b) J. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2009, 108, 2265 CrossRef PubMed; (c) B. S. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099 RSC.
- L. A. Anderson, A. Redden and K. D. Moeller, Green Chem., 2011, 13, 1652 RSC.
- The photovoltaic-cells employed in these experiments are available from Solar-Made.
- For the specific cyclization reaction shown see: G. Xu and K. D. Moeller, Org. Lett., 2010, 12, 2590 CrossRef CAS PubMed.
- For the oxidation of the t-Boc protected methylproline see: Y. M. Fobian and K. D. Moeller, Methods Mol. Med., 1999, 23(Peptidomimetic Protocols), 259 CAS.
- For reviews of indirect electrochemical methods see: (a) Y. N. Ogivin, M. N. Elinson and G. I. Nikishin, Russ. Chem. Rev., 2009, 78, 89 CrossRef; (b) J. Simonet and J.-F. Pilard, in Organic Electrochemistry: Fourth Edition, Revised and Expanded, ed. H. Lund and O. Hammerich, Marcel Dekker, New York, 2001, p. 1163 Search PubMed; (c) E. Steckhan, Angew. Chem., Int. Ed. Engl., 1986, 25, 683 CrossRef.
- S. Torii, P. Liu and H. Tanaka, Chem. Lett., 1995, 319 CrossRef CAS.
- K. Schnatbaum and H. J. Schäfer, Synthesis, 1999, 864 CrossRef CAS.
- S. Torri, T. Inokuchi and T. Sugiura, J. Org. Chem., 1986, 51, 155 CrossRef.
- T. Inokuchi, L. Ping, F. Hamaue, M. Izawa and S. Torii, Chem. Lett., 1994, 121 CrossRef CAS.
- C. Amatore, C. Cammoun and A. Jutand, Adv. Synth. Catal., 2007, 349, 292 CrossRef CAS.
- H. Tanaka, M. Kuroboshi, H. Takeda, H. Kanada and S. Torii, J. Electroanal. Chem., 2001, 507, 75 CrossRef CAS.
- For the analogous recycling of Ce(IV) at an anode see: D. Kesselring, K. Maurer and K. D. Moeller, J. Am. Chem. Soc., 2008, 130, 11290 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A sample experimental procedure is included for the oxidation reaction along with characterization data for all new compounds. See DOI: 10.1039/c3gc41650j |
| This journal is © The Royal Society of Chemistry 2014 |