Common processes drive the thermochemical pretreatment of lignocellulosic biomass†
Paul
Langan
abc,
Loukas
Petridis
def,
Hugh M.
O'Neill
ac,
Sai Venkatesh
Pingali
ac,
Marcus
Foston‡
g,
Yoshiharu
Nishiyama
h,
Roland
Schulz
def,
Benjamin
Lindner
def,
B. Leif
Hanson
b,
Shane
Harton§
i,
William T.
Heller
a,
Volker
Urban
ac,
Barbara R.
Evans
j,
S.
Gnanakaran
k,
Arthur J.
Ragauskas
g,
Jeremy C.
Smith
def and
Brian H.
Davison
*d
aBiology and Soft Matter Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
bDepartment of Chemistry, University of Toledo, Toledo, OH 43606, USA
cCenter for Structural Molecular Biology, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
dBioscience Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: davisonbh@ornl.gov
eDepartment of Biochemistry and Cellular and Molecular Biology, University of Tennessee, Knoxville, TN 37996, USA
fUT/ORNL Center for Molecular Biophysics, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
gInstitute of Paper Science and Technology, School of Chemistry and Biochemistry and Georgia Institute of Technology, Atlanta, GA 30332, USA
hCentre de Recherches sur les Macromolécules Végétales (CERMAV-CNRS), BP 53, F-38041 Grenoble Cedex 9, France
iMaterials Science and Technology Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
jChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
kTheoretical Biology & Biophysics Group, Los Alamos National Laboratory, Los Alamos, NM 87545, USA
First published on 29th October 2013
Abstract
Lignocellulosic biomass, a potentially important renewable organic source of energy and chemical feedstock, resists degradation to glucose in industrial hydrolysis processes and thus requires expensive thermochemical pretreatments. Understanding the mechanism of biomass breakdown during these pretreatments will lead to more efficient use of biomass. By combining multiple probes of structure, sensitive to different length scales, with molecular dynamics simulations, we reveal two fundamental processes responsible for the morphological changes in biomass during steam explosion pretreatment: cellulose dehydration and lignin-hemicellulose phase separation. We further show that the basic driving forces are the same in other leading thermochemical pretreatments, such as dilute acid pretreatment and ammonia fiber expansion.
Lignocellulosic biomass, the major plant cell-wall material, is composed mainly of aligned bundles of crystalline cellulose fibrils embedded in a hydrated matrix of less ordered, covalently coupled, hemicellulose and lignin polymers. This complex cell wall architecture evolved in plants for strength and resistance to degradation, and consequently makes biomass recalcitrant to industrial hydrolysis to glucose for the production of fuels and chemicals via microbial fermentation or chemical catalysis.1–4 Expensive chemical and enzymatic pretreatments are currently required prior to fermentation to disrupt morphological, dynamic, and mechanical barriers that limit accessibility of cellulases to their cellulose substrates and subsequently their catalytic activities.5–7 We assembled a platform of experimental techniques that allowed us to image cell wall morphology, and its response to pretreatment, over the large range of relevant length scales. The experimental results were interpreted using simulation techniques, thereby revealing underlying processes that drive these changes.
In order to understand the hierarchical cell wall structure of biomass responsible for its recalcitrance, we chose to analyze wood chips from aspen trees (Populus tremuloides) due to their potential as an abundant, low cost, feedstock.8 Compositional analysis shows the chips contain mostly cellulose (Fig. S1†). At the molecular level, X-ray studies reveal aligned, partly crystalline cellulose fibrils, Fig. 1A, containing crystalline domains of 2–3 nm in diameter (Table S1, Fig. S2†). Previous electron microscopic analysis of wood indicated that the fibrils are 3–4 nm in diameter.9 These results are consistent with a model in which the cellulose chains pack in a well-ordered manner in the fibril core with a progressive loss of order from the center out, so that the chains become more disordered toward their hydrated surfaces.10 Small angle neutron scattering (SANS) from chips, soaked in heavy water to improve the scattering contrast of cellulose, revealed an equatorial diffraction feature due to side-by-side packing of fibrils bundled tightly together in macrofibrillar bundles, Fig. 1C and 1E. A similar approach was recently used to characterize lateral packing of elementary cellulose fibrils in spruce wood.11
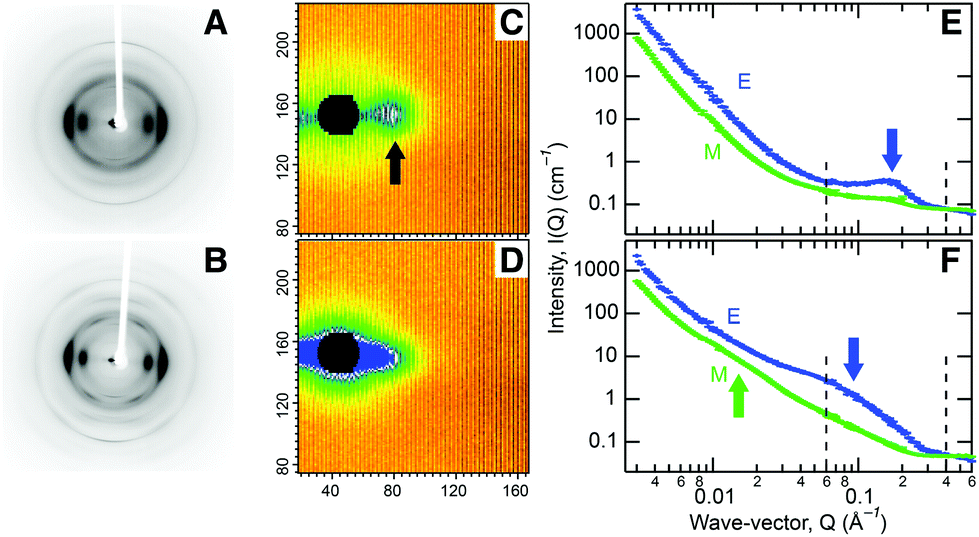 | ||
| Fig. 1 X-ray fiber diffraction (A and B) and Small Angle Neutron scattering (C and D) patterns collected from P. tremuloides chips that were (A and C) untreated Aspen and (B and D) steam explosion pretreated (SEP). The equatorial and meridional directions are horizontal and vertical, respectively. X-ray diffraction spots become sharper after pretreatment, indicating an increase in cellulose crystalline domain size. An equatorial neutron diffraction feature, indicated by the black arrow (C), corresponds to side-by-side packing of cellulose fibrils. Scans through the neutron diffraction patterns collected from untreated and SEP treated samples are shown in E and F, respectively. Scans in the equatorial direction are colored blue and labeled E and scans in the meridional direction are colored green and labeled M. The equatorial scans show that the diffraction feature, indicated by a blue arrow, broadens and moves to smaller scattering angle. The meridional scans show that a scattering shoulder, indicated by a green arrow, appears after pretreatment that corresponds to lignin aggregates. This scattering shoulder is relatively isotropic and is also present in the equatorial scan, although it is difficult to see because of the dominant presence of the diffraction feature. The wave-vector (Q) along the horizontal axes in E and F is related to the distance in Angstroms (d) by the expression Q = 2π/d. | ||
Our compositional analysis (Fig. S1†), the measured moisture content (59%) of never-dried chips kept at 100% relative humidity, and the ease of exchange of labile hydrogen atoms by deuterium, together indicate that equal parts of hemicellulose and lignin form a hydrated, homogeneous matrix. Taken together these results tell us that cellulose is biosynthesized as elementary fibrils, with well-ordered cores and hydrated, disordered surfaces, with regions in which they are packed together tightly in macrofibrillar bundles with little room for interpenetration of matrix polymers (Fig. 4A). The cellulose is surrounded by a disordered hydrated matrix of covalently linked hemicellulose and lignin polymers which contains voids but which physically shields cellulose from cellulases.
We then examined the chips as they were subjected to steam explosion pretreatment (SEP) to characterize the resulting morphological changes. The invariance of the SANS data in the very low scattering angle region indicates that cell wall morphology does not change at corresponding length scales of a few hundred nanometers (Fig. 1D). However, we see X-ray diffraction features becoming sharper (Fig. 1B) due to an increase in the size of crystalline regions to 4 nm, matching the diameter of the cellulose fibrils (Table S1†). Further, the strong SANS equatorial diffraction feature broadens and moves to lower scattering angle, indicating an increase in the lateral side-by-side packing distance of cellulose fibrils to 9 nm (Fig. 1D and 1F), matching the macrofibrillar bundle diameter observed by microscopy.12 This diffraction feature remained after bleaching to remove lignin (Fig. S3E†), verifying its association with cellulose.
The origin of the above changes was investigated using molecular dynamics (MD) simulation of a bundle of seven fibrils with two hydration shells initially separating them (Fig. 2A). At room temperature (RT) the hydration shells remain for the duration of the simulation, with the number of core water molecules, W, that lie within 7 Å of the central fibril remaining approximately constant (Fig. 2D). W sharply decreases in simulations at SEP temperatures (Fig. 2D), indicating that water is expelled from the fibril interface. The fibrils begin coalescing into macrofibrils as surface cellulose chains lose their hydration and pack closely (Fig. 2A–2C). After decreasing the temperature back to RT, W remains approximately constant, indicating water expulsion is a non-equilibrium process. This behavior explains the increase in side-by-side ordering of cellulose fibrils we observed by SANS, and the increase in crystalline order within fibrils we observed by X-rays. Diffraction peaks calculated from the simulated model cellulose bundle show an increase in sharpness after pretreatment supporting this interpretation (Fig. S5A†). Initially, the core water molecules have significantly lower translational and rotational entropy (∼51 J mol−1 K−1) than the bulk (∼67 J mol−1 K−1) (see Fig. S5 and Table S2†), so their confinement is entropically unfavorable. On the other hand, core water molecules form stronger hydrogen bonds than bulk, indicating cellulose hydration is enthalpically favored (Fig. 2E). The entropic penalty of confinement increases at SEP temperatures, thus leading to the release of water and to the fibril bundle collapsing to a dehydrated state. Our simulations do not determine whether the wet core or dry core state is thermodynamically more stable at RT. However, the irreversible nature of the core water removal indicates that large barriers could be separating these two states.
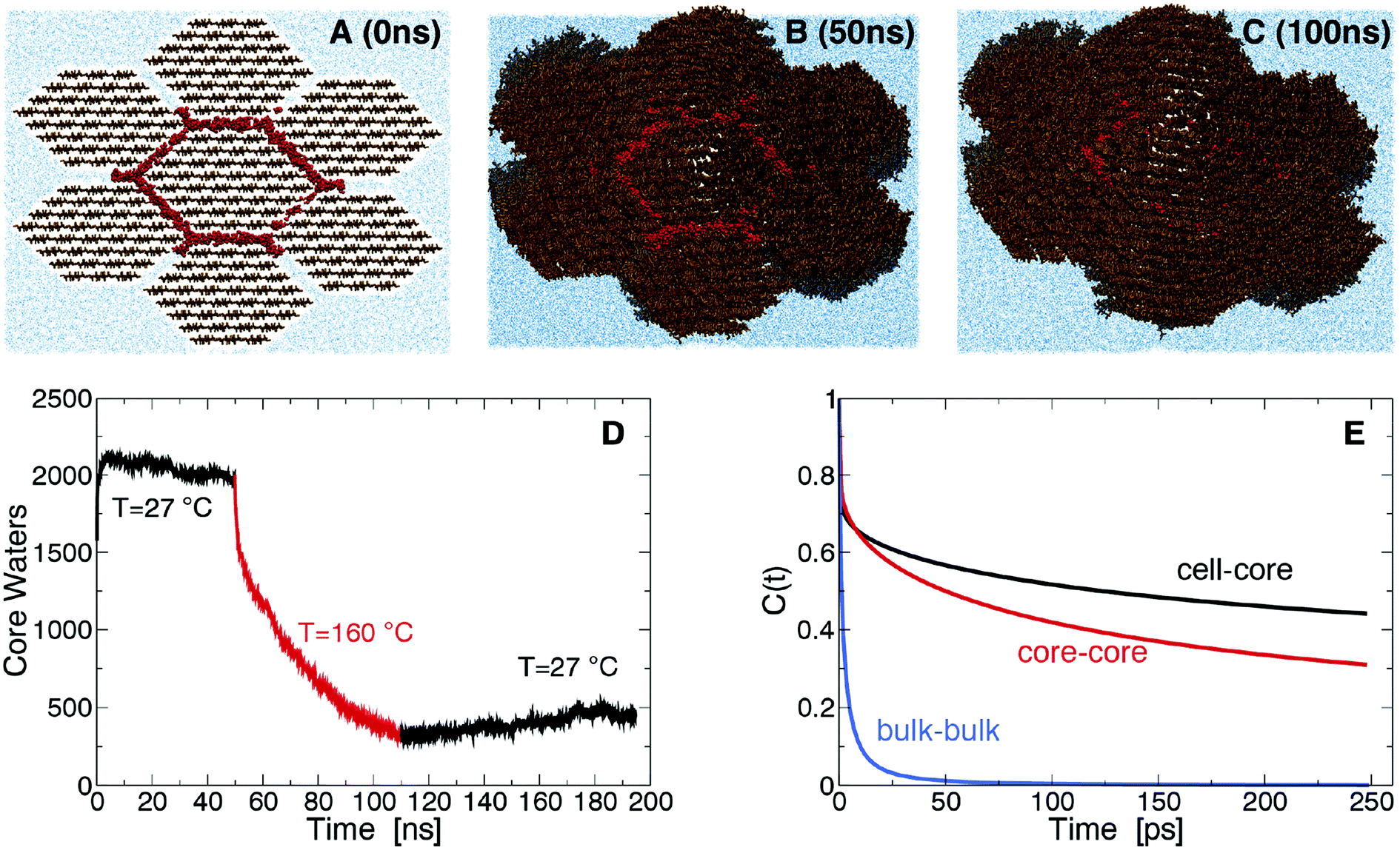 | ||
| Fig. 2 Snapshots of the macrofibrillar cellulose bundle system simulated after 0 ns (A), 50 ns (B) and 100 ns (C). Cellulose is shown in brown, water in blue and core water molecules in red. The number of core water molecules as a function of time, t, is plotted in D, for t < 50 ns and t > 110 ns the simulations were run at 27 °C and for 50 ns < t < 110 ns at 160 °C. Hydrogen bond autocorrelation function (eqn (S1)†) is shown in E. | ||
Significant changes also occur in the encrusting matrix during pretreatment. A feature can be seen in our off-equatorial SANS data after SEP arising from the emergence of 30–40 nm diameter globular aggregates of lignin (Fig. 1F). This feature diminishes after bleaching to remove lignin, confirming its origin (Fig. S3E†). The relative hemicellulose content decreases from 24 to 18% (Fig. S1†) because of auto-catalyzed hydrolysis; results reported by others show enzymatic isolated lignin from steam pretreated poplar has a residual sugar content and suggest after pretreatment hemicellulose remains associated with lignin most likely through covalent lignin–carbohydrate complexes (LCC).13
To understand this behavior an entangled block lignin-hemicellulose copolymer was constructed (Fig. 3A) for MD simulation in aqueous solution. At RT, after ∼100 ns of simulation the fraction, f, of lignin monomers in contact with hemicellulose decreased to a constant ∼0.58 ± 0.06 (Fig. 3B). In contrast, at SEP temperatures f decreased to ∼0.25 ± 0.12 (Fig. 3C), indicating that these components are able to separate into lignin-rich and hemicellulose-rich phases. After returning the system to RT (time > 400 ns), the lignin and hemicellulose remain loosely coupled, f ∼ 0.29 ± 0.05. At both RT and the elevated SEP temperatures the hemicellulose component behaves as a coil polymer in good solvent. In contrast, at RT lignin behaves in a manner consistent with theoretical predictions for collapsed “equilibrium” globules, in which monomers distant along the chain have a relatively high probability of being close in space.14,15 At SEP temperatures, the lignin acquires a “crumpled” globule conformation in which the lignin is collapsed, but the monomers that are distant along the chain are also distant in space (15). The phase separation of the relatively hydrophobic lignin and hydrophilic hemicellulose occurs while the two polymers remain cross-linked, and is consistent with the emergence of SANS scattering contrast between lignin globules and more dispersed hemicellulose.
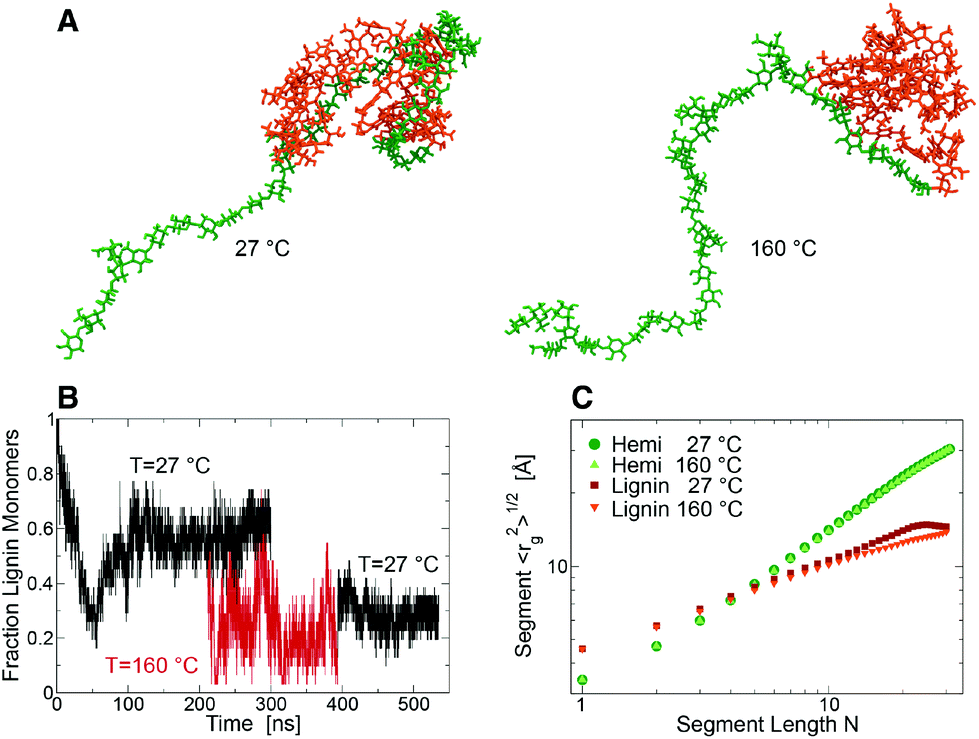 | ||
| Fig. 3 (A) Snapshots of the simulated lignin (orange)-hemicellulose (green) copolymer at 27 °C (138 ns) and 160 °C (366 ns). (B) Fraction of lignin monomeric units forming at least one atomic contact with hemicellulose at 27 °C (black) and 160 °C (red). The configuration of the system at 211 ns at 27 °C was the starting configuration of the 160 °C simulation. (C) Root mean square of the radius of gyration rg(N) of lignin and hemicellulose polymer segments comprising (N + 1) monomers calculated at 27 and 160 °C. At 27 °C data are averaged over the last 230 ns of the simulation. | ||
There is not a large increase in the calculated density of lignin within the globules that are formed during these simulations. However, as hemicellulose untangles and separates from the globules, hydrated hemicellulose chains are extruded of increasing length, and the cell wall must therefore swell in volume. This will cause neighboring lignin globules to be more distant, or at least exert a repulsive pressure. Calculations indicate that the end-to-end distance of the extruded chain of hemicellulose not in contact with lignin, Re, increases from ∼5 nm to ∼9 nm as the simulation temperature is cycled from 27 °C to 160 °C and back to 27 °C (Fig. S6†). In an unconstrained hydrated system of closely packed globules, this would cause the globules to separate with the density of globules varying as ∼Re−3i.e. the density of globules decreases six-fold, the latter being a qualitative measure of cellulose accessibility. In the constrained environment of the cell wall this phase separation and the pressure of lignin globules to separate, will cause swelling and will expand the size of small voids by a few nanometers.
The above results provide a new conceptual understanding of the underlying processes that change biomass morphology during SEP, Fig. 4. As the temperature is increased during SEP core water is released to the surrounding matrix in a thermodynamically entropy-driven process, causing the fibrils to coalesce into bundles with more confined, and therefore ordered, surface chains (Fig. 4B). Lignin transitions to conformations that give rise to fewer lignin hemicellulose entanglements, allowing a phase separation, with lignin aggregating into crumpled globules and with some loss of hemicellulose through auto-hydrolysis (Fig. 4B). As hemicellulose untangles and extrudes itself from the lignin globules there is a pressure for the lignin globules to be pushed apart as the cell wall swells. When the temperature is brought back down to RT, the fibril bundles reach a more thermodynamically stable dehydrated state (Fig. 4C). The lignin globules collapse due to the unfavorable entropy of lignin hydration compared to bulk (Fig. 4C), but they are separated and pushed apart by the untangled and extruded hemicellulose chains, which increases the size of small water-filled voids within the cell wall matrix by a few nanometers. The thermodynamic balance between the entropy and enthalpy of hydration is a key driver for overcoming kinetic barriers associated with the metastable state into which the cell wall is biosynthesized.
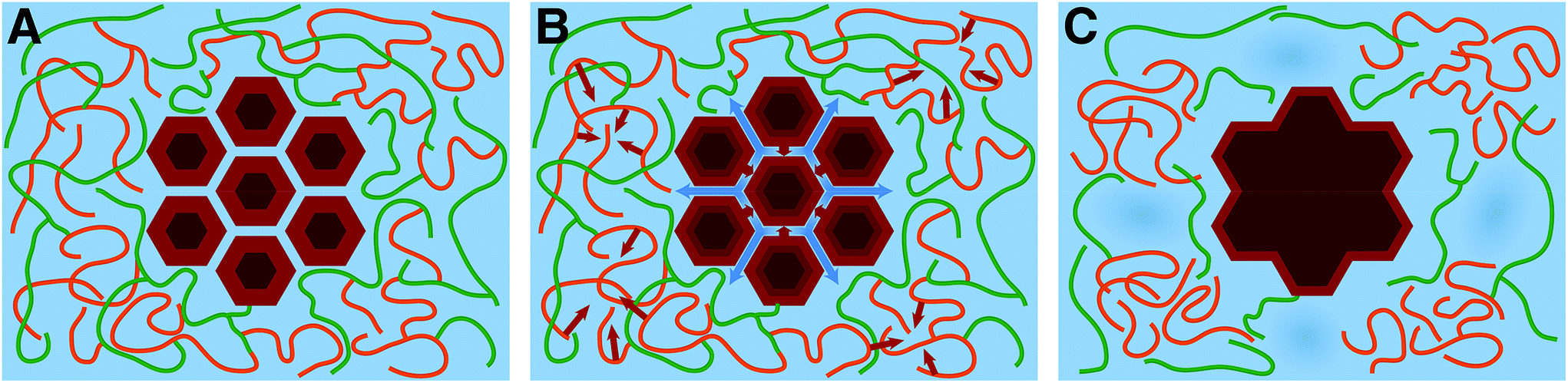 | ||
| Fig. 4 Schematic cartoon of SEP pretreatment process. (A) Native biomass consists of cellulose fibrils, shown with their well-ordered cores in dark brown and their more disordered and hydrated surface regions in light brown. The fibrils are packed together tightly in macrofibrillar bundles into which water (blue) penetrates. Fibril bundles are surrounded by a hydrated matrix of covalently linked hemicellulose (green) and lignin (orange). (B) As the temperature is increased during SEP pretreatment the core water molecules are expelled (the movement of water is represented by blue arrows) from the fibril bundles and the surrounding matrix goes through a phase separation, with the more hydrophobic lignin component aggregating (lignin aggregation is represented by orange arrows) into crumpled globules. (C) When the temperature is reduced back down to RT, the cellulose fibril bundles remain dehydrated and more ordered and the lignin globules collapse due to the unfavorable entropy of lignin hydration compared to bulk, producing an increase in the size of water-filled pores in the matrix. | ||
We then examined two other common thermochemical pretreatments, dilute acid pretreatment (DAP) and an ammonia fiber expansion (AFEX) that uses anhydrous ammonia,10,16 to see if they could also be understood by these newly revealed physical drivers. We found that DAP increases the sizes of crystalline domains within elementary fibrils (Table S1, Fig. S4†) and the aggregation of fibrils by dehydration (Fig. S3B†). DAP also causes lignin aggregation (Fig. S3B†), but more hemicellulose is lost (from 24% to 12%) than with SEP due to acid-catalyzed hydrolysis (Fig. S1†). The changes to the matrix can therefore be explained by the same phase separation process, but it is enhanced by increased hydrolysis of hemicellulose and covalent LCCs which then facilitates hemicellulose removal and the formation of larger pores.
During AFEX pretreatment, we found that ammonia molecules penetrate the cellulose structure and cause a solid state phase transition from the naturally occurring crystal forms of cellulose I17,18 to cellulose IIII![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19,20 that has enhanced sensitivity to hydrolysis.21 The crystalline domains within the elementary fibrils increase to 6–7 nm, which is greater than the original diameter of the fibrils (4 nm), indicating their coalescence (Table S1, Fig. S4†). The processes driving cellulose transformation in AFEX differ from SEP and DAP, but as with SEP and DAP, AFEX produces lignin aggregation (Fig. S3D†) through a separation of phases, with no significant loss of hemicellulose (Fig. S1†). Again, the rapid decompression occurring as a result of AFEX would also have a significant effect on cell wall structure.
19,20 that has enhanced sensitivity to hydrolysis.21 The crystalline domains within the elementary fibrils increase to 6–7 nm, which is greater than the original diameter of the fibrils (4 nm), indicating their coalescence (Table S1, Fig. S4†). The processes driving cellulose transformation in AFEX differ from SEP and DAP, but as with SEP and DAP, AFEX produces lignin aggregation (Fig. S3D†) through a separation of phases, with no significant loss of hemicellulose (Fig. S1†). Again, the rapid decompression occurring as a result of AFEX would also have a significant effect on cell wall structure.
The physical processes driving changes in cellulose structure during pretreatment observed here may also be involved in hornification; the irreversible stiffening and shrinking of wood or paper pulp after it has been dried.22 It has been proposed that hornification, occurring when water is removed from intra-lamellar microreticular pores,23 is related to an esterification process with covalent cross-links being formed between elementary fibrils during heating and drying that change mechanical properties of the macrofibrils and prevent their re-swelling in water.24 However, the results presented here suggest that coalescence of elementary cellulose fibrils into larger fibrils, with an increase in the size of ordered domains, as water is irreversibly released from the core of the macrofibrils after a kinetic barrier is overcome, could also contribute to hornification. Indeed, recent NMR results have shown than cellulose co-crystallization contributes to hornification and that there is a decrease in accessible cellulose surfaces as the cellulose fibrils aggregate.25,26 This would appear to support some aspects of earlier explanations of hornification that involve hydrated fibrils being brought together with the removal of water during drying, with the fibril surfaces therefore becoming inaccessible to water.27
Conclusions
We have discovered that some of the underlying physical processes responsible for the morphological changes in biomass during SEP and DAP are the same. The balance between the entropy and enthalpy of hydration is a unifying principle for driving the material over kinetic barriers. Further, although in AFEX pretreatment ammonia changes the morphology of cellulose fibrils differently, the matrix undergoes a phase separation that appears to be a fundamental physical mechanism. This phase separation may increase cell wall porosity across length scales. This is in agreement with other studies that have reported increases in cell wall porosity and accessible surface area as a result of pretreatments.28 Our findings suggest that new pretreatments and plant modifications that promote lignin and hemicellulose phase separation and increase the porosity of the cell wall matrix while preventing increases in cellulose crystallization as a result of dehydration will improve biomass conversion.Materials and methods
Details of materials and methods are provided as ESI.† Briefly, quaking aspen (Populus tremuloides) samples were obtained by Benchmark International in High Level, Alberta, Canada and intact chips were subjected to SEP, DAP and AFEX pretreatment, and lignin removal by bleaching. X-ray fiber diffraction data were collected with an in-house Rigaku FR-E with R-Axis IV++ detector. SANS data were collected at the CG-2 instrument at the High Flux Isotope Reactor (HFIR) facility of Oak Ridge National Laboratory. HPLC based anion-exchange chromatography was used for compositional analysis. MD simulations were performed employing the carbohydrate29,30 and lignin31 CHARMM force fields and the TIP3P water model.32Acknowledgements
The authors are grateful for the assistance and equipment provided by Constance Schall, University of Toledo, for preparation of ammonia fiber explosion and dilute acid samples. This research is funded by the Genomic Science Program, Office of Biological and Environmental Research, U. S. Department of Energy. The Center for Structural Molecular Biology (CSMB) and the Bio-SANS beam line is supported by the Office of Biological and Environmental Research, using facilities supported by the U. S. Department of Energy, managed by UT-Battelle, LLC under contract no. DE-AC05-00OR22725. This Research at Oak Ridge National Laboratory's High Flux Isotope Reactor was sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, US Department of Energy. This research used resources of NERSC, supported by the Office of Science of DOE under contract no. DE-AC02-05CH11231.Notes and references
- M. E. Himmel, S. Y. Ding, D. K. Johnson, W. S. Adney, M. R. Nimlos, J. W. Brady and T. D. Foust, Science, 2007, 315, 804–807 CrossRef CAS PubMed
.
- R. E. H. Sims, W. Mabee, J. N. Saddler and M. Taylor, Bioresour. Technol., 2010, 101, 1570–1580 CrossRef CAS PubMed
- M. Knauf and M. Moniruzzaman, Int. Sugar J., 2004, 106, 147–150 CAS
- H. Jorgensen, J. B. Kristensen and C. Felby, Biofuels Bioprod. Biorefin.-Biofpr, 2007, 1, 119–134 CrossRef
- P. Alvira, E. Tomas-Pejo, M. Ballesteros and M. J. Negro, Bioresour. Technol., 2010, 101, 4851–4861 CrossRef CAS PubMed
- B. S. Donohoe, M. J. Selig, S. Viamajala, T. B. Vinzant, W. S. Adney and M. E. Himmel, Biotechnol. Bioeng., 2009, 103, 480–489 CrossRef CAS PubMed
- R. Kumar and C. E. Wyman, Biotechnol. Bioeng., 2009, 102, 1544–1557 CrossRef CAS PubMed
- P. Sannigrahi, A. J. Ragauskas and G. A. Tuskan, Biofuels Bioprod. Biorefin.-Biofpr, 2010, 4, 209–226 CrossRef CAS
- T. Saito, S. Kimura, Y. Nishiyama and A. Isogai, Biomacromolecules, 2007, 8, 2485–2491 CrossRef CAS PubMed
- S. P. S. Chundawat, G. Bellesia, N. Uppugundla, L. D. Sousa, D. H. Gao, A. M. Cheh, U. P. Agarwal, C. M. Bianchetti, G. N. Phillips, P. Langan, V. Balan, S. Gnanakaran and B. E. Dale, J. Am. Chem. Soc., 2011, 133, 11163–11174 CAS
- A. N. Fernandes, L. H. Thomas, C. M. Altaner, P. Callow, V. T. Forsyth, D. C. Apperley, C. J. Kennedy and M. C. Jarvis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E1195–E1203 CrossRef PubMed
- K. Abe, S. Iwamoto and H. Yano, Biomacromolecules, 2007, 8, 3276–3278 CrossRef CAS PubMed
- S. Nakagame, R. P. Chandra and J. N. Saddler, Biotechnol. Bioeng., 2010, 105, 871–879 CAS
- A. Grosberg, Y. Rabin, S. Havlin and A. Neer, Europhys. Lett., 1993, 23, 373–378 CrossRef CAS
- L. Petridis, R. Schulz and J. C. Smith, J. Am. Chem. Soc., 2011, 133, 20277–20287 CrossRef CAS PubMed
- S. P. S. Chundawat, B. S. Donohoe, L. D. Sousa, T. Elder, U. P. Agarwal, F. C. Lu, J. Ralph, M. E. Himmel, V. Balan and B. E. Dale, Energy Environ. Sci., 2011, 4, 973–984 CAS
- Y. Nishiyama, P. Langan and H. Chanzy, J. Am. Chem. Soc., 2002, 124, 9074–9082 CrossRef CAS PubMed
- Y. Nishiyama, J. Sugiyama, H. Chanzy and P. Langan, J. Am. Chem. Soc., 2003, 125, 14300–14306 CrossRef CAS PubMed
- M. Wada, H. Chanzy, Y. Nishiyama and P. Langan, Macromolecules, 2004, 37, 8548–8555 CrossRef CAS
- M. Wada, Y. Nishiyama, G. Bellesia, T. Forsyth, S. Gnanakaran and P. Langan, Cellulose, 2011, 18, 191–206 CrossRef CAS
- K. Igarashi, M. Wada and M. Samejima, FEBS J., 2007, 274, 1785–1792 CrossRef CAS PubMed
- G. Jayme, Wochenbl. Papierfabr., 1944, 6, 187–194 Search PubMed
- A. Ostlund, T. Kohnke, L. Nordstierna and M. Nyden, Cellulose, 2010, 17, 321–328 CrossRef CAS PubMed
- J. Diniz, M. H. Gil and J. Castro, Wood Sci. Technol., 2004, 37, 489–494 CrossRef
- A. Idstrom, H. Brelid, M. Nyden and L. Nordstierna, Carbohydr. Polym., 2013, 92, 881–884 CrossRef PubMed
- R. H. Newman, Cellulose, 2004, 11, 45–52 CrossRef CAS
- G. Jayme and G. Hunger, Monatsh. Chem., 1954, 87, 8–23 CrossRef
- X. Zhao, L. Zhang and D. Liu, Biofuels Bioprod. Biorefin., 2012, 6, 561–579 CrossRef CAS
- O. Guvench, S. N. Greene, G. Kamath, J. W. Brady, R. M. Venable, R. W. Pastor and A. D. Mackerell, J. Comput. Chem., 2008, 29, 2543–2564 CrossRef CAS PubMed
- G. Kamath, O. Guvench and A. D. MacKerell, J. Chem. Theory Comput., 2008, 4, 765–778 CrossRef CAS
- L. Petridis and J. C. Smith, J. Comput. Chem., 2009, 30, 457–467 CrossRef CAS PubMed
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3gc41962b |
| ‡ Current address: Department of Energy, Environmental & Chemical Engineering, Washington University, St. Louis, MO 63130, USA. |
| § Current address: Pall Corporation, 25 Harbor Park Drive, Port Washington, NY 11050, USA. |
| This journal is © The Royal Society of Chemistry 2014 |