Rapid screening of boron isotope ratios in nuclear shielding materials by LA-ICPMS – a comparison of two different instrumental setups†
Christoph
Kurta
a,
Ladina
Dorta
b,
Florian
Mittermayr
c,
Karl
Prattes
d,
Bodo
Hattendorf
b,
Detlef
Günther
b and
Walter
Goessler
*a
aInstitute of Chemistry – Analytical Chemistry, University of Graz, Stremayrgasse 16, 8010 Graz, Austria. E-mail: walter.goessler@uni-graz.at
bLaboratory of Inorganic Chemistry, ETH Zürich, Wolfgang-Pauli-Strasse 10, 8093 Zürich, Switzerland
cInstitute of Technology and Testing of Building Materials, Graz University of Technology, Inffeldgasse 24, 8010 Graz, Austria
dBöhler-Edelstahl GmbH, Mariazellerstraße 16, 8605 Kapfenberg, Austria
First published on 26th November 2013
Abstract
10B enriched resources are widely used in shielding materials such as boron-alloyed steels in nuclear reactors and storage containers. During production, quality control is an indispensable part of the process. In this study two methods for the rapid screening of 10B enriched special steel samples with a 10B isotopic fraction from 90 to 97% – taken from the production process – were developed and compared. We used both ns-LA-ICPMS and fs-LA-MC-ICPMS and evaluated the different instrumental setups for this application. Even though the micro-inhomogeneous boron distribution in the steel samples was determined by LA-ICPMS and EPMA no effect on the isotope ratios could be observed. Besides matrix- and isotope-ratio-matched standardization with in-house reference materials, a non-matrix- and non-isotope-ratio-matched calibration using NIST 610 glass SRM was applied successfully.
Introduction
Boron has two stable natural isotopes, 10B (19.9%) and 11B (80.1%). The neutron absorption properties of boron alloyed stainless steels depend on the content of the 10B isotope with an absorption cross-section for thermal neutrons of more than 3.8 kilobarn (10−25 m2). This isotope is an important neutron absorber used in shielding materials such as boron-alloyed steels for storage of spent nuclear fuel, reactor shielding and control, and instruments detecting neutrons.1,2The production of shielding materials is challenging economically as well as metallurgically. During steel production, internal recycling of materials is part of the production process. Inadvertent mix-up of 10B enriched materials with natural low 10B materials tremendously increases reject costs. To avoid such mix-up during the recycling process the sampling of liquid steel and recycling materials is an integral part of process control.
Traditionally, wet chemical mineralization methods are employed to digest steel samples for the determination of the trace element content and the isotopic composition.3 Different mass spectrometric techniques like secondary ion mass spectrometry (SIMS),4 isotope dilution thermal ionization mass spectrometry (ID-TIMS),5–7 spark source mass spectrometry (SS-MS),8 glow discharge mass spectrometry (GD-MS),9 quadrupole inductively coupled plasma mass spectrometry (Q-ICPMS),9–12 single collector sector field inductively coupled plasma mass spectrometry (SF-ICPMS),13 or multi-collector inductively coupled plasma mass spectrometry (MC-ICPMS)7,14–16 have been used for the determination of boron isotope ratios, mainly in geological samples and reference materials. Unconventional approaches like electrospray mass spectrometry (ES-MS)17 or high resolution continuum source flame atomic absorption spectrometry (HR-CS-FAAS)3 have been used for assessing boron isotope ratios as well.
However, all of these methods are either work-extensive in terms of mineralization (HR-CS-FAAS, ICPMS),3,13 matrix removal (ICPMS),10,18 extraction19 and sample preparation (TIMS)20 or require rather complex instrumental setups (SIMS).21
LA-ICPMS is an attractive alternative because of its high lateral and depth resolution, low limits of detection, a minimum of sample preparation and short analysis time compared to classical sample preparation methods which are tedious and time consuming.22 Moreover, the sampling process is not limited by physical or chemical properties of the materials of interest.21 Sample preparation of steel for LA-ICPMS analysis is not critical to achieve stable signals23 but it is indispensable for EPMA and SEM.24
In 2006 Možná et al. investigated the quantification capabilities of iron-based samples by different UV-ns (ArF excimer, Nd:YAG) and femtosecond (fs) (Ti-sapphire) laser ablation systems coupled to ICPMS. Evaluation of different wavelengths and laser pulse width for the analysis of minor and trace elements using 57Fe as an internal standard has been performed. Matrix- (within metallic samples) and non-matrix-matched (using silicate glass standard reference materials) calibrations have been applied for quantification showing improved analytical results in terms of precision and accuracy using matrix matched calibration and fs-LA-ICPMS.25 In 2011 Wiltsche and Günther used fs-LA-ICPMS for the quantification of 23 metallurgical relevant elements (including boron) in unalloyed, alloyed, highly alloyed steels, and super alloys. Scanning mode ablation with large ablation spot diameters (250 μm) and calibration with matrix- and non-matrix-matched standards provided similar results agreeing within the uncertainty of the certified values.26
LA with multi-collector-ICPMS for the analysis of boron isotopes (δ11B) at <1‰ 2σ precision at the nanogram level in natural and synthetic glasses was first presented by le Roux et al. in 2004.27 Tiepolo et al. applied LA-MC-ICPMS for the δ11B characterization of geological materials in 2006.28 In 2010 Fietzke et al. proposed the use of NIST glasses for the non-matrix-matched calibration of a new in situ method using LA-MC-ICPMS (193 nm excimer laser) for the determination of stable boron isotopes in carbonates.29
In order to demonstrate the benefits and limitations of different LA-ICPMS systems, we developed a fast and simple method for the screening of boron isotope ratios in special steels for quality and production control using standard equipment (ns-LA coupled to Q-ICPMS) as well as more advanced instrumentation (fs-LA-MC-ICPMS).
Experimental
Samples
NIST SRM 612 (nominal trace element concentration 50 mg kg−1) “Trace Elements in a Glass Matrix” (Gaithersburg, MA, USA) was used for daily instrument tuning (ns-LA-ICPMS). For non-matrix-matched isotope ratio determination and tuning of fs-LA-MC-ICPMS NIST SRM 610 (nominal trace element concentration 500 mg kg−1) “Trace Elements in a Glass Matrix” (Gaithersburg, MA, USA) with a boron content of 356 ± 7 mg kg−1 (ref. 30) and an isotope ratio of 10B/11B = 0.2472 ± 0.0003 (ref. 27) was employed.Currently no steel reference material with a certified boron isotope ratio is available on the market.31 For this reason in-house reference materials from Böhler Edelstahl GmbH, Kapfenberg, Austria were used for the experiments. Major constituents in the steel in-house reference materials S20, S35 and S97 are 19.7% Cr, 12.3% Ni, 1.8% B, 1.1% Mn, 0.02% C and a balance of Fe.3,12 Boron isotope ratios in these materials have previously been determined by Q-ICPMS, MC-ICPMS and TIMS.3,12 The isotopic ratios are presented in Table 1.
| S20 | S35 | S97 | |
|---|---|---|---|
| 10B/11B | 0.2477 ± 0.0010 | 0.5314 ± 0.0010 | 33.13 ± 0.01 |
| Abundance of 10B (%) | 19.85 ± 0.07 | 34.70 ± 0.09 | 97.07 ± 0.03 |
| Total B (%) | 1.80 ± 0.05 | 1.84 ± 0.05 | 1.79 ± 0.05 |
Additionally, three samples (N1, N2 and N3) with approximately 1.8% total B content and different 10B enrichment levels (>90%) were taken from the production process and analysed as unknowns.
Sample preparation
LA-ICPMS is only requiring a minimum of sample preparation (e.g. cutting and/or grinding). For wet chemical analysis of steel the sample has to be cut, reduced to chip form, digested in concentrated acids, and diluted prior to analysis – which is usually taking at least 30 minutes, compared to 5 minutes of sample preparation for LA-ICPMS. However, due to the EPMA experiments performed in this study sample preparation (60 minutes for cutting, embedding, grinding, and polishing) cannot be considered negligible any more.Samples were taken from metal sheet plates (except in-house reference material S20, which was only available as metal chips), roughly ground and cut into pieces of approximately 3 × 6 mm in size. The samples were used for both LA-ICPMS and microprobe experiments and thus had to be embedded in epoxy resin for LA and EPMA sample holders (1 inch diameter), ground down with 600 and 1200 grid SiC powder and polished with both Buehler METADI® 3 and 1 micron diamond suspension (Lake Bluff, IL, USA) (ESI, Fig. 1†).
Imaging by LA-ICPMS and electron probe microanalysis (EPMA)
In order to visualize the boron distribution in the steel samples several 2000 μm long line scans with a spot size of 100 μm, a scan speed of 10 μm s−1, a laser energy of 4.0 J cm−2 and a repetition rate of 10 Hz were performed by ns-LA-ICPMS. After a 30 second laser warm-up (laser shutter closed but laser firing) a line scan of 200 seconds was started, followed by 60 seconds of washout.Additionally, elemental distribution images of boron were recorded by electron probe microanalysis using a JEOL JXA-8200 Superprobe (JEOL, Tokyo, Japan). The wavelength-dispersive analytical mode with 20 kV acceleration voltage and a beam current of 180 nA was used. Boron intensities were recorded at the peak position of the B Kα line at 129.29 mm on a layered dispersion element (LDEB) diffracting crystal. 1024 × 1024 point analyses and a step size of 0.5 μm yielded elemental distribution mappings of 512 × 512 μm (ESI, Fig. 2†).
Instrumentation
Results and discussion
Boron distribution in special steels
Mean isotope ratios for sequential ablation experiments using ns-LA and quadrupole based ICPMS analyses showed relative standard deviations (RSDs) of less than 3%. RSDs for 10B or 11B signals, however, were as high as 40% within an individual line scan or a single spot ablation. In order to obtain information about the boron distribution in the sample single line scans were performed by ns-LA-ICPMS in time resolved analysis (TRA) mode. Evidently an inhomogeneous distribution at the micro-scale for both isotopes was present in the special steel sample in-house reference material S97. Similarity for the two boron isotopes was confirmed and thus the boron distribution appeared to be homogeneous (3% RSD) with respect to isotope ratios (Fig. 1). Similar micro-inhomogeneity of the boron distribution was observed in in-house reference materials S20 and S35.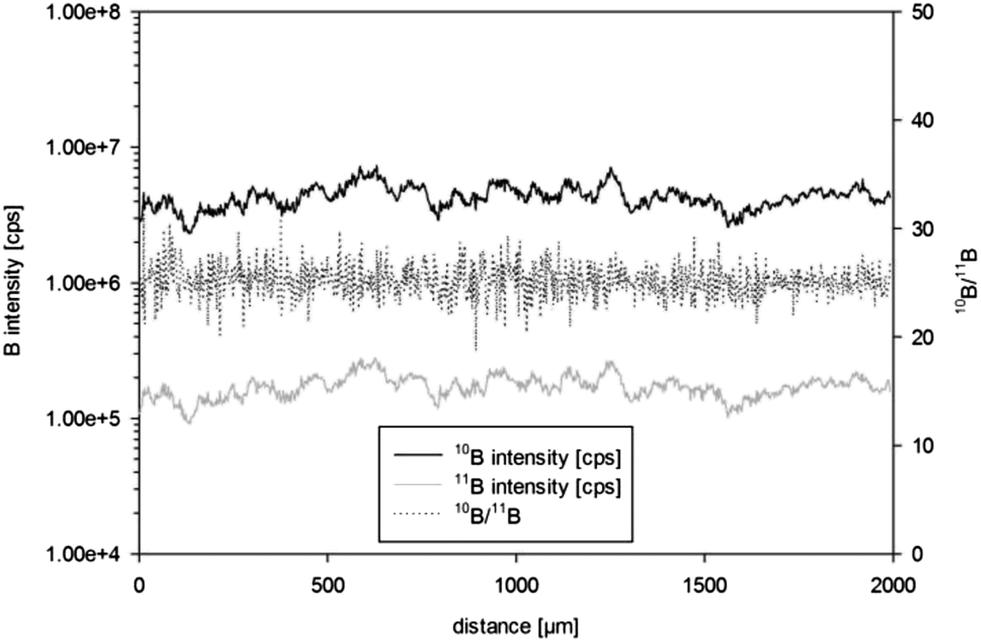 | ||
| Fig. 1 Time resolved analysis of a 2000 μm line scan (ns-LA-ICPMS, 100 μm spot size) on the polished surface of in-house reference material S97 – apparent micro-inhomogeneity in the boron distribution with similar pattern of both boron isotopes but stable boron isotope ratio. | ||
Optimization of LA and ICPMS
| ns-LA-ICPMS | fs-LA-MC-ICPMS | |
|---|---|---|
| LA system | New Wave UP-213 | Coherent Legend (795 nm) |
| Scan pattern | Single spot | Single spot |
| Pulse width | 4 ns | ∼150 fs |
| Energy | ∼0.13 mJ | ∼1.0 mJ (steel) |
| ∼1.7 mJ (glass) | ||
| Repetition rate | 10 Hz | 10 Hz (guidance level) |
| Spot size | 100 μm | 200 μm |
| Laser warm-up – background | 30 s | 30 s |
| Laser analysis time | 100 s | 130 s |
| Laser washout | 300 s | 300 s |
| He carrier gas flow | 0.95 l min−1 | 0.95 l min−1 |
| Ar carrier gas flow | 0.65 l min−1 | 0.74 l min−1 |
Elemental mapping using EPMA confirmed the inhomogeneous distribution of boron at the micro-scale in the samples (ESI, Fig. 2†), explaining the 40% relative standard deviations during LA-ICPMS line scans or single spot ablations.
Some considerations when analyzing boron with ICPMS have to be taken into account. Due to its first ionization potential of 8.3 eV (ref. 1) boron is only about 60% ionized in the plasma.36 Because of the high boron concentration in the samples (up to 1.8%) elevated blank levels for boron can be expected due to carry-over effects in a liquid sample introduction system13 and might also lead to elevated background in LA-ICPMS. Additionally an elevated carbon background can occur from the carbon content of the steel samples, from impurities in the carrier gas helium, and due to degassing of the tube transport system,37 leading to potential spectral interference on 11B+ due to tailing of 12C+.38 According to previous studies by Wang et al. optimization of the sampling depth and carrier gas flow rates is essential for high sensitivity and reduced fractionation effects in LA-ICPMS.39 Andrén et al. additionally reported that variation of the instrumental mass bias can be reduced to a minimum when optimizing the sampling depth before isotope ratio measurements by ICPMS.16 Therefore optimization parameters for ICPMS consisted of sampling depth and extraction ion lens settings. Optimization was performed aiming at highest intensities for boron and lowest boron isotope ratio RSD. Details can be found in the ESI, Fig. 10 and 11.† LA-ICPMS settings were optimized daily in terms of signal intensity and oxide ratios using NIST SRM 612, containing 35 ± 3 mg kg−1 boron.30 Daily pulse/analog (P/A) detector tuning for both boron isotopes using in-house reference material S35 was performed. Additionally, a dead time calibration according to manufacturer recommendation was carried out with no significant effect on the results. Optimized operating conditions for LA and ICPMS are summarized in Tables 2 and 3.
| ICPMS system | Agilent 7700x | Nu plasma HR |
|---|---|---|
| Sampling depth | 4.5 mm | 1.5 mm |
| Extraction lens (HV 1) | — | 6 kV |
| Ω lens | 4.0 V | — |
| Ω bias | −70 V | — |
| m/z measured | 10, 11 | 10, 11 |
| Detector | Auto (pulse and analog counting) | Faraday cups (L5, H6) |
| Integration time | 100 ms per mass | 200 ms per mass |
| Analysis mode | Isotope ratio (IR) | Time resolved analysis (TRA) |
| B sensitivity (100 μm spot size) | ∼6500 cps per mg kg−1 B | ∼1 × 107 cps per mg kg−1 B |
In the MC-ICPMS tailing of 12C+ on 11B+ was not observed while a spectral overlap from 40Ar4+ on the 10B+ signal was avoided by selecting a measurement position of about 0.02 amu higher than the 10B+ centre mass, where the interference could be resolved.
LA-ICPMS settings were optimized daily for highest sensitivity using either NIST SRM 610, containing 356 ± 7 mg kg−1 boron30 or in-house reference material S97 containing 1.8% boron. Optimized settings for LA and ICPMS are summarized in Tables 2 and 3.
Data acquisition
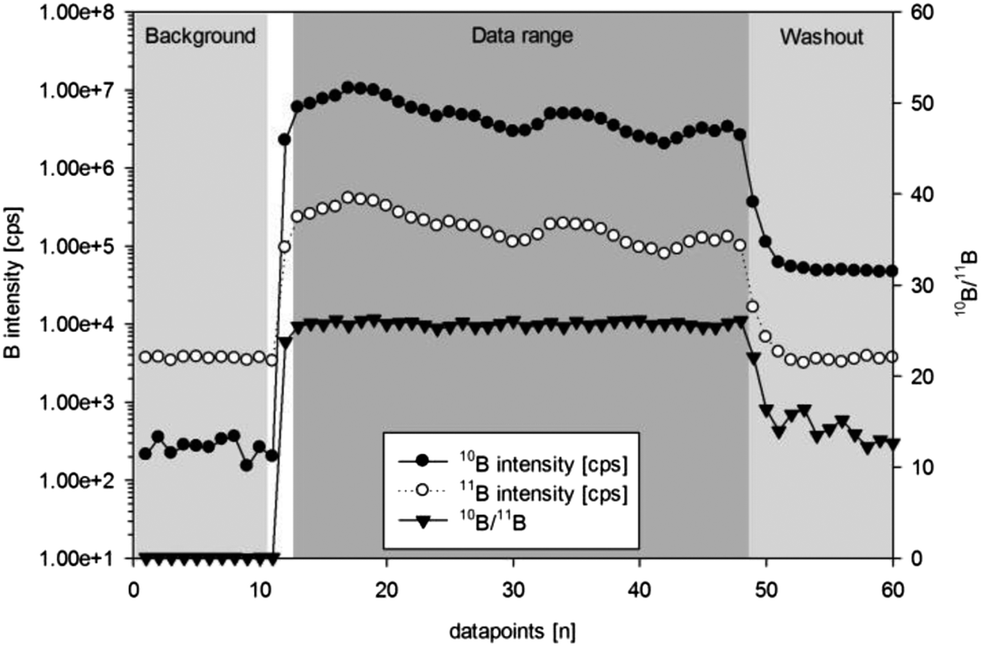 | ||
| Fig. 2 ns-LA-ICPMS: time resolved signals from a typical dataset (consisting of 60 data points) from a single spot experiment on in-house reference material S97 – for data evaluation 35 points (point 13–47) are extracted, for background correction the first ten points are used. | ||
The instrument provides three acquisition modes of the detector, namely pulse, analog, and mixed mode (with automatic selection of the two modes). Analog mode extends the dynamic range of the detection system but requires an accurate calibration of the electron current vs. pulse count, which may introduce an additional uncertainty when high signal intensities are acquired in analog mode, while low intensities are registered by pulse counting (<106 cps).
For example, the 10B intensity in in-house reference material S20 (Fig. 3) was measured in analog mode for the first 18 data points and in pulse mode for the remaining 17 data points. In order to remain in one detector mode (either pulse or analog) for each isotope, data points used for calculations were filtered manually (mode extracted) – selecting the detector mode giving more accurate isotope ratio values (e.g. analog/pulse for process samples N1 and N2). Even though consequent daily calibration of “pulse to analog” (P/A) factors for both boron isotopes using in-house reference material S35 has been performed, the accuracy of the P/A factor calibration was insufficient and leads to wrong estimation of the isotope signal measured in analog mode compared to pulse mode. This caused the 10B/11B intensity ratio to shift to lower values.
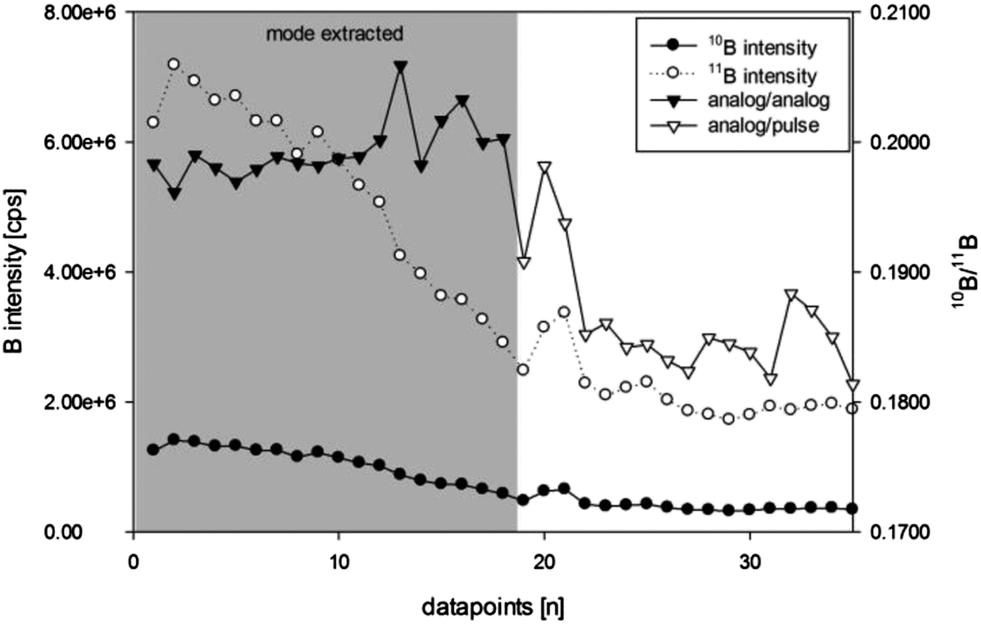 | ||
| Fig. 3 Boron intensities and isotope ratio of in-house reference material S20 in a mixed mode (pulse count and analog) acquisition run (ns-LA-ICPMS) – evident change in the calculated isotope ratio when pulse counts are evaluated for the 10B isotope (data points 18 to 19). | ||
Table 4 presents an overview about the detector modes used for different samples.
| Sample | 10B | 11B | Mode extracted |
|---|---|---|---|
| NIST 610 | Pulse | Pulse | |
| S20 | Mixed | Analog | Analog (10B) |
| S35 | Analog | Analog | |
| S97 | Analog | Pulse | |
| N1 | Analog | Mixed | Pulse (11B) |
| N2 | Analog | Mixed | Pulse (11B) |
| N3 | Analog | Pulse |
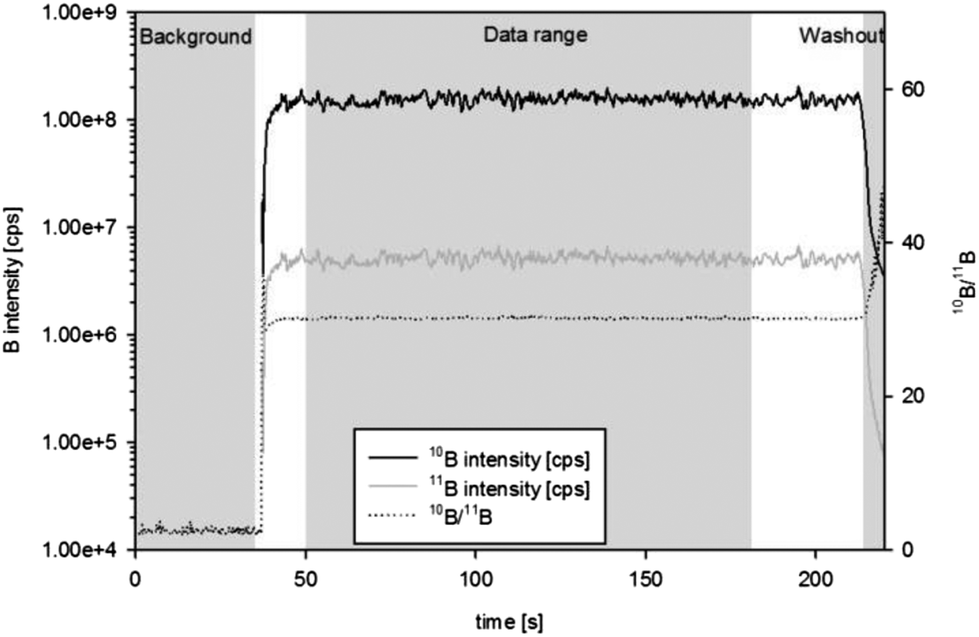 | ||
| Fig. 4 fs-LA-MC-ICPMS: time resolved signals of a typical dataset (consisting of approximately 1000 data points) from a single spot experiment on in-house reference material S97 – for data evaluation, data points between 50 s and 180 s are evaluated. | ||
Mass bias correction
The sample–standard bracketing approach provides the most applicable method to correct for mass bias in this case. Measurements in the order of standard – sample – standard – … were performed and the arithmetic mean of two mass bias values obtained for the standards was used for correction of the isotope ratio of the unknown sample measured in between using eqn (1):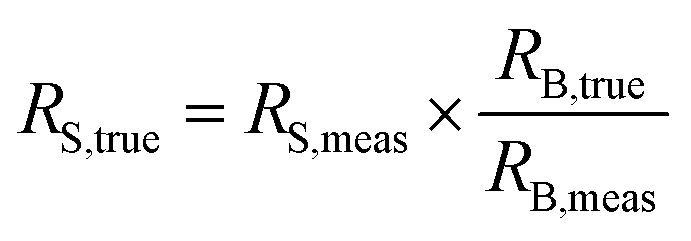 | (1) |
In the first step the three matrix-matched in-house reference materials (Table 1) – where substantial differences in the boron isotope ratios occur (e.g. in-house reference material S20 as the standard and in-house reference material S97 as unknown) – were compared and in a second step additionally non-matrix-matched correction using NIST SRM 610 as the standard and the three steel in-house reference materials as unknowns was investigated.
All in-house reference materials (S20, S35 and S97) and NIST SRM 610 were analyzed in sequences consisting of 6 times standard analysis (e.g. S20), interspersed by 5 samples (e.g. S97) (ESI, Fig. 14†).
Boron isotope ratios in reference materials by LA-ICPMS
By applying the sample–standard bracketing approach, all in-house reference materials and NIST SRM 610 were evaluated as unknowns as well as reference and their respective boron isotope ratios were determined. In this way the dependence of accuracy and precision on sample matrix and isotope ratio was evaluated. The isotope ratio reference values and results for both ns-LA-ICPMS and fs-LA-MC-ICPMS experiments are summarized in Table 5. For process control the needed accuracy and precision was defined by the reference values of the in-house reference materials given in Table 1.| ns-LA-ICPMS | NIST 610 | S20 | S35 | S97 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference sample | 10B/11B | SDa | RSD% | 10B/11B | SDa | RSD% | 10B/11B | SDa | RSD% | 10B/11B | SDa | RSD% |
| NIST 610 | Bracketing standard | 0.2525 | 0.0016 | 0.62 | 0.5320 | 0.0033 | 0.62 | 33.43 | 0.11 | 0.33 | ||
| S20 | 0.2429 | 0.0027 | 1.10 | Bracketing standard | 0.5259 | 0.0022 | 0.43 | 33.05 | 0.15 | 0.44 | ||
| S35 | 0.2467 | 0.0015 | 0.62 | 0.2499 | 0.0005 | 0.21 | Bracketing standard | 33.58 | 0.27 | 0.81 | ||
| S97 | 0.2447 | 0.0009 | 0.38 | 0.2482 | 0.0010 | 0.40 | 0.5254 | 0.0037 | 0.71 | Bracketing standard | ||
| fs-LA-MC-ICPMS | NIST 610 | S20 | S35 | S97 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference sample | 10B/11B | SDa | RSD% | 10B/11B | SDa | RSD% | 10B/11B | SDa | RSD% | 10B/11B | SDa | RSD% |
| NIST 610 | Bracketing standard | 0.2477 | 0.0002 | 0.08 | 0.5317 | 0.0010 | 0.19 | 33.18 | 0.17 | 0.51 | ||
| S20 | 0.2465 | 0.0002 | 0.08 | Bracketing standard | 0.5311 | 0.0005 | 0.10 | 33.37 | 0.02 | 0.06 | ||
| S35 | 0.2470 | 0.0004 | 0.16 | 0.2479 | 0.0004 | 0.16 | Bracketing standard | 33.39 | 0.03 | 0.09 | ||
| S97 | 0.2471 | 0.0012 | 0.49 | 0.2459 | 0.0001 | 0.04 | 0.5273 | 0.0002 | 0.04 | Bracketing standard | ||
Overall, ns-LA-ICPMS was able to yield a reasonable estimate of the boron isotope ratio in the samples. The precision of the determined isotope ratios was in between 0.21 and 1.1%. Several publications over the last few years have shown that determination of boron isotope ratios by applying Q-ICPMS is possible, even though trade-offs with respect to accuracy and precision have to be made.7,10,11
Boron isotope ratios in process samples
All three samples N1, N2, and N3 were analyzed against either NIST SRM 610 or one of the three in-house reference materials S20, S35, and S97 as the bracketing standard (Table 6). The relative standard deviations of the boron isotope ratios of the different samples amount to 0.22–0.59% when using ns-LA-ICPMS. For fs-LA-MC-ICPMS, RSDs between 0.04 and 0.16% were obtained, except for N3 with bracketing standard NIST SRM 610 (1.66% RSD). The elevated RSD for NIST SRM 610 most likely resulted from insufficient washout, leading to carry-over from the steel sample (N3) to the bracketing standard NIST SRM 610. However, overall precision was improved by a factor of five using fs-LA-MC-ICPMS.| Sample | N1 | N2 | N3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 10B/11B | SDa | RSD% | 10B/11B | SDa | RSD% | 10B/11B | SDa | RSD% | |
| a Standard deviation of 5 independent determinations. | |||||||||
| ns-LA-ICPMS | |||||||||
| NIST 610 | 9.134 | 0.046 | 0.50 | 11.98 | 0.04 | 0.35 | 37.24 | 0.08 | 0.21 |
| S20 | 8.994 | 0.036 | 0.40 | 11.90 | 0.08 | 0.65 | 36.69 | 0.11 | 0.30 |
| S35 | 9.148 | 0.02 | 0.22 | 12.04 | 0.04 | 0.32 | 37.58 | 0.08 | 0.21 |
| S97 | 9.153 | 0.054 | 0.59 | 12.07 | 0.03 | 0.25 | 37.82 | 0.08 | 0.21 |
| fs-LA-MC-ICPMS | |||||||||
| NIST 610 | 9.118 | 0.015 | 0.16 | 12.19 | 0.05 | 0.41 | 38.56 | 0.64 | 1.66 |
| S20 | 9.210 | 0.004 | 0.04 | 12.20 | 0.05 | 0.41 | 38.09 | 0.03 | 0.08 |
| S35 | 9.206 | 0.007 | 0.08 | 12.18 | 0.01 | 0.08 | 38.02 | 0.05 | 0.13 |
| S97 | 9.272 | 0.011 | 0.12 | 12.26 | 0.01 | 0.08 | 37.69 | 0.02 | 0.05 |
Irrespective of the bracketing standard used, isotope ratios determined by ns-LA-ICPMS were lower than those determined by fs-LA-MC-ICPMS (Table 6), which could result from different detector modes (pulse/analog) and an insufficient accuracy of the P/A factor calibration. ANOVA analysis revealed significant differences (95% confidence interval) between ns-LA-ICPMS and fs-LA-MC-ICPMS results for all samples except for process sample N1 with NIST SRM 610 as the bracketing standard. When expanding the confidence interval to 99% also N1 and N3 with in-house reference material S97 as the standard were not showing significant deviations. Since no additional analytical data of these process samples were available, results from fs-LA-MC-ICPMS with in-house reference material S97 as the bracketing standard were assumed to be closest to the true values.
Conclusions
In the present study we evaluated different LA-ICPMS setups for the determination of boron isotope ratios in special steel samples. The micro-inhomogeneous boron distribution in the steel samples was apparent from EPMA analyses and also appears in the LA-ICPMS transient signals but had no significant effect on the isotope ratios when large ablation craters were employed. ns-LA-ICPMS has some limitations concerning precision (by about a factor of five less than fs-LA-MC-ICPMS) and accuracy, especially when the isotopic composition and concentration of standard and sample are substantially different. A major limitation in this case is caused by the detection system. The sequential signal acquisition of quadrupole based ICPMS is inherently affected by slight variations of the aerosol density inside the ICP, caused by the pulsed laser ablation event. Additionally, the signal detection using the electron multiplier can cause significant bias in the isotope ratio when the isotopes are detected in different modes (pulse counting vs. analog detection). Thus the selection of the right detector mode is of great importance for ns-LA-ICPMS. Nevertheless, with some special considerations (P/A factor calibration, background correction, extended washout times, and the use of matrix- and isotope-ratio-matched standards), this approach can be useful for the rapid screening of steel samples. The costs for ns-laser ablation and Q-ICPMS equipment are affordable and thus this technique could find or has already found its way into the routine laboratory of steel manufacturers. For high precision measurements however fs-LA-MC-ICPMS is the method of choice. Even though the use of NIST SRM 610 as the bracketing standard is possible, results are improved by the use of matrix- and isotope-ratio-matched standards. For this application fs-LA-MC-ICPMS is comparable to TIMS in terms of accuracy and precision (Table 5). When appropriate standards are available, this technique is even superior because sample preparation can be reduced to a minimum and shorter measurement times are required. Due to the relatively long washout times (5 minutes per analysis) the total measurement times however were substantially extended. An optimization of the sample introduction system especially using a shorter transport tubing should still improve this situation. In this study, an extensive sample preparation procedure has been applied to make the samples suitable for EPMA. For routine application by LA-ICPMS alone, however, the benefit of almost no sample preparation can be sustained and would make the technique interesting for high-throughput analysis.References
- Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, 2002 Search PubMed.
- H. Möllinger, Themen zur Chemie des Bors, Dr. Alfred Hüthig Verlag, Heidelberg, 1976 Search PubMed.
- H. Wiltsche, K. Prattes, M. Zischka and G. Knapp, Spectrochim. Acta, Part B, 2009, 64, 341–346 CrossRef PubMed.
- I. Marchetti, L. Menichetti, C. Kusmic, L. A. De las Heras, P. Salvadori, R. Fuoco, F. Belloni, A. L'Abbate and M. Betti, Spectrochim. Acta, Part B, 2009, 64, 911–920 CrossRef PubMed.
- K. G. Heumann and H. Zeininger, Int. J. Mass Spectrom. Ion Processes, 1985, 67, 237–252 CrossRef CAS.
- K. G. Heumann, S. Eisenhut, S. Gallus, E. H. Hebeda, R. Nusko and A. Vengosh, Analyst, 1995, 120, 1291–1299 RSC.
- J. Aggarwal, F. Böhm, G. Foster, S. Halas, B. Hönisch, S.-Y. Jiang, J. Kosler, A. Liba, I. Rodushkin, T. Sheehan, J. J.-S. Shen, S. Tonarini, Q. Xie, C.-F. You, Z.-Q. Zhao and E. Zuleger, J. Anal. At. Spectrom., 2009, 24, 825–831 RSC.
- R. A. Lukaszew, J. G. Marrero, R. F. Cretella and C. J. Noutary, Analyst, 1990, 115, 915–917 RSC.
- F. Chartier and M. Tabarant, J. Anal. At. Spectrom., 1997, 12, 1187–1193 RSC.
- D. C. Gregoire, Anal. Chem., 1987, 59, 2479–2484 CrossRef CAS.
- A. Al-Ammar, E. Reitzernová and R. M. Barnes, Spectrochim. Acta, Part B, 2000, 55, 1861–1867 CrossRef.
- K. Prattes, G. Hochoertler and W. Wegscheider, in Proceedings of the Chemists' Conference, Corus Research, Development & Technology, 2000, vol. 51, pp. 12–14 Search PubMed.
- J. Vogl, M. Rosner and W. Pritzkow, J. Anal. At. Spectrom., 2011, 26, 861–869 RSC.
- H.-E. Gäbler and A. Bahr, Chem. Geol., 1999, 156, 323–330 CrossRef.
- J. K. Aggarwal, K. Mezger, E. Pernicka and A. Meixner, Int. J. Mass Spectrom., 2004, 232, 259–263 CrossRef CAS PubMed.
- H. Andrén, I. Rodushkin, A. Stenberg, D. Malinovsky and D. C. Baxter, J. Anal. At. Spectrom., 2004, 19, 1217–1224 RSC.
- M. C. B. Moraes, J. G. A. Brito Neto and C. L. Do Lago, J. Anal. At. Spectrom., 2001, 16, 1259–1265 RSC.
- A. G. Coedo, T. Dorado, B. J. Fernandez and F. J. Alguacil, Anal. Chem., 1996, 68, 991–996 CrossRef CAS.
- K. E. Burke, Analyst, 1972, 97, 19–28 RSC.
- D. J. Scott and G. Gauthier, Chem. Geol., 1996, 131, 127–142 CrossRef CAS.
- J. Pisonero, B. Fernández and D. Günther, J. Anal. At. Spectrom., 2009, 24, 1145–1160 RSC.
- J. S. Becker, Spectrochim. Acta, Part B, 2002, 57, 1805–1820 CrossRef.
- A. Bengtson, M. Granfors, I. Gustavsson, M. Sedlakova, A. Gómez Coedo, I. Padilla, H. Dillen, K. Van den Bergh, D. Günther and C. Latkoczy, High spatial resolution analysis of steel samples using laser ablation ICP-MS, 2006, EUR 21995 Search PubMed.
- S. J. B. Reed, Electron Microprobe Analysis and Scanning Electron Microscopy in Geology, Cambrige University Press, Cambridge, 2005 Search PubMed.
- V. Možná, J. Pisonero, M. Holá, V. Kanický and D. Günther, J. Anal. At. Spectrom., 2006, 21, 1194–1201 RSC.
- H. Wiltsche and D. Günther, Anal. Bioanal. Chem., 2011, 399, 2167–2174 CrossRef CAS PubMed.
- P. J. le Roux, S. B. Shirey, L. Benton, E. H. Hauri and T. D. Mock, Chem. Geol., 2004, 203, 123–138 CrossRef CAS PubMed.
- M. Tiepolo, C. Bouman, R. Vannucci and J. Schwieters, Appl. Geochem., 2006, 21, 788–801 CrossRef CAS PubMed.
- J. Fietzke, A. Heinemann, I. Taubner, F. Bohm, J. Erez and A. Eisenhauer, J. Anal. At. Spectrom., 2010, 25, 1953–1957 RSC.
- N. J. G. Pearce, W. T. Perkins, J. A. Westgate, M. P. Gorton, S. E. Jackson, C. R. Neal and S. P. Chenery, Geostand. Newsl., 1997, 21, 115–144 CrossRef CAS.
- J. Vogl and W. Pritzkow, J. Anal. At. Spectrom., 2010, 25, 923–932 RSC.
- D. Bleiner and D. Günther, J. Anal. At. Spectrom., 2001, 16, 449–456 RSC.
- W. Müller, M. Shelley, P. Miller and S. Broude, J. Anal. At. Spectrom., 2009, 24, 209–214 RSC.
- J. Koch, M. Wälle, J. Pisonero and D. Gunther, J. Anal. At. Spectrom., 2006, 21, 932–940 RSC.
- M. B. Fricker, D. Kutscher, B. Aeschlimann, J. Frommer, R. Dietiker, J. Bettmer and D. Günther, Int. J. Mass Spectrom., 2011, 307, 39–45 CrossRef CAS PubMed.
- K. E. Jarvis, A. L. Gray and R. S. Houk, Handbook of inductively coupled plasma mass spectrometry, Chapman and Hall, New York, 1992 Search PubMed.
- R. Kovacs and D. Günther, J. Anal. At. Spectrom., 2008, 23, 1247–1252 RSC.
- J. A. Moreton and H. T. Delves, J. Anal. At. Spectrom., 1999, 14, 1545–1556 RSC.
- Z. Wang, B. Hattendorf and D. Günther, J. Am. Soc. Mass Spectrom., 2006, 17, 641–651 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimization of LA and ICPMS parameters. See DOI: 10.1039/c3ja50194a |
| This journal is © The Royal Society of Chemistry 2014 |