Disease-on-a-chip: mimicry of tumor growth in mammary ducts
Pierre-Alexandre
Vidi
a,
Teimour
Maleki†
b,
Manuel
Ochoa
b,
Lei
Wang
a,
Sara M.
Clark
a,
James F.
Leary
*abcd and
Sophie A.
Lelièvre
*ad
aDepartment of Basic Medical Sciences, Purdue University, 625 Harrison Street, Lynn Hall, West Lafayette, IN 47907-2026, USA. E-mail: lelievre@purdue.edu; jfleary@purdue.edu; Fax: +1 765 494 0781; Tel: +1 765 496 7793
bBirck Nanotechnology Center, Purdue University, West Lafayette, IN, USA
cWeldon School of Biomedical Engineering, Purdue University, West Lafayette, IN, USA
dCenter for Cancer Research, Purdue University, West Lafayette, IN, USA
First published on 29th October 2013
Abstract
We present a disease-on-a-chip model in which cancer grows within phenotypically normal breast luminal epithelium on semicircular acrylic support mimicking portions of mammary ducts. The cells from tumor nodules developing within these hemichannels are morphologically distinct from their counterparts cultured on flat surfaces. Moreover, tumor nodules cocultured with the luminal epithelium in hemichannels display a different anticancer drug sensitivity compared to nodules cocultured with the luminal epithelium on a flat surface and to monocultures of tumor nodules. The mimicry of tumor development within the epithelial environment of mammary ducts provides a framework for the design and test of anticancer therapies.
Reproducing human pathologies in a tissue context in vitro is paramount to the development of strategies to specifically identify and treat diseased cells amidst a complex organ system. Organs-on-chips that recapitulate tissue architectures1 have a large potential for theranostic developments. In the breast, glandular units (acini) are grouped into lobules and connected via a branched ductal system to the nipple. The vast majority of breast cancers arise from the inner layer of luminal epithelial cells in the terminal mammary ducts connected to the lobules.2 We previously developed a breast-on-a-chip based on polydimethylsiloxane (PDMS) that mimicked the luminal portion of the branched epithelium with channels of decreasing diameters, similar to those found in the breast.3 Importantly, polarization of the epithelial cells lining the ducts was recapitulated in response to the addition of the extracellular matrix (ECM) component laminin 111. The second stage of the breast-on-a-chip model, presented here, is the addition of the diseased element – the tumor.
Coculture systems combining various cell types have been implemented to engineer normal organs, for instance by layering different cell types and matrices to produce arteries, bladders and portions of liver among other organs or systems.4 Cocultures of non-neoplastic epithelial and cancer cells have also been reported, but the majority of these models used cell monolayers lacking epithelial tissue architecture or culture inserts preventing direct contact between cancer and noncancer cells. Few systems allowing contacts between healthy and diseased epithelia have been described, notably to evaluate the influence of non-neoplastic cells on treatment responses by tumor cells,5 but these systems do not recreate the physiological architecture of the normal tissue. We provide, to the best of our knowledge, the first model for the growth of cancer nodules in the presence of their normal counterpart as it occurs in vivo, in the luminal epithelial tissue context. This step is indispensable towards the preclinical assessment of realistic novel therapies.
PDMS elastomers have numerous advantages for microfabrication, including optical transparency, gas permeability, and straightforward molding of microsized patterns by soft lithography.6,7 PDMS surfaces are compatible with cell growth,8 yet may cause artifacts. The effect on cell behavior of uncrosslinked polymers leaching from PDMS structures is unknown and adsorption of hydrophobic molecules – including steroid hormones – on PDMS surfaces is a matter of concern, especially for the culture of mammary epithelial cells.9 For models of the breast ductal system, another limitation is that PDMS hemichannels produced by conventional microfabrication techniques have a rectangular cross-section. Non-neoplastic mammary epithelial HMT-3522 S1 cells cultured on laminin-coated PDMS are organized into a polarized layer with apical tight junctions like in the luminal (outer) portion of the breast epithelium;3 however, they usually form multilayers in the corners of the channels (Fig. 1A). This issue had to be resolved because cell multilayering is one of the characteristics of solid tumor development.
 | ||
| Fig. 1 Impact of hemichannel geometry and substrate texture on the arrangement of non-neoplastic mammary epithelial HMT-3522 S1 cells. A. Multilayer of cells in the corner of PDMS channels with a rectangular section (see inset). B. Cell multilayering in acrylic channels with a rough texture. C. Monolayer of cells on a smooth semicircular acrylic channel. SEM images (left) show the appearance of each type of hemichannel before seeding the cells. Nuclei are stained with DAPI (blue). Scale bars, 10 μm. | ||
We have employed laser micromachining to produce acrylic-based hemichannels with a circular cross section. This method has high development efficiency (cost/time ratio) and offers the flexibility of adjusting the microchannel's depth across the chip.10 Microchannels were engraved on Shape Products® acrylic sheets (Menomonie, WI) using a CO2 laser cutting and engraving system (Universal Laser System, Inc.) operating in continuous wave mode at 30 watts power and 1 mm ms−1. Focusing the laser on the acrylic surface yielded V-shaped cross-sections. To achieve semicircular cross-sections, the laser was focused 0.5–1 mm above the acrylic surface (depending on the desired diameter). Rough surfaces obtained after laser micromachining promoted cell growth as multilayers rather than the desired single-layer coating of the channel walls usually found in vivo (Fig. 1B). This issue was solved by smoothing the surfaces by spin coating polymethyl methacrylate (PMMA; A4, Microchem, MA; 2000 rpm, 30 s; Fig. 1C). Treating the devices with air plasma increased hydrophilicity, as observed from water contact angle measurements, which facilitated coating with laminin 111 (BD Biosciences, Ultrapure; 3 μg cm−2). As previously observed on laminin 111-coated PDMS surfaces,3 S1 epithelial cells in acrylic hemichannels were basoapically polarized with basal distribution of the polarity marker α6-integrin and the apical localization of the tight-junction marker ZO-1 (Fig. 2A). Compared with PDMS surfaces, S1 cells covered acrylic hemichannels more uniformly (Fig. 2B).
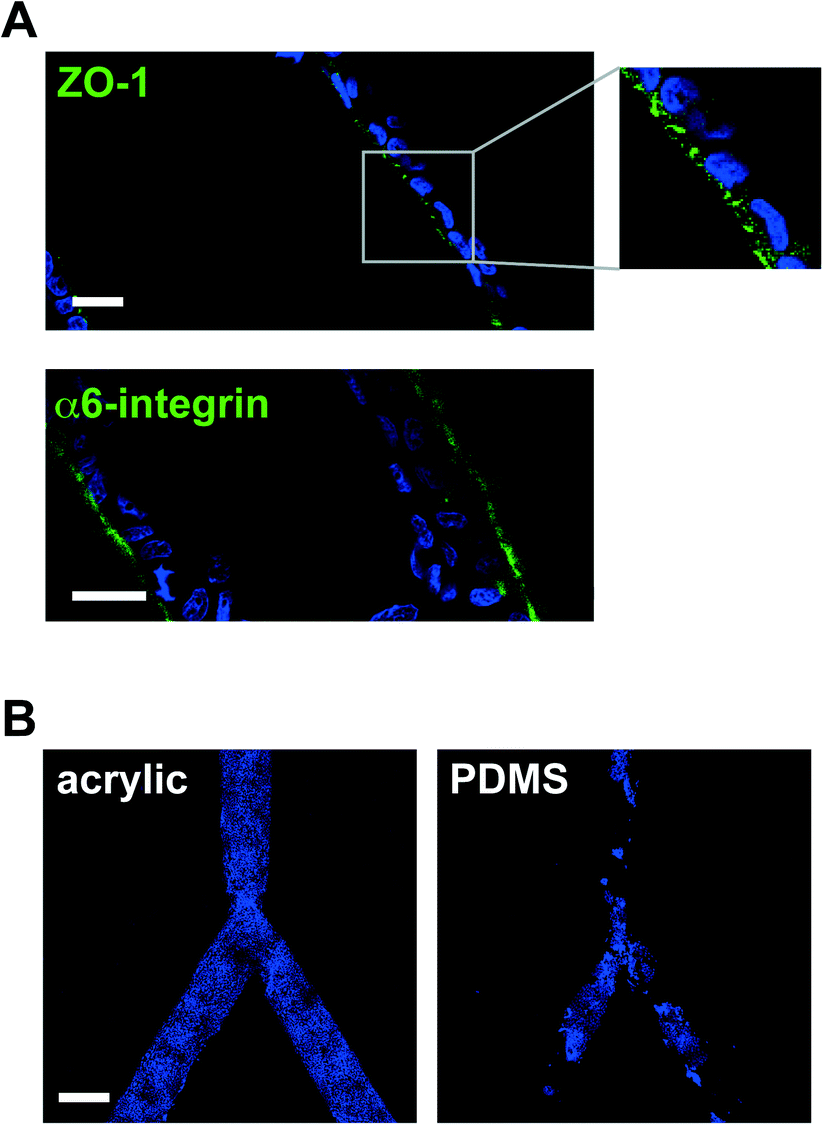 | ||
| Fig. 2 S1 epithelium polarization and substrate coverage in hemichannels. A. Basoapical polarization on laminin 111-coated acrylic hemichannels shown by immunostaining for apical tight junction marker ZO1 (top, in green; see region magnification in inset) and basal hemidesmosome marker α6-integrin (bottom, in green). Nuclei were counterstained with DAPI (blue). Scale bars, 20 μm. B. Cell coverage on acrylic (left) and PDMS (right) hemichannels. Projections of confocal images of DAPI signals are shown. Scale bar, 200 μm. | ||
In solid cancers, tumors develop within the normal cell bed. In the breast, the cell bed encompasses the epithelium that delineates the ducts, and tumors initially develop via the accumulation of cells within the lumen of the ductal channel. It has been reported that the presence of non-neoplastic cells prevents the proliferation of tumor cells in culture.11,12 In these experiments, the non-neoplastic cells were not mimicking the luminal epithelial architecture. Therefore, we assessed in a first approach whether cancer cells could form tumors in the presence of predifferentiated acini using classical embedded three-dimensional (3D) cell culture.13 Normal acini are basoapically polarized, spherical structures made of myoepithelial cells surrounding the inner luminal epithelium organized into one layer around a central lumen.14 Groups of connected acini form the terminal ductal lobular units (TDLUs) prolonging mammary ducts (Fig. 3A). S1 cells were induced to differentiate into the luminal portion of acinar structures for 10 days in the presence of Matrigel™ (BD Biosciences), then released from Matrigel™ with dispase, washed in medium, and mixed in Matrigel™ with small nodules containing a few S1-derived HMT3522 T4-2 cancer cells.15 The T4-2 nodules grew in coculture and, surprisingly, T4-2 cancer cells were attracted to acini and came in contact with acinar cells. Once in contact, they engulfed the acini completely, suggesting that, in an appropriate 3D microenvironment, small tumors strive in the presence of phenotypically normal epithelium (Fig. 3B). This behavior reflects the capacity of invasive cancer cells to colonize normal territories. However, this coculture system does not represent the in vivo tissue context in which a tumor develops within the lumen of the terminal ducts.
 | ||
| Fig. 3 Coculture of tumor nodules and breast acini. A. Left: drawing of the organization of a TDLU. The glandular epithelium is arranged into a grape-like structure opening into the terminal duct. A glandular unit (acinus) is marked by a green shadow. Luminal cells (pale gray) are lined by myoepithelial cells (dark gray). Right: hematoxylin–eosin staining of a histological section through a breast TDLU. Acini and the terminal duct appear as round circles and a hollow channel, respectively. Cell nuclei are in blue. The section plane is indicated by the red dashed line on the drawing. L, lumen; magnification, 20×. B. S1 acini and small T4-2 tumor nodules produced separately in 3D culture were mixed together, embedded in a layer of Matrigel™, and followed by time-lapse microscopy during 10 days. Stable GFP expression in S1 cells (green) and DiI staining of T4-2 cells prior to coculture (red) allowed us to discriminate between the cell types. Nuclei were stained with DAPI (blue). Images show a tumor nodule touching an acinus at day 6 of coculture, tumor nodules covering acini at day 8, and a large nodule that has engulfed two acini at day 10. Magnification, 40×. | ||
We therefore used the acrylic hemichannels described above to recapitulate the physiological context of mammary tumor development. First, we determined optimal coculture conditions for this system. When individual T4-2 cancer cells were seeded on a layer of polarized S1 cells, only a fraction of cells (<10%) developed into small tumor nodules (2,870 ± 289 μm2 in cross section; n = 16 from two biological replicates) and the majority of the cells remained as single entities on top of the S1 layer. T4-2 cells were also observed as a flat monolayer (2D culture) in regions of the acrylic devoid of S1 cells. In contrast, T4-2 cells seeded as small nodules (3–5 cells) on the S1 cell monolayer developed larger tumors (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 731 ± 1,522 μm2; n = 65), hence confirming our initial observations with the embedded coculture system that small tumor nodules thrive in the presence of the non-neoplastic cells. With this approach, more than 80% of T4-2 colonies exhibited 3D growth patterns after four days in coculture (Fig. 4A). Of note, tumor nodules also thrive in the presence of a monolayer of S1 cells that lack apical polarity (e.g., S1 cells cultured on a substratum devoid of laminin 111 coating3), indicating that apical tight junctions between S1 cells are not a major factor influencing T4-2 cell proliferation (data not shown). Rather, the use of small clusters of cancer cells (instead of single cells) for seeding appears determinant for tumor growth in the coculture system. Tumor nodules cocultured with S1 cells within the hemichannels were fixed with 4% paraformaldehyde and immunostained for CD44, a cell surface marker of cancer stem cells abundantly present in the T4-2 cell population. Confocal microscopy imaging revealed tumor nodules developing at the bottom or at the side of the hemichannels. In some nodules, cancer cells appeared to rest against the ECM-coated acrylic substratum instead of the non-neoplastic epithelium. The same observation was made when staining cell membranes of tumor nodules with DiI (Fig. 4B). This tumor pattern mimics the organization observed in sections of biopsies of cancerous tissue, with cancer cells infiltrating the epithelium, thereby contacting the basement membrane (Fig. 4C).
731 ± 1,522 μm2; n = 65), hence confirming our initial observations with the embedded coculture system that small tumor nodules thrive in the presence of the non-neoplastic cells. With this approach, more than 80% of T4-2 colonies exhibited 3D growth patterns after four days in coculture (Fig. 4A). Of note, tumor nodules also thrive in the presence of a monolayer of S1 cells that lack apical polarity (e.g., S1 cells cultured on a substratum devoid of laminin 111 coating3), indicating that apical tight junctions between S1 cells are not a major factor influencing T4-2 cell proliferation (data not shown). Rather, the use of small clusters of cancer cells (instead of single cells) for seeding appears determinant for tumor growth in the coculture system. Tumor nodules cocultured with S1 cells within the hemichannels were fixed with 4% paraformaldehyde and immunostained for CD44, a cell surface marker of cancer stem cells abundantly present in the T4-2 cell population. Confocal microscopy imaging revealed tumor nodules developing at the bottom or at the side of the hemichannels. In some nodules, cancer cells appeared to rest against the ECM-coated acrylic substratum instead of the non-neoplastic epithelium. The same observation was made when staining cell membranes of tumor nodules with DiI (Fig. 4B). This tumor pattern mimics the organization observed in sections of biopsies of cancerous tissue, with cancer cells infiltrating the epithelium, thereby contacting the basement membrane (Fig. 4C).
 | ||
| Fig. 4 Development of tumor nodules in the presence of the luminal epithelium. A. Left panel: T4-2 cancer cells stained with DiI (red) and seeded as single cells (top) or small cell clusters (bottom) onto a preformed S1 cell luminal epithelium, as illustrated in the drawings, and imaged after four days of coculture. DiI fluorescence and corresponding bright field (BF) images are shown. The asterisk indicates the 2D spread of cancer cells and the arrowhead points to a single cancer cell in the coculture following seeding of single T4-2 cells. Right panel: confocal images after fixation with 4% paraformaldehyde and staining of S1 and T4-2 nuclei with DAPI (blue). Orthogonal views (xz) correspond to the positions indicated by the dashed lines. B. Confocal images of CD44 immunostained (left) and DiI-stained (right) T4-2 cells in hemichannels. Images were taken four days after seeding small T4-2 cell clusters onto preformed S1 monolayers. Orthogonal views (xz) at the level of the tumor nodules and in regions devoid of cancer cells are shown at the bottom. C. Hematoxylin–eosin staining of a breast cancer tissue section showing a duct lined with myoepithelial and luminal cells as well as larger cells forming a tumor inside the lumen (magnification, 40×). The arrow indicates a site where the tumor is against the channel wall (note that a similar phenomenon is apparent in the orthogonal views of the coculture system, see B). The drawing indicates a possible location for the section plane. D. Quantification of morphological features of T4-2 cells in the hemichannels and on flat surfaces of the chip after four days of coculture with S1 cells. Top: representative confocal images of β-catenin immunostaining (green) and DAPI signals (blue) used to delineate cell boundaries and nuclei, respectively. Bottom: cellular and nuclear cross sections and circularity determined using the ImageJ software (http://rsbweb.nih.gov/ij/). For each of the confocal images corresponding to optical sections of nodules, cross section and circularity values were averaged. These per nodule-averages are represented in box-and-whisker plots. P value is from unpaired t-test. The inset represents average ± SEM of variance; F = flat surface and H = hemichannel. The number of nodules analyzed (n) and coefficients of variation (CV) are indicated on the graphs in E. Apoptotic indexes from T4-2 nodules treated with bleomycin (BLM, 10 mU ml−1; unpaired t-tests, n = 15 nodules from three biological replicates) or with doxorubicin (DOX, 10 μM; n ≥ 15 nodules from an experiment with three culture triplicates) for 24 hours. Nodules were cocultured with S1 epithelium in hemichannels and on flat surfaces of the chip or were produced on the same growth substratum in the absence of S1 epithelium. Scale bars, 100 μm (A) and 50 μm (B, D). | ||
To further characterize the on-chip coculture system, we compared tumor nodules developing within hemichannels to nodules on the flat surfaces, both lined with the luminal epithelium. To ease comparisons, the Y-shaped design (Fig. 2B) was replaced with parallel hemichannels at 2 mm intervals on 22 × 22 mm acrylic sheets. Morphological features of cancer cells are routinely analyzed in the clinic as part of the pathological evaluation of tumors.16 Hence, we measured the size and shape of the T4-2 cells and their nuclei. β-Catenin immunostaining of the plasma membranes was used to delineate cell boundaries whereas nuclear morphological features were extracted from the DAPI images. Averaged cellular size and shape parameters were not different between nodules developing within the hemichannels vs. neighboring nodules on a flat surface. The average cross section (area) of nuclei was also comparable in both conditions (Fig. 4D). In contrast, the circularity of T4-2 nuclei differed depending on the location of the nodules. Nodules on flat surfaces had significantly less round nuclei compared to nodules in the hemichannels. Also, nodules on flat surfaces displayed significantly higher variability for this shape parameter – both among nodules (F test, P = 0.025; see coefficient of variation Fig. 4D) and within nodules (see comparison of averaged variances Fig. 4D). Similar observations were made for other nuclear shape descriptors including aspect ratio and solidity (i.e., area/convex area; data not shown). Aggressive tumors are characterized by highly heterogeneous cellular features. In particular, nuclear pleomorphism (i.e., marked variations in nuclear sizes and shapes within tumors), is typically associated with high-grade breast cancers characterized by poorer prognosis.17,18
The findings described above prompted us to test the sensitivity of T4-2 tumor nodules to the chemotherapeutic drugs bleomycin (BLM, a radiomimetic agent used in the laboratory to study cellular responses to treatments that compromise genome integrity) and doxorubicin (DOX, an anthracycline used for the treatment of breast cancer). In this pilot study, apoptosis was utilized as an endpoint to evaluate drug responses and was quantified using the TUNEL assay19 combined with visual scoring for pyknotic and karyorrhectic nuclei via DAPI staining (i.e. nuclear condensation and fragmentation, respectively20). Three types of T4-2 nodules were compared: monocultures, cocultures with S1 luminal epithelium on the flat surface of the chip, and cocultures with S1 epithelium in hemichannels. The results revealed increased sensitivity to BLM and DOX of tumor nodules cocultured with the non-neoplastic breast epithelium on flat surfaces compared to monocultures of tumor nodules (Fig. 4E). The low toxicity of BLM to T4-2 nodules is expected since the drug concentration chosen is normally used to ease the detection of increased sensitivity to genotoxic stress and it should minimally affect cell survival.21
Interestingly, the sensitivity of cancer cells to DOX and BLM was significantly less for nodules that were located within hemichannels vs. on flat surfaces (Fig. 4E). This difference is intriguing and could not be explained by the proliferation status within nodules on flat surfaces and in hemichannels (27.5% ± 1.8% and 24.2% ± 2.5% Ki67-positive cells, respectively; P = 0.292, unpaired t-test, n > 10 nodule sections corresponding to >700 nuclei). Moreover, similar effects were observed in cells treated with 3 Gy of ionizing radiations (data not shown), suggesting that differences in apoptotic indexes did not merely result from altered drug penetration rates. It is possible that the microenvironment or the geometry in the hemichannels promotes modifications in the nuclear organization of tumor cells, which may influence cellular responses to cytotoxic drugs. This view is supported by the measured differences in nuclear circularity reported above, and is in agreement with early recommendations that nuclear morphometric descriptors, including roundness, should be investigated as possible predictors of response to therapy.22 The breast-on-a-chip used for coculture with cancer cells provides a model with potential for improved understanding of cancer development and drug assessment in tissue context.
This work brings a proof-of-concept of a model for the growth of cancer nodules amidst a non-neoplastic luminal epithelial environment. The acrylic support was chosen for the possibility of rapidly creating smooth semicircular surfaces enabling expansion of a population of breast epithelial cells as a polarized monolayer. We anticipate applications in drug evaluation for this novel disease-on-a-chip model. Testing patient tumor samples for their response to anticancer drugs is an emerging direction for individualized therapy,23–25 and disease-on-chips models might help determine tumor responses to therapies with increased accuracy by placing cancer cells within a proper architectural context. Future developments of disease-on-chips will include compartmentalization with microfluidics to permit simultaneous testing of multiple tumors or of different drug regimens. The concept presented here may also serve in the initial development steps of new anticancer approaches, notably for the detection and treatment of neoplasia with nanoparticles delivered via the breast ductal system.
Acknowledgements
We thank Dr. Kurt Hodges for comments on the manuscript and for providing images of histological sections of breast tumors and breast lobules, and Dr. Jun Xie for helpful discussion regarding the statistical analyses of cellular and nuclear features of tumor nodules. We are grateful to Christy Cooper and Laurie Mueller for assistance with electron microscopy. This project was supported by the Congressionally-Directed Materials Research Command/Breast Cancer Research Program (W81XWH-09-1-0354 to SAL and JFL) and the National Institute of Health (K99CA163957 to PAV).References
- D. Huh, Y. S. Torisawa, G. A. Hamilton, H. J. Kim and D. E. Ingber, Lab Chip, 2012, 12, 2156–2164 RSC.
- H. M. Jensen, Am. J. Obstet. Gynecol., 1986, 154, 1280–1284 CrossRef CAS.
- M. M. Grafton, L. Wang, P. A. Vidi, J. Leary and S. A. Lelièvre, Integr. Biol., 2011, 3, 451–459 RSC.
- A. Soto-Gutierrez, J. A. Wertheim, H. C. Ott and T. W. Gilbert, J. Clin. Invest., 2012, 122, 3817–3823 CrossRef CAS PubMed.
- L. M. Merlo, R. E. Kosoff, K. L. Gardiner and C. C. Maley, BMC Cancer, 2011, 11, 461 CrossRef CAS PubMed.
- L. Kim, Y. C. Toh, J. Voldman and H. Yu, Lab Chip, 2007, 7, 681–694 RSC.
- E. Berthier, E. W. Young and D. Beebe, Lab Chip, 2012, 12, 1224–1237 RSC.
- J. N. Lee, X. Jiang, D. Ryan and G. M. Whitesides, Langmuir, 2004, 20, 11684–11691 CrossRef CAS PubMed.
- K. J. Regehr, M. Domenech, J. T. Koepsel, K. C. Carver, S. J. Ellison-Zelski, W. L. Murphy, L. A. Schuler, E. T. Alarid and D. J. Beebe, Lab Chip, 2009, 9, 2132–2139 RSC.
- C. G. Khan Malek, Anal. Bioanal. Chem., 2006, 385, 1362–1369 CrossRef CAS PubMed.
- B. C. Spink, R. W. Cole, B. H. Katz, J. F. Gierthy, L. M. Bradley and D. C. Spink, Cell Biol. Int., 2006, 30, 227–238 CrossRef CAS PubMed.
- N. Kosaka, H. Iguchi, Y. Yoshioka, K. Hagiwara, F. Takeshita and T. Ochiya, J. Biol. Chem., 2012, 287, 1397–1405 CrossRef CAS PubMed.
- P. A. Vidi, M. J. Bissell and S. Lelièvre, in Methods in Molecular Biology, 2013, pp. 193–219 Search PubMed.
- O. W. Petersen, L. Ronnov-Jessen, A. R. Howlett and M. J. Bissell, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9064–9068 CrossRef CAS.
- P. Briand, K. V. Nielsen, M. W. Madsen and O. W. Petersen, Cancer Res., 1996, 56, 2039–2044 CAS.
- E. A. Rakha, J. S. Reis-Filho, F. Baehner, D. J. Dabbs, T. Decker, V. Eusebi, S. B. Fox, S. Ichihara, J. Jacquemier, S. R. Lakhani, J. Palacios, A. L. Richardson, S. J. Schnitt, F. C. Schmitt, P. H. Tan, G. M. Tse, S. Badve and I. O. Ellis, Breast Cancer Res., 2010, 12, 207 Search PubMed.
- J. P. Baak, P. H. Kurver, A. J. De Snoo-Niewlaat, S. De Graef, B. Makkink and M. E. Boon, Histopathology, 1982, 6, 327–339 CrossRef CAS.
- P. Kronqvist, T. Kuopio and Y. Collan, Br. J. Cancer, 1998, 78, 800–805 CrossRef CAS.
- Y. Gavrieli, Y. Sherman and S. A. Ben-Sasson, J. Cell Biol., 1992, 119, 493–501 CrossRef CAS.
- K. Shirai, T. Mizui, Y. Suzuki, Y. Kobayashi, T. Nakano and T. Shirao, Neurosci. Lett., 2006, 399, 57–60 CrossRef CAS PubMed.
- P. A. Vidi, G. Chandramouly, M. Gray, L. Wang, E. Liu, J. J. Kim, V. Roukos, M. J. Bissell, P. V. Moghe and S. A. Lelièvre, J. Cell Sci., 2012, 125, 350–361 CrossRef CAS PubMed.
- A. W. Partin, A. C. Walsh, J. I. Epstein, B. G. Leventhal and J. P. Gearhart, J. Urol., 1990, 144, 952–954 CAS.
- B. Rubio-Viqueira, H. Mezzadra, M. E. Nielsen, A. Jimeno, X. Zhang, C. Iacobuzio-Donahue, A. Maitra, M. Hidalgo and S. Altiok, Mol. Cancer Ther., 2007, 6, 515–523 CrossRef CAS PubMed.
- S. L. Suchy, L. M. Hancher, D. Wang, P. R. Ervin, Jr. and S. L. Brower, Cancer Biol. Ther., 2011, 11, 1059–1064 CrossRef CAS.
- S. J. Conde, A. Luvizotto Rde, M. T. de Sibio and C. R. Nogueira, Eur. J. Cancer Prev., 2012, 21, 333–335 CrossRef PubMed.
Footnote |
| † Currently at Bruker Nano Surfaces Division, 3601 Calle Tecate Ave. Ste C, Camarillo, CA 93012. |
| This journal is © The Royal Society of Chemistry 2014 |