Effect of oxindolimine copper(II) and zinc(II) complexes on human topoisomerase I activity†
Prafulla
Katkar
a,
Andrea
Coletta
a,
Silvia
Castelli
a,
Gustavo L.
Sabino
b,
Ricardo A. Alves
Couto
b,
Ana M.
da Costa Ferreira
b and
Alessandro
Desideri
*a
aDipartimento di Biologia, Università Tor Vergata di Roma, 00173 Roma, Italy. E-mail: desideri@uniroma2.it
bInstituto de Química, Universidade de São Paulo, 05508-000 São Paulo, SP, Brazil
First published on 30th October 2013
Abstract
The ability of oxindolimine copper(II) and zinc(II) complexes, known to have antitumor activity, to inhibit human topoisomerase IB has been tested through enzymatic kinetic assays and molecular docking simulations. These copper and zinc compounds are able to inhibit remarkably the cleavage reaction and only partially the religation step, the copper compound being more efficient than the zinc one. A complete inhibition activity of the cleavage is only obtained when the enzyme is pre-incubated with the compound, the inhibition being irreversible and reversible for the copper and zinc compounds, respectively. The relative stability of such complexes was estimated by competitive equilibria with human serum albumin (HSA), monitored by CD spectroscopy. The copper species shows a log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KCuL = 17.2, while the analogous zinc complex exhibits a log KZnL = 7.2. Molecular docking simulation studies show that the almost square planar geometry of the copper compound allows a direct coordination of the metal with two amino acids (Glu492, Asp563) of the enzyme at variance of the zinc compound which has a more tetrahedral geometry. Altogether, the data indicate that the different coordination geometry achieved by the two transition metal ions has an important role in modulating their efficiency as topoisomerase I inhibitors.
KCuL = 17.2, while the analogous zinc complex exhibits a log KZnL = 7.2. Molecular docking simulation studies show that the almost square planar geometry of the copper compound allows a direct coordination of the metal with two amino acids (Glu492, Asp563) of the enzyme at variance of the zinc compound which has a more tetrahedral geometry. Altogether, the data indicate that the different coordination geometry achieved by the two transition metal ions has an important role in modulating their efficiency as topoisomerase I inhibitors.
1. Introduction
Human topoisomerases are ubiquitous enzymes that catalyze the topological changes of DNA during replication, transcription, recombination, repair, chromatin assembly and chromosome segregation by triggering, breaking and re-joining of the DNA strand.1 Strand cleavage by all topoisomerase involves nucleophilic attack by the catalytic tyrosine residue on the scissile phosphodiester bond that culminates in the formation of a covalent bond between the enzyme and one end of the broken strand.2 There are two types of human topoisomerase enzymes i.e. type I and type II. Type II enzymes are dimeric and cleave both DNA strands, while type I enzymes mediate the catalytic reaction creating a transient single strand break in the backbone of DNA.3 The catalytic cycle of the topoisomerase I enzyme begins at the active site tyrosine (tyr723) through a nucleophilic attack on DNA, creating breakage in one strand with the enzyme covalently attached to the 3′-phosphate to form the cleaved complex. After changing the linking number, a second nucleophilic attack, driven by the 5′hydroxyl DNA end, restores an intact double stranded DNA, and the enzyme is released. Human topoisomerase I consists of 765 amino acids with four distinct domains: the N-terminal, the core, the linker and the C-terminal domain.4 The bilobed structure of the enzyme formed by the core and C-terminal domains fully wraps around the DNA during the catalytic cycle.5Topoisomerase IB is the target of a vast number of drugs that depending on their action are divided as poisons and inhibitors.6,7 The poisons reversibly bind to the DNA–enzyme cleavage complex, camptothecin is the best characterized representative. Camptothecin converts topoisomerase I into a DNA damaging agent by prolonging the lifetime of the cleavage intermediate, thus creating an obstacle to the advancement of replication machinery that ultimately results into DNA damage and cell death.8–10 Two main analogs of camptothecin, topotecan and irinotecan, have been successfully used in the treatment of several human cancers and have been approved by the US food and Drug administration for the clinical purpose.11,12 Catalytic inhibitors are compounds that prevent the binding of topoisomerase I to DNA or inhibit the cleavage reaction of the enzyme and so inhibit the DNA relaxation.13–16 Some compounds are able to inhibit both cleavage and religation.17,18 A few metal complexes have also been found to target topoisomerases, including essential and non-essential metals.19–23 The here presented compounds are particularly interesting since their geometrical arrangement can be finely tuned by the coordination properties of each metal and structural features of each ligand. Some oxindole–Schiff base copper(II) complexes have been synthesized and characterized, and have shown a significant cytotoxicity towards the human neuroblastoma SH-SY5Y cells.24 Additionally, these complexes were shown to have a potential role in the activation of apoptosis in different tumor cell lines.25,26
In the present work we have investigated the ability of oxindolimine copper(II) and zinc(II) complexes, hereafter called [Cu(isapn)]2+ and [Zn(isapn)]+, to interact with human topoisomerase IB, analyzing the different steps of the enzyme catalytic cycle. The results indicate that both the metal complexes inhibit significantly the cleavage reaction and slow down the religation process. A possible mechanism for the inhibition, based on the different coordination properties of the copper and zinc metal ions, is proposed from the results of molecular docking simulations of both compounds over the protein.
2. Experimental
2.1. Syntheses of ligand and metal complexes
The free oxindolimine ligand (isapn) was synthesized by condensation of isatin (or 2,3-dioxoindole, 10 mmol) and 1,3-diaminepropane (5 mmol) in ethanol, at pH 5.5, under constant stirring for 6 hours, and was isolated as a yellow powder, after filtration and drying in a desiccator, under vacuum, with 79% yield (1.31 g; 3.95 mmol). Found: C, 67.64; H, 4.72; N, 16.52%. Calc. for C19H16N4O2: C, 68.66; H, 4.85; N, 16.86%. FT-IR (cm−1, KBr): 3450 m, ν(OH); 3232 s, ν(NH); 1728 s, ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1593 s, ν(CN); 759 s, δ (CHar). 1H NMR (300 MHz, DMSO-d6, δ ppm): 2.32 (q, 2H, CH2), 4.15 (t, 4H, CH2), 6.90 (d, 2H, CHar), 7.04 (t, 2H, CHar), 7.41 (t, 2H, CHar), 7.88 (d, 2H, CHar), 10.80 (s, 2H, NH) (ESI,† Fig. S-1). MS (ESI+): m/z = 333.1, [M + 1]+ in CH3OH, MW = 332.36 for C19H16N4O2 (ESI,† Fig. S-2).
O); 1593 s, ν(CN); 759 s, δ (CHar). 1H NMR (300 MHz, DMSO-d6, δ ppm): 2.32 (q, 2H, CH2), 4.15 (t, 4H, CH2), 6.90 (d, 2H, CHar), 7.04 (t, 2H, CHar), 7.41 (t, 2H, CHar), 7.88 (d, 2H, CHar), 10.80 (s, 2H, NH) (ESI,† Fig. S-1). MS (ESI+): m/z = 333.1, [M + 1]+ in CH3OH, MW = 332.36 for C19H16N4O2 (ESI,† Fig. S-2).
This isolated ligand was then metallated in an ethanolic solution with zinc(II) ions added as perchlorate salt, at pH 7.5, under constant stirring for 2 hours. An orange solid was collected, filtered off, washed with cooled ethanol and ethyl ether, and dried in a vacuum. Yield: 85% (1.67 g; 3.36 mmol), [Zn(isapn)]ClO4. Found: C, 45.71; H, 3.48; N, 11.31%. Calc. for C19H15N4O2Zn(ClO4): C, 45.99; H, 3.05; N, 11.29%. FT-IR (cm−1, KBr): 3469 m, ν(OH); 1728 s, ν(CO); 1595 s, ν(CN); 1105 and 624 s, ν(ClO4 non coordinated). 1H NMR of the protonated form, at an apparent pH 5 (500 MHz, MeOH-d4, δ ppm): 6.96 (d, 2H, CHar), 7.11 (t, 2H, CHar), 7.55 (d, 2H, CHar), 7.59 (t, 2H, CHar) (ESI,† Fig. S-3). The NMR spectrum at an apparent pH 7 shows an equilibrium with the keto–eno and the eno–eno species, as indicated by the presence of multiple signals (ESI,† Fig. S-4). MS (ESI+): m/z = 396.9, in CH3OH–H2O, MW = 396.05, fragment monocation [C19H16N4O264Zn]; 398.9 [MW = 398.05 for the isotopic pattern of the (66Zn) monocation] (ESI,† Fig. S-5).
The analogous copper(II) complex has already been synthesized by condensation of the precursors isatin and 1,2-diaminopropane, followed by metallation with copper(II) perchlorate as previously described.27 A brown solid was isolated, with 85% yield, [Cu(isapn)](ClO4)2. Found: C, 39.33 H, 2.88; N, 9.69; Cu, 10.58%. Calc. for C19H16N4O2Cu(ClO4)2: C, 38.36; H, 2.71; N, 9.42; Cu, 10.72%. FT-IR (cm−1, KBr): 3447 m, ν(OH or NH); 1591 s, ν(CN); 1459 m, ν(CO); 1102 and 625 s, ν(ClO4 non coordinated). EPR parameters: g⊥ = 2.091; A⊥ = 14.2; g∥ = 2.301; A∥ = 126 × 104 cm−1; g∥/A∥ = 183 cm.27
2.2. Physical methods
Elemental analyses were performed at the Central Analítica – USP using a Perkin-Elmer 2400 CHN Elemental Analyzer. Infrared spectra were recorded in a BOMEM 3.0 (diffuse reflectance) instrument, in the range 4000–400 cm−1. Conductivity measurements with 1.0 mmol L−1 solution of the complexes were carried out on a Digimed DM-31 instrument using a 1.0 mmol L−1 KCl solution as standard (specific conductivity = 146.9 μS cm−1, at 25 °C). 1H NMR spectra were recorded in a Bruker DRX-500 instrument, operating at 500 MHz, at Central Analitica of IQ-USP. The spectrum of the ligand isapn was registered at room temperature in dmso-d6 solution and those of the complex [Zn(isapn)]ClO4 in MeOH-d4, at apparent pH 5 and 7. Interactions of both copper(II) and zinc(II) complexes with DNA were verified by using Circular dichroism spectroscopy. CD spectra were registered in a JASCO J-720 spectropolarimeter, using a quartz cuvette of 0.1 cm path length, at room temperature, in the range 200–300 nm. The initial experimental DNA concentration was 800 μM, and the spectra were registered in the absence or in the presence of increasing concentrations (up to 160 μM) of each complex studied. Additionally, the relative stability constants of these metal complexes were estimated by CD measurements, using human serum albumin as a competitive ligand. Simultaneous equilibria of the protein with the zinc(II) complex allowed the estimation of logKZnL, analogous to the previous determination of relative stability constants of similar copper(II) complexes.28 Titration experiments of HSA with the zinc(II) or copper(II) complex solutions up to 0.3 mmol L−1, in phosphate buffer (50 mM, pH 7.4), have been monitored by CD spectroscopy, at 354 or 564 nm, respectively. A zinc–aqua complex, [Zn(H2O)4]2+, was used in the control experiments in order to monitor and calculate the concentration of the formed [Zn(HSA)]+ species at 354 nm. Based on the CD spectral data, the relative thermodynamic stability constants of the different complexes were estimated, according to the previously described method.27
2.3. Purification of human topoisomerase IB
The human topoisomerase IB was expressed under the galactose inducible promoter in a multi-copy plasmid, YEpGAL1-e-wild type and YEpGAL1-e-Y723F, used for the transformation of EKY3 cells, as described previously.29 The epitope-tagged constructs contain the N-terminal sequence FLAG: DYKDDDY (indicated with “e”), recognized by the M2 monoclonal antibody. The purification was carried out using an ANTI-FLAG M2 Affinity Gel (Sigma) column. The FLAG-fusion topoisomerase IB was eluted by competition with five column volumes of a solution containing a 100 μg ml−1 FLAG peptide in 50 mM Tris–HCl, 150 mM KCl, pH 7.4. Glycerol was added into each fraction collected up to a final concentration of 33%. All the fractions were stored at −20 °C. Integrity of the protein was verified by the immunoblot assay. The purified protein was resolved on SDS-PAGE, transferred to nitrocellulose paper and immunoblotted with specific monoclonal antibody (Sigma-A9469). An immunoreactive band, corresponding to topoisomerase I, was detected with the BCIP/NBT substrate (Sigma-B3804) (ESI,† Fig. S-6).2.4. Topoisomerase IB activity in vitro: DNA relaxation assay
Human topoisomerase IB activity was observed in 30 μl reaction volume containing 0.5 μg of negatively supercoiled pBlueScript KSII(+) DNA and reaction buffer (20 mM Tris-HCl, 0.1 mM Na2EDTA, 10 mM MgCl2, 50 μg ml−1 acetylated BSA and 150 mM KCl, pH 7.5).The effect of [Cu(isapn)]2+ and [Zn(isapn)]+ on enzyme activity was tested adding different compound concentrations at different time intervals. The reaction was stopped by the addition of 0.5% SDS to each time course point incubated at 37 °C. The samples were electrophoresed in a 1% horizontal agarose gel in 50 mM Tris, 45 mM boric acid, and a 1 mM EDTA buffer system. The gel was stained with ethidium bromide (5 μg ml−1), destained with water and photographed under UV illumination. Where indicated, enzyme and the inhibitor were pre-incubated 5 min prior to the addition of the substrate. The stability of the inhibitory effect of [Cu(isapn)]2+ and [Zn(isapn)]+ was monitored as a function of time. Each assay was performed at least three times.
2.5. Cleavage kinetics
The oligonucleotide substrate CL14 (5′-GAAAAAAGACTTAG-3′) radiolabelled with [γ-32P] ATP at its 5′ end and the CP25 complementary strand (5′-TAAAAATTTTTCTAAGTCTTTTTTC-3′), phosphorylated at its 5′ end with unlabeled ATP, were annealed at a 2-fold molar excess of CP25 over CL14, creating the so called “suicide substrate”, which contains only a partial duplex.The suicide cleavage reactions were carried out incubating 20 nM of this partial duplex substrate with an excess of enzyme in reaction buffer at 37 °C and in the presence of 50 μM [Cu(isapn)]2+ and 300 μM [Zn(isapn)]+. DMSO was added to no-drug control. Where indicated [Cu(isapn)]2+ or [Zn(isapn)]+ was pre-incubated with enzyme for 5 min before the addition of a DNA substrate. Before the addition of the enzyme, a 5 μl sample of the reaction mixture was removed and used as the zero time point. At different time points 5 μl aliquots were removed and the reaction stopped with 0.5% SDS. After ethanol precipitation samples were re-suspended in 6 μl of 1 mg ml−1 trypsin and incubated at 37 °C for 1 hour. Samples were analyzed using denaturing urea/polyacrylamide gel electrophoresis. The experiment was replicated at least three times and a representative gel is shown. The percentage of cleavage at the preferential site (CL1) was quantified through PhosphoImager and ImageQuant software, comparing the percentage of the CL1 product obtained in each line to the maximal CL1 percentage obtained at the longest times.18
2.6. Religation kinetics
A suicide CL14/CP25 substrate (20 nM), prepared as above, was incubated with an excess of topoisomerase IB enzyme for 30 min at 37 °C in reaction Buffer. A 5 μl sample of the reaction mixture was removed and used as the zero time point. Religation reactions were initiated by adding a 200-fold molar excess of R11 oligonucleotide (5′-AGAAAAATTTT-3′) over the duplex CL14/CP25 in the presence or absence of 50 μM [Cu(isapn)]2+ and 300 μM [Zn(isapn)]+.At different times 5 μl aliquots were removed and the reaction stopped with 0.5% SDS. After ethanol precipitation samples were re-suspended in 5 μl of 1 mg ml−1 trypsin and incubated at 37 °C for 30 min. Samples were analyzed by denaturing urea/polyacrylamide gel electrophoresis. The experiment was replicated three times and a representative gel is shown.
The percentage of religation was determined using PhosphoImager and ImageQuant software, normalized on the total amount of radioactivity in each lane and relatively to the highest amount of substrate converted to the reaction product by human topoisomerase IB in the experiments.
2.7. Electrophoretic mobility shift assay
The shift in DNA mobility due to topoisomerase binding was carried out with a 25 mer full duplex oligonucleotide CL25/CP25. The reaction was performed with the Y723F topoisomerase IB mutant which is catalytically inactive. The inactive enzyme was incubated under standard reaction conditions [20 mM Tris–HCl, pH 7.5, 0.1 mM Na2EDTA, 10 mM MgCl2, 50 μg ml−1 acetylated BSA and 150 mM KCl] in the presence of 1% (v/v) DMSO or 50 μM [Cu(isapn)]2+ and 300 μM [Zn(isapn)]+ at 37 °C for 30 min, in a final volume of 30 μl. The binding reaction was performed at 37 °C for 30 min. Reactions were stopped by the addition of 5 μl of dye [0.125% Bromophenol Blue and 40% (v/v) glycerol]. Samples were loaded onto 6% (v/v) native polyacrylamide gels and electrophoresed at 40 V in TBE3 (12 mM Tris, 11.4 mM boric acid and 0.2 mM EDTA) at 4 °C for 4 hours. Products were visualized by phosphor imaging.2.8. Molecular docking
The input files were prepared with MglTools ver.1.5.6.rc3 (ref. 30) (http://mgltools.scripps.edu/). The receptor structures of the topoisomerase IB in the absence of DNA were obtained using the PDB entry 1A36. DNA was removed and the missing lateral chain of protein residues was modeled with UCSF-Chimera 1.6.1 (ref. 31) (http://www.cgl.ucsf.edu/chimera/) and were subjected to 1000 steps of steepest descent minimization with the same software. The receptor structure of the covalent complex was obtained using the PDB entry 1A31, where the linker domain 3D structure is not resolved. In order to include this domain in the simulation a model was built using the linker structure of PDB entry 1A36. This model structure was refined by 1000 steps of steepest descent minimization. The 3D structure of the metal compounds was obtained at the PB86/TZVP level of theory as previously described.28 The molecular docking simulations were performed using an in house modified version of AutoDock ver.4.2.2.1 (ref. 30) (http://autodock.scripps.edu/) on a grid of 140 × 140 × 140 bins with a step of 0.250 Å, centered on the Tyr723 (catalytic Tyrosine). 250 binding poses were generated for both [Cu(isapn)]2+ and [Zn(isapn)]+ using the Genetic Lamarckian algorithm, with the default parameters (medium depth of search). The poses were then subjected to an RMSD-based clusterization using a 2.0 Å cut-off.3. Results and discussion
3.1. Preparation and stability of the metal complexes
The free ligand, isapn, and the corresponding copper(II) and zinc(II) complexes were prepared according to the procedure described in the Experimental section. Both metal complexes were characterized by UV/Vis and IR spectroscopies, in addition to elemental analysis and conductivity measurements. The copper(II) complex has also been previously characterized by EPR and ESI-MS.27Conductivity measurements on the metal complex solutions in dimethyl formamide (DMF) at 25 °C indicate values of ΛM = 40.1 Scm2 mol−1 for [Zn(isapn)]+, which points to a 1:1 electrolyte, and ΛM = 101.4 Scm2 mol−1 for [Cu(isapn)]2+, indicative of a 2:1 electrolyte.32 These results are consistent with keto–enolic equilibria of the indole rings at the ligand isapn, as shown in Fig. 1, being the keto–enol form of the predominant structure of [Zn(isapn)]+ and keto–keto form of the [Cu(isapn)]2+, as isolated species in the solid state and as ionic species in DMF solution. However, in the 1H NMR spectra of the zinc complex, registered in methanol-d4 solution, the form A (keto–keto) is the main species, according to the more intense peaks attributed in comparison to the free ligand (ESI,† Fig. S-3 and S-4). At pH 5 the predominant species is still A, but additional peaks indicate the presence of corresponding keto–enol species B and enol–enol species C, which are more significant at pH 7 (see ESI,† Fig. S-4) or pH 10 (data not shown, indicating also decomposition). Therefore at physiological pH the B species is as well represented.
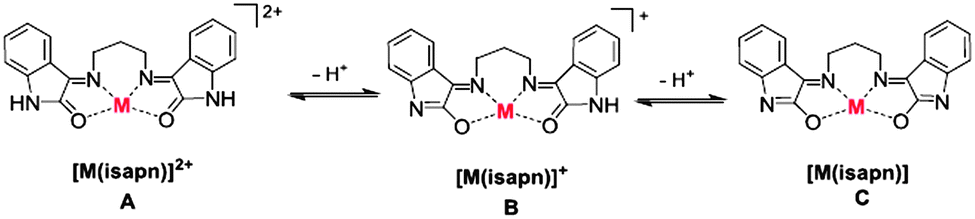 | ||
| Fig. 1 Equilibria of copper(II) and zinc(II) complexes with the isapn ligand in solution. (A) Keto–keto species, (B) keto–enol species, (C) enol–enol species. M: Cu2+ or Zn2+. Both complexes have perchlorate as a counter-ion. | ||
In order to be sure that the possible effect on the topoisomerase IB is due to the metal complex and not to the only free metal or the free ligand the thermodynamic stability of the complexes was studied by CD experiments. In particular, the relative stability constant of the zinc(II) complex, [Zn(isapn)]+, was estimated by CD experiments using HSA as a competitive ligand, similar to what had been previously done for different copper complexes, including [Cu(isapn)]2+.28 In Fig. 2, the corresponding CD curves obtained during titration of HSA by using each metal complex are shown. The obtained value for logKZn(isapn) was 7.2, which is very close to that reported in the literature for logKZn(HSA) = 7.1 (see Table 1). Therefore, the [Zn(isapn)]+ complex is as stable as the zinc ion coordinated by albumin. Analogously, copper(II) complex [Cu(isapn)]2+ displayed a logKCu(isapn) = 18.1, in comparison to a logKCu(HSA) = 16.2.33 In the case of copper ions, the insertion is preferential at the N-terminal metal binding site of the protein, while for zinc ions there are at least two more different sites described in the literature for this metal binding, one at Cys34 and another one in an inter-domain region, formed by two amino acid residues from domain I (Asn99 and His67) and two from domain II (His247 and Asp249).34,35
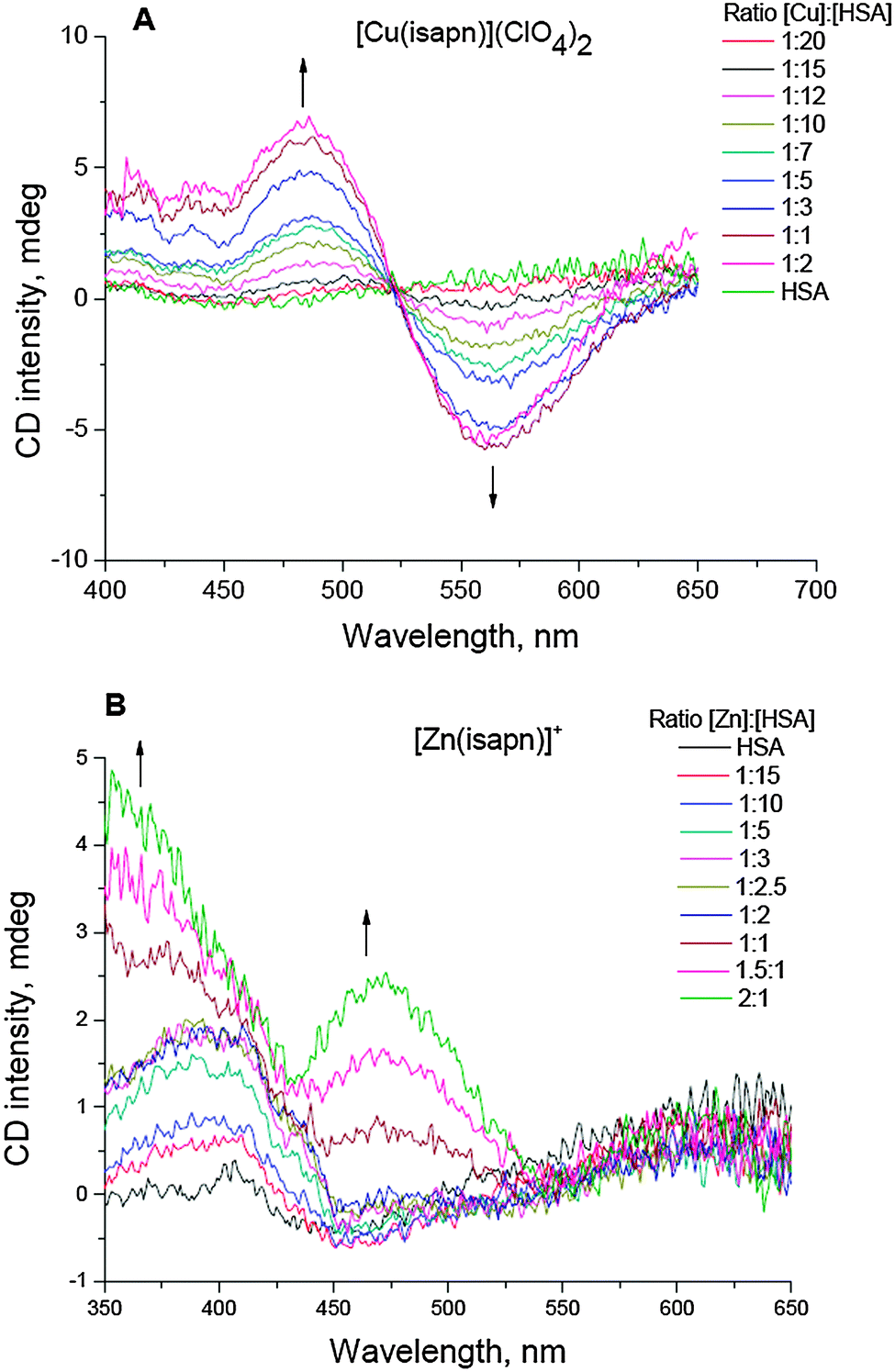 | ||
| Fig. 2 CD spectra registered during titration of HSA (0.10 mM) with: (A) complex [Cu(isapn)](ClO4)2, and (B) complex [Zn(isapn)]ClO4 solutions up to 0.3 mmol L−1 in phosphate buffer (50 mM, pH 7.4). The corresponding stability constants were determined using the absorbance changes at 564 nm and 354 nm, respectively. | ||
3.2. Inhibition of topoisomerase IB relaxation activity by [Cu(isapn)]2+ and [Zn(isapn)]+
The effect of [Cu(isapn)]2+ and [Zn(isapn)]+ in inhibiting the relaxation activity of topoisomerase IB has been assayed by plasmid relaxation assay, reacting a supercoiled plasmid incubated with the enzyme in the absence or the presence of different concentrations of the compound (Fig. 3A). The relaxation activity monitored at 30 min indicates that both [Cu(isapn)]2+ and [Zn(isapn)]+ inhibit the human topoisomerase IB activity in a dose dependent manner, [Cu(isapn)]2+ being more active since a full inhibition is achieved at 50 μM (Fig. 3A, lane 3), while for [Zn(isapn)]+ a full inhibition occurs at 300 μM (Fig. 3A, lane 18). Since both [Cu(isapn)]2+ and [Zn(isapn)]+ are dissolved in DMSO, an assay of the enzyme in the presence of an identical concentration of DMSO without the compounds has been carried out to show that, under these conditions, the relaxation activity of topoisomerase I is not affected (Fig. 3A, lane 19).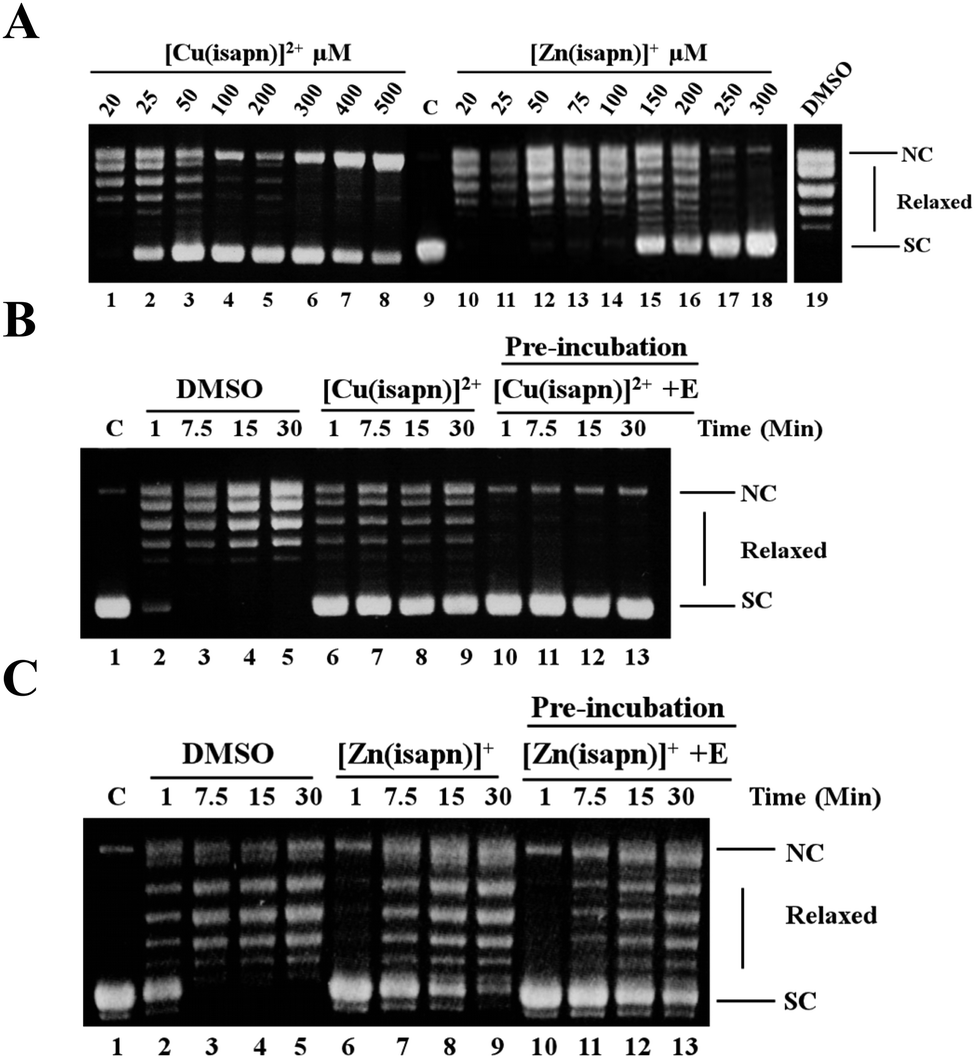 | ||
| Fig. 3 (A) Relaxation of negative supercoiled plasmid DNA by topoisomerase IB in the presence of increasing concentration of [Cu(isapn)]2+ (lanes 1–8) and [Zn(isapn)]+ (lanes 10–18). The reaction product were resolved in an agarose gel and visualized with ethidium bromide. Lane 9, no protein added. Lane 19, control reaction with DNA and DMSO without [Cu(isapn)]2+ and [Zn(isapn)]+. NC, nicked circular plasmid DNA. SC, supercoiled plasmid DNA. (B) Relaxation of negative supercoiled plasmid DNA in a time course experiment with DMSO (lanes 2–5), in the presence of 25 μM [Cu(isapn)]2+ (lanes 6–9), after pre-incubation of 25 μM [Cu(isapn)]2+ with the enzyme for 5 minutes at 37 °C (lanes 10–13). Lane 1, no protein added. (C) Relaxation of negative supercoiled plasmid DNA in a time course experiment with DMSO (lanes 2–5), in the presence of 150 μM [Zn(isapn)]+ (lanes 6–9), after preincubation of 150 μM [Zn(isapn)]+ with the enzyme for 5 minutes at 37 °C (lanes 10–13). Lane 1, no protein added. | ||
A lower concentration of [Cu(isapn)]2+ is required to have a complete inhibition of topoisomerase I, if the compound is pre-incubated with the enzyme. The results show that 25 μM of [Cu(isapn)]2+ fully inhibits the topoisomerase I when pre-incubated with the enzyme for 5 min before the addition of the substrate (Fig. 3B, lane 10–13). It is interesting to notice that the inhibition effect displayed by [Cu(isapn)]2+ persists as a function of time in the 1 to 30 min time window, indicating that under these conditions the [Cu(isapn)]2+ inhibitory effect is found to be irreversible. In the case of [Zn(isapn)]+ a full inhibition of the relaxation activity is observed only within the first minute even if a large excess of compound is used (150 μM) independently if the compound is concomitantly added or it is preincubated with the enzyme (Fig. 3C), indicating that under both conditions the effect is reversible.
3.3. Metal complex–DNA interactions
The CD DNA-signals are sensitive to the interaction with external compounds. The spectra usually consist of two bands, the positive one at 275 nm is due to the stacking between the compounds and DNA bases, whilst the negative one at 245 nm is due to the right-handed helicity of the B-DNA form.36 Titration of CT-DNA with [Cu(isapn)]2+ and [Zn(isapn)]+ shows only small perturbations in the negative and positive bands of the CD spectra (ESI,† Fig. S-7), indicating that the interaction between these complexes and DNA is negligible, as similarly observed with copper complexes with other oxindolimine ligands.37 In line, a perturbation of the DNA electrophoretic band is observed in the absence of the enzyme, at very high compound concentration (500 μM), remarkably above the concentrations needed to inhibit the enzyme, 50 μM and 300 μM for the copper and the zinc complex, respectively (data not shown). These results permit us to exclude a significant direct DNA–compound interaction at the concentration used in the topoisomerase inhibition assay.3.4. Analysis of cleavage and religation kinetics
The cleavage and the religation reactions have been followed in separate experiments to clarify if the metal compound inhibits the relaxation reaction affecting one of the two processes or both of them. The cleavage kinetics has been studied reacting an excess of topoisomerase I with the suicide substrate (Fig. 4A), in the absence and the presence of the compound. The PAGE analysis of the reaction products is shown in Fig. 4B and C, where the band corresponding to the cleaved DNA fragment is indicated as CL1. The cleavage kinetics is fast in the absence of the compound, whilst it is strongly reduced in the presence of 50 μM [Cu(isapn)]2+ and it is completely inhibited if the same concentration of the copper compound is pre-incubated with the enzyme before the substrate addition (Fig. 4B). The same trend has been observed in the case of the zinc compound, although a concentration of 300 μM is necessary in this case and a full inhibition is never observed even after pre-incubation (Fig. 4C).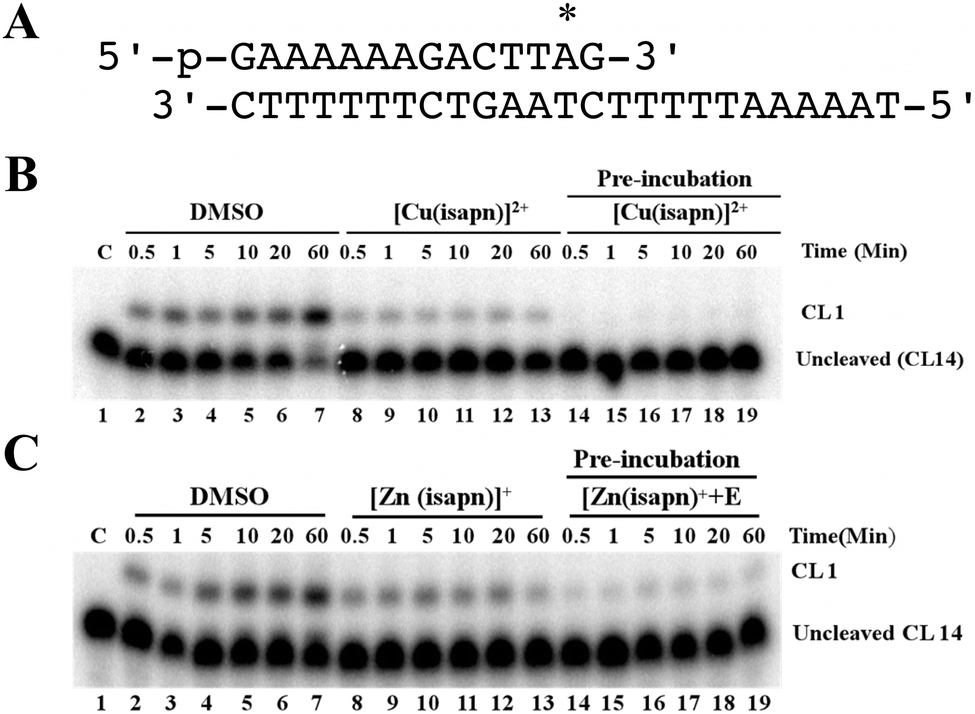 | ||
| Fig. 4 (A) The CL14/CP25 suicide substrate used to measure the cleavage kinetics of the enzyme. The preferred topoisomerase I binding site is indicated by an asterisk. (B) Cleavage kinetics of topoisomerase IB alone (lanes 2–7), in the presence of 50 μM [Cu(isapn)]2+ (lanes 8–13), and after 5 min pre-incubation of 50 μM [Cu(isapn)]2+ with the enzyme (lanes 14–19). Lane 1, no protein added. CL1 represents the DNA fragment cleaved at the preferred enzyme site. (C) Cleavage kinetics of topoisomerase IB alone (lanes 2–7), in the presence of 300 μM [Zn(isapn)]+ (lanes 8–13), and after 5 min pre-incubation of 300 μM [Zn(isapn)]+ with the enzyme (lanes 14–19). Lane 1, no protein added. CL1 represents the DNA fragment cleaved at the preferred enzyme site. | ||
The religation kinetics has been carried out incubating the suicide cleavage substrate with an excess of native enzyme for 30 min to produce the cleaved complex, followed by subsequent addition of 200-fold molar excess of the R11 complementary oligonucleotide (Fig. 5A) in the presence of DMSO, [Cu(isapn)]2+ or [Zn(isapn)]+ respectively. The data show that the religation kinetics of the native protein (Fig. 4B, lane 2–8) is slowed down in the presence of 50 μM [Cu(isapn)]2+ (Fig. 5B, lane 9–15). The difference is clear in the first part of the reaction, where the copper compound is able to almost completely inhibit the religation reaction (Fig. 5C). A similar trend is observed in the case of [Zn(isapn)]+ (Fig. 5D and E), although in this case a much higher concentration of the compound has been used (300 μM).
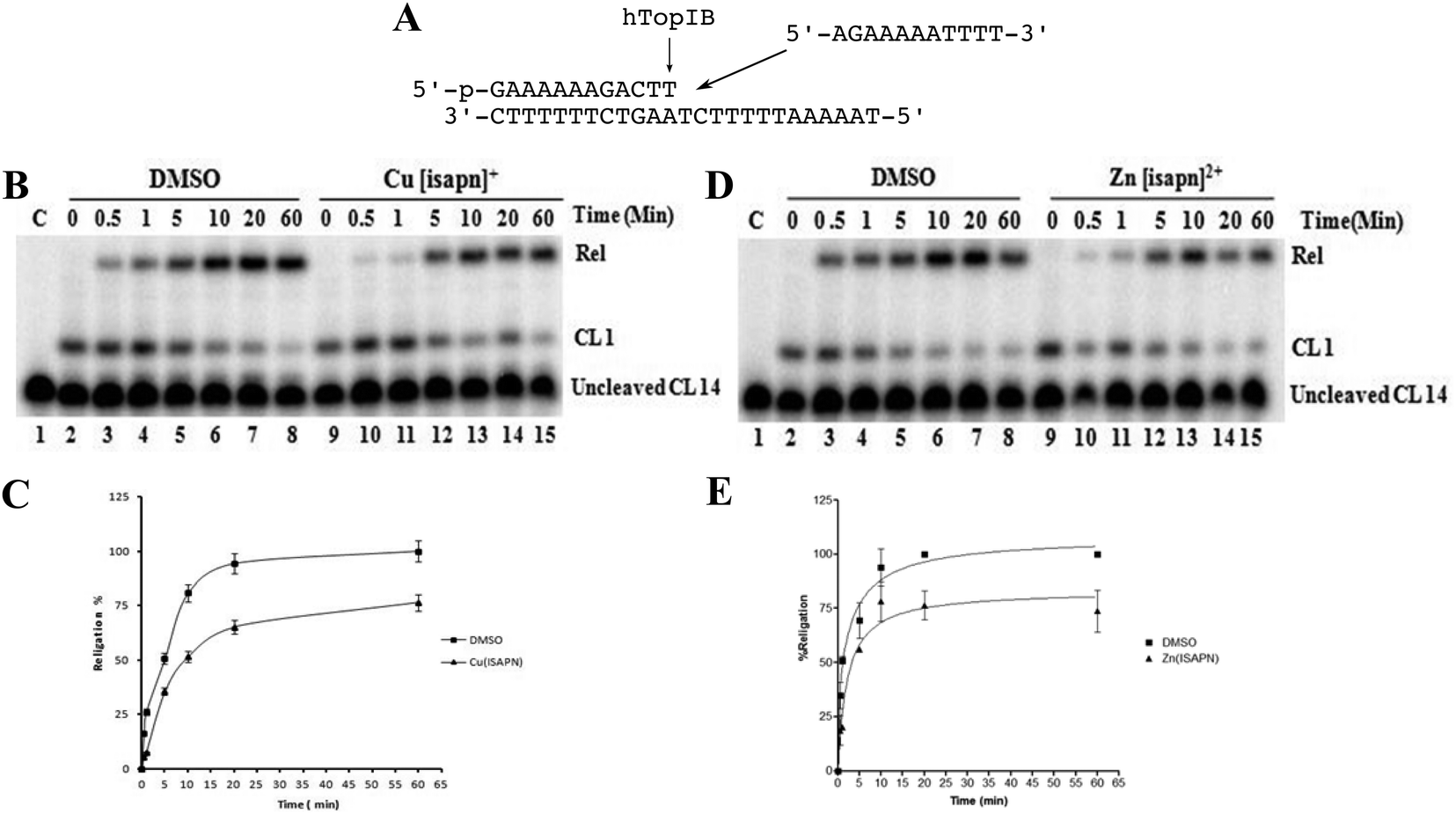 | ||
| Fig. 5 (A) The suicide substrate CL14/CP25 and the R11 complementary oligonucleotide used to measure the religation kinetics of the enzyme. (B) Urea-polyacrylamide gel electrophoretic analysis of the religation kinetics of topoisomerase IB in the absence (lanes 2–8) or in the presence of 50 μM [Cu(isapn)]2+ (lanes 9–15). Lane 1 represents the substrate alone. (C) Percentage of religation products plotted at different time intervals for topoisomerase IB in the absence (square) or in the presence of 50 μM [Cu(isapn)]2+ (triangle). The data reported are the average ± SD of three independent experiments. (D) Urea-polyacrylamide gel electrophoretic analysis of the religation kinetics of topoisomerase IB in the absence (lanes 2–8) or in the presence of 300 μM [Zn(isapn)]+ (lanes 9–15). Lane 1 represents the substrate alone. (E) Percentage of religation products plotted at different time intervals in the absence (square) or in the presence of 300 μM [Zn(isapn)]+ (Triangle). The data reported are the average ± SD of three independent experiments. | ||
3.5. Topoisomerase IB–DNA interaction study
The ability of the metal compounds to affect the enzyme–DNA binding has been monitored through a DNA binding assay carried out with the inactive Y723F mutant. The data reported in Fig. 6 indicate that a band corresponding to the DNA–topoisomerase IB complex is observed when the substrate is incubated with the enzyme alone or in the presence of [Zn(isapn)]+, whilst no band is observed in the presence of the [Cu(isapn)]2+ compound. These data indicate that the two compounds have a different inhibition mechanism, since [Cu(isapn)]2+ does not permit the binding of the enzyme to DNA at the variance of [Zn(isapn)]+.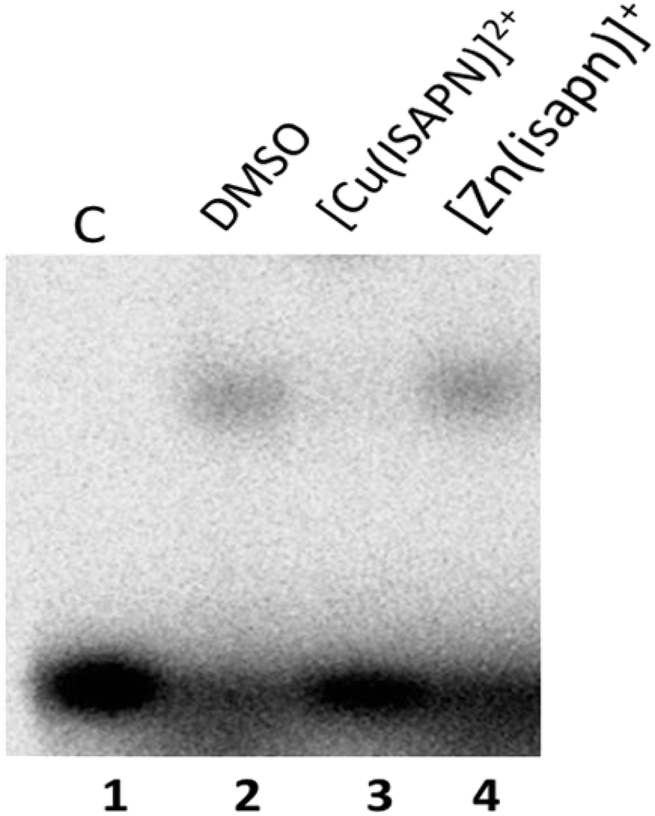 | ||
| Fig. 6 Electrophoretic mobility shift assay of the [γ−32P] radiolabelled substrate CL25/CP25 alone (lane 1), in the presence of inactive mutant Y723F enzyme (lane 2), in the presence of the mutant and 50 μM [Cu(isapn)]2+ (lane 3), in the presence of the mutant and 300 μM [Zn(isapn)]+ (lane 4). The asterisk indicates the band due to the DNA–enzyme complex. | ||
3.6. Molecular docking
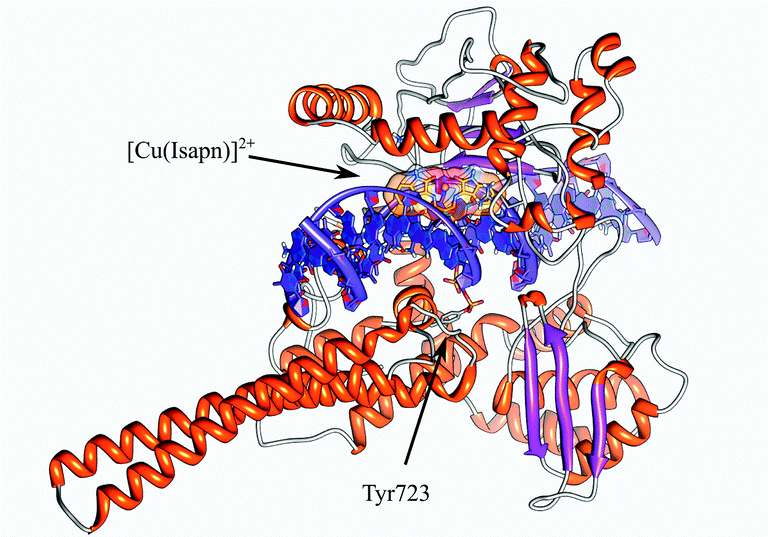
Using the molecular geometries of the two metal complexes obtained at the PB86/TZVP level of theory, molecular docking experiments have been performed for the [Cu(isapn)]2+ and for the [Zn(isapn)]+ compounds on topoisomerase IB. In detail since the compounds have been shown to inhibit the cleavage (Fig. 3B and C), a first docking has been done with the enzyme alone in the absence of DNA. The docking gives rise to 250 structures that have been subjected to an RMSD-based clusterization using a 2.0 Å cutoff. For both compounds no binding sites are found in the internal surface that is rich of positively charged amino acids, due to the positive total charge of the two compounds (2+ and 1+ for [Cu(isapn)]2+ and [Zn(isapn)]+ respectively). In the case of [Cu(isapn)]2+ the energies of binding of the 250 complexes, obtained by molecular docking, range from −9.7 to −5.3 kcal mol−1 and more than 50% of the complexes (130 out of 250) are grouped in a single cluster with a mean binding energy of −9.6 kcal mol−1. The representative structure of this cluster is reported in Fig. 7 where it can be observed that the Cu atom is at a coordinating distance with oxygen of the lateral chain of Asp563 and Glu492 of topoisomerase IB. Glu492 is part of one of the two loops called “Lips” that are known to stabilize the topoisomerase IB clamp around DNA through a salt bridge interaction between Glu497 and Lys369.38,39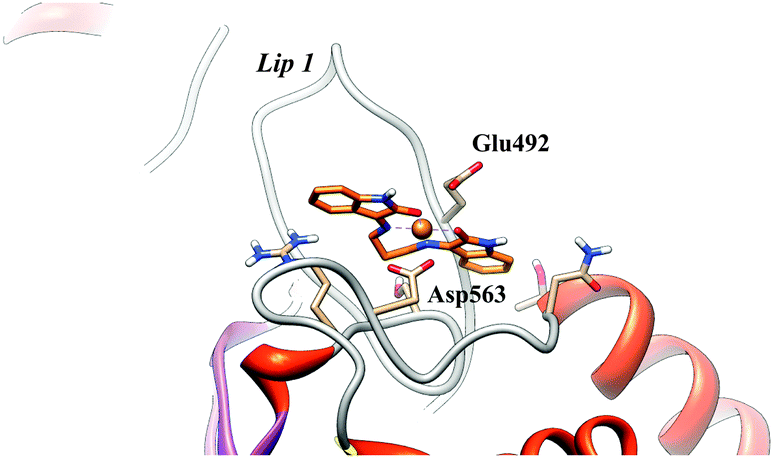 | ||
| Fig. 7 Representative structure of the most populated cluster for the [Cu(isapn)]2+-topoisomerase I docking. The Glu492 and Asp563 residues located on lip1 and coordinating the [Cu(isapn)]2+ compound are labeled and represented as sticks. | ||
The energies of binding of the complexes obtained from the docking with [Zn(isapn)]+ range from −9.1 to −6.2 kcal mol−1. The most populated cluster (89 out of 250) is again located on the external surface near the “Lips” region but has an energy of −7.5 kcal mol−1, larger than the one observed for [Cu(isapn)]2+. Due to the non-planarity of the [Zn(isapn)]+ compound, that displays a tetrahedral geometry, no direct coordination with the protein is observed (Fig. 8).
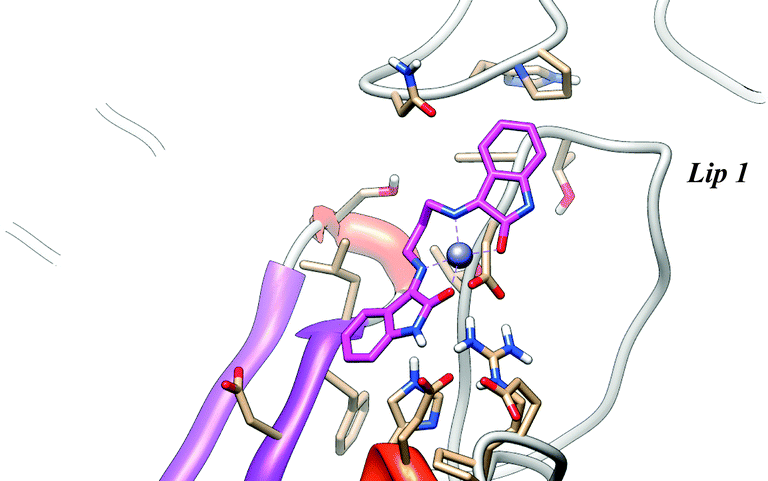 | ||
| Fig. 8 Representative structure of the most populated cluster for the [Zn(isapn)]+-topoisomerase I docking. The energy of binding is −7.45 kcal mol−1. [Zn(isapn)]+, is represented as magenta sticks. | ||
A molecular docking experiment has also been performed on the topoisomerase IB–DNA covalent complex, since the compounds have been shown to be able to partially inhibit the religation (Fig. 5B and D). In all the complexes the compounds bind on the DNA minor groove, downstream of the cleavage site without a direct interaction with the enzyme. The energies of binding of [Cu(isapn)]2+ range from −14.9 to −13.0 kcal mol−1 and almost all of the docking poses (192 out of 250) are in the first and most populated cluster with an energy of −14.9 kcal mol−1. A representative structure is shown in Fig. 9. The energies of binding of [Zn(isapn)]+ range from −12.1 to −11.3 kcal mol−1. Almost all of the docking poses (186 out of 250) are in the first and most populated cluster with a mean binding energy of −12.1 kcal mol−1, in a position almost identical to that observed for the copper compound (figure not shown).
 | ||
| Fig. 9 Representative structure of the most populated cluster for the docking of [Cu(isapn)]2+ on the binary (topoisomerase I + DNA) covalent complex docking. The [Cu(isapn)]2+ compound is labeled and represented as orange sticks. The protein and the DNA backbone are represented as ribbons. Catalytic Tyr723 of topoisomerase I is labeled and represented as sticks. | ||
4. Conclusion
The plasmid relaxation assay indicates that both the [Cu(isapn)]2+ and [Zn(isapn)]+ metal ligand complexes inhibit the topoisomerase IB relaxation activity (Fig. 3A), the effect being enhanced when the compound is pre-incubated with the enzyme before substrate addition (Fig. 3B and C). The copper complex fully inhibits the relaxation of topoisomerase IB at 50 μM concentration, while the analogous zinc compound shows a similar effect at around 300 μM. The cleavage reaction is also inhibited by both compounds and again the effect is more intense when the compounds are pre-incubated with the enzyme (Fig. 4B and C). An interesting point is that [Cu(isapn)]2+ is more efficient than [Zn(isapn)]+, its inhibition occurs at a concentration about 3–4 times lower than that of the zinc compound. The molecular docking experiments suggest that this difference is related to the different coordination geometry displayed by the [Cu(isapn)]2+ and the [Zn(isapn)]+ compounds. The copper in fact displays a quite squared planar coordination while the zinc shows a more tetrahedral one. The different coordination geometry permits the copper to form a stable complex with topoisomerase IB in terms of cluster population size and energy of binding, since its planar coordination permits to fulfill a distorted octahedral geometry coordinating with the Glu492 and Asp563 residues of topoisomerase IB (Fig. 7). The Glu492 is located in one of the two “lips” that clamp DNA during the cleavage reaction, and this explains the efficiency of the copper compound in inhibiting the enzyme activity. The direct coordination of the copper to the enzyme also explains the copper compound irreversible inhibitory effect that persists at least up to 30 minutes (Fig. 3B). On the other hand the tetrahedral zinc geometry only permits a loose interaction with the protein, explaining the need for larger [Zn(isapn)]+ concentrations in order to inhibit similarly the cleavage and the relaxation reaction (Fig. 2A and 4A). Moreover, the lack of a direct coordination of the zinc ion to the protein explains its reversible inhibitory effect (Fig. 3C). The different interaction displayed by the two metal compounds also explains the different effect exerted on the DNA binding (Fig. 6). The direct coordination of the copper compound to a residue located on a lip clamping of the protein inhibits the protein–DNA binding as indicated by the DNA shift experiment (Fig. 6), whilst the protein still binds the DNA in the presence of [Zn(isapn)]+ (Fig. 6) in line with the absence of a direct protein coordination to the zinc ion (Fig. 8). On the other hand, both the copper and zinc compounds are able to slightly inhibit the religation, but in this case the effect is reversible and is lost after a couple of minutes (Fig. 5B and D). This behavior can be again explained by the docking experiments since in the protein–DNA covalent complex, the two compounds bind in the minor groove downstream of the cleavage site (Fig. 9) partially destabilizing the protein–DNA interaction.It can be interesting to compare the effect of the here reported metal compounds with the effect, reported in the literature, of other compounds containing metals.19,40–45 In detail a series of α-heterocyclic-N4-substituted thiosemicarbazones and their corresponding copper(II) complexes are described as inhibitors of topoisomerase IIα, in the range of IC50 0.3–7.2 μM.46 These copper(II) complexes are shown to be more potent than the thiosemicarbazone ligands, and they act as antineoplastic agents towards two different breast cancer cells, expressing high and low levels of Topo-IIα. Their mechanism of action was not completely elucidated, although they are reported as catalytic inhibitors rather than poisons of Topo-IIα. A more recent study describes an active copper(II) complex, with a bis-(thiosemicarbazone) ligand derived from glyoxal, capable of inhibiting Topo-IIα up to 90% at concentration of 100 μM, while other complexes of the same series are only moderately active (50–60%).47 Some zinc(II) ternary complexes, [Zn(bpy)L], where bpy is 2,2′-bipyridine, and L is a doubly deprotonated thiosemicarbazone, are able to inhibit topoisomerase I,48 the inhibition being more remarkable when the complex is previously incubated with the protein, before the addition of DNA. This behavior is indicative of a direct binding of the complex with the enzyme that may be the dominant pathway in the mechanism of action. In another work two ternary zinc(II) complexes, with 1,10-phenanthroline (phen) and with a dipicolinate or L-threoninate ligand, have been shown to inhibit topoisomerase I at 120 μM.49
It must be noticed that many of those studies report the enzyme inhibition only studying the relaxation activity and do not go into the dissection of the different catalytic steps as reported in our work, making difficult a direct comparison with our compounds. A comparison of the inhibitory effect indicates that the here reported Cu[isapn]2+ complex is more efficient to inhibit topoisomerase I activity than the other metal complex and it is 5–6 times more efficient than the analogous zinc compound (around 300 μM).
In conclusion, the here reported results indicate that the use of metal compounds can be an interesting strategy to target topoisomerase I and the ability of different transition metal ions capable of imposing different coordination geometry can be exploited to modulate their inhibition effects.
Abbreviations
| Topo | Topoisomerase |
| CD | Circular dichroism |
| HAS | Human serum albumin |
| isapn | Imine ligand prepared from isatin and 1,3-diaminopropane |
| CT-DNA | Calf thymus DNA |
Acknowledgements
A.M.D.C. Ferreira and A. Desideri are grateful to the Brazilian entity FAPESP for grants (Proc. 11/50318-1 and 12/03080-2). A.M.D.C. Ferreira is also thankful to INCT-Redox Processes in Biomedicine (Redoxoma), and NAP-Redoxoma/USP, and A. Desideri to the Italian Association for Cancer Research (AIRC) for financing the project No. 10121.References
- J. J. Champoux, Annu. Rev. Biochem., 2001, 70, 369–413 CrossRef CAS PubMed.
- J. C. Wang, Annu. Rev. Biochem., 1996, 65, 635–692 CrossRef CAS PubMed.
- J. J. Champoux, J. Biol. Chem., 1981, 256, 4805–4809 CAS.
- M. R. Redinbo, L. Stewart, X. Qiu, W. G. Hol and J. J. Champoux, Science, 1998, 279, 1534–1541 CrossRef.
- J. B. Leppard and J. J. Champoux, Chromosoma, 2005, 114, 75–85 CrossRef CAS PubMed.
- D. A. Burden and N. Osheroff, Biochim. Biophys. Acta, 1998, 1400, 139–154 CrossRef CAS.
- J. A. Holden, Curr. Med. Chem.: Anti-Cancer Agents, 2001, 1, 1–25 CrossRef CAS PubMed.
- Y. H. Hsiang, M. G. Lihou and L. F. Liu, Cancer Res., 1989, 49, 5077–5082 CAS.
- Y. Yonezawa, T. Hada, K. Uryu, T. Tsuzuki, K. Nakagawa, T. Miyazawa, H. Yoshida and Y. Mizushina, Int. J. Oncol., 2007, 30, 1197–1204 CAS.
- S. Castelli, S. Vieira, I. D'Annessa, P. Katkar, L. Musso, S. Dallavalle and A. Desideri, Arch. Biochem. Biophys., 2013, 530, 7–12 CrossRef CAS PubMed.
- Y. Pommier, P. Pourquier, Y. Fan and D. Strumberg, Biochim. Biophys. Acta, 1998, 1400, 83–105 CrossRef CAS.
- (a) Y. Pommier, Chem. Rev., 2009, 109, 2894–2902 CrossRef CAS PubMed; (b) Y. Pommier, ACS Chem. Biol., 2013, 8(1), 82–95 CrossRef CAS PubMed.
- S. Castelli, A. Campagna, O. Vassallo, C. Tesauro, P. Fiorani, P. Tagliatesta, F. Oteri, M. Falconi, H. K. Mujumder and A. Desideri, Arch. Biochem. Biophys., 2009, 486, 103–110 CrossRef CAS PubMed.
- C. Bailly, J. F. Riou, P. Colson, C. Houssier, E. Rodrigues-Pereira and M. Prudhomme, Biochemistry, 1997, 36, 3917–3927 CrossRef CAS PubMed.
- D. Montaudon, K. Palle, L. P. Rivory, J. Robert, C. Douat-Casassus, S. Quideau, M. Bjornsti and P. Pourquier, J. Biol. Chem., 2007, 282, 14403–14412 CrossRef CAS PubMed.
- S. Castelli, A. Coletta, I. D'Annessa, P. Fiorani, C. Tesauro and A. Desideri, Biol. Chem., 2012, 393(11), 1327–1340 CrossRef CAS PubMed.
- C. Tesauro, P. Fiorani, I. D'annessa, G. Chillemi, G. Turchi and A. Desideri, Biochem. J., 2010, 425, 531–539 CrossRef CAS PubMed.
- S. Castelli, P. Katkar, O. Vassallo, M. Falconi, S. Linder and A. Desideri, Anti-Cancer Agents Med. Chem., 2013, 13, 356–363 CrossRef CAS PubMed.
- S. Castelli, O. Vassallo, P. Katkar, C. Che, R. Sun and A. Desideri, Arch. Biochem. Biophys., 2011, 516, 108–112 CrossRef CAS PubMed.
- A. Casini, G. Kelter, C. Gabbiani, M. A. Cinellu, G. Minghetti, D. Fregona, H. H. Fiebig and L. Messori, JBIC, J. Biol. Inorg. Chem., 2009, 14, 1139–1149 CrossRef CAS PubMed.
- N. Farrell, J. Inorg. Biochem., 2013, 119, 54–64 CrossRef PubMed.
- X. Wu, J. C. Yalowich and B. B. Hasinoff, J. Inorg. Biochem., 2011, 105, 833–838 CrossRef CAS PubMed.
- Z.-F. Chen, M.-X. Tan, L.-M. Liu, Y.-C. Liu, H.-S. Wang, B. Yang, Y. Peng, H.-G. Liu, H. Liang and C. Orvig, Dalton Trans., 2009, 10824–10833 RSC.
- G. Cerchiaro, K. Aquilano, G. Filomeni, G. Rotilio, M. R. Ciriolo and A. M. D. C. Ferreira, J. Inorg. Biochem., 2005, 99, 1433–1440 CrossRef CAS PubMed.
- V. C. Silveira, J. S. Luz, C. C. Oliveira, I. Graziani, M. R. Ciriolo and A. M. D. C. Ferreira, J. Inorg. Biochem., 2008, 102, 1090–1103 CrossRef PubMed.
- G. Filomeni, G. Cerchiaro, A. M. D. C. Ferreira, J. Z. Pedersen, A. De Martino, G. Rotilio and M. R. Ciriolo, J. Biol. Chem., 2007, 282, 12010–12021 CrossRef CAS PubMed.
- (a) G. Cerchiaro, P. L. Saboya and A. M. D. C. Ferreira, Transition Met. Chem., 2004, 29, 495–504 CrossRef CAS; (b) G. Cerchiaro, G. A. Micke, M. F. M. Tavares and A. M. D. C. Ferreira, J. Mol. Catal. A: Chem., 2004, 221, 29–39 CrossRef CAS.
- V. C. Silveira, G. F. Caramori, M. P. Abbott, M. B. Gonçalves, H. M. Petrilli and A. M. D. C. Ferreira, J. Inorg. Biochem., 2009, 103, 1331–1341 CrossRef PubMed.
- G. Chillemi, P. Fiorani, S. Castelli, A. Bruselles, P. Benedetti and A. Desideri, Nucleic Acids Res., 2005, 33, 3339–3350 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- W. J. Geary, Coord. Chem. Rev., 1972, 7, 81–122 CrossRef.
- M. Valko, H. Morris, M. Mazur, J. Telser, E. J. L. McInnes and F. E. Mabbs, J. Phys. Chem. B, 1999, 103, 5591–5597 CrossRef CAS.
- Y. Zhang and D. E. Wilcox, JBIC, J. Biol. Inorg. Chem., 2002, 7, 327–337 CrossRef CAS PubMed.
- C. A. Blindauer, I. Harvey, K. E. Bunyan, A. J. Stewart, D. Sleep, D. J. Harrison, S. Berezenko and P. J. Sadler, J. Biol. Chem., 2009, 284, 23116 CrossRef CAS PubMed.
- V. I. Ivanov, L. E. Minchenkova, A. K. Schyolkina and A. I. Poletayer, Biopolymers, 1973, 12, 89–110 CrossRef CAS PubMed; B. Macías, M. V. Villa, B. Gómez, J. Borrás, G. Alzuet, M. González-Álvarez and A. Castiñeiras, J. Inorg. Biochem., 2007, 101, 444–451 CrossRef PubMed.
- V. C. Silveira, H. Benezra, J. S. Luz, R. C. Georg, C. C. Oliveira and A. M. D. C. Ferreira, J. Inorg. Biochem., 2011, 105, 1692–1703 CrossRef PubMed.
- M. R. Redinbo, L. Stewart, P. Kuhn, J. J. Champoux and W. G. Hol, Science, New York, N.Y, 1998, vol. 279, pp. 1504–1513 Search PubMed.
- G. Chillemi, A. Bruselles, P. Fiorani, S. Bueno and A. Desideri, Nucleic Acids Res., 2007, 35, 3032–3038 CrossRef CAS PubMed.
- S. Tabassum, W. M. Al-Asbahy, Md. Afzal, F. Arjmandand and V. Bagchi, Dalton Trans., 2012, 41, 4955–4965 RSC.
- K. W. Tan, H. L. Seng, F. S. Lim, S.-C. Cheah, C. H. Ng, K. S. Koo, M. R. Mustafa, S. W. Ng and Md. J. Maah, Polyhedron, 2012, 38, 275–284 CrossRef CAS.
- H.-L. Seng, S.-T. Von, K.-W. Tan, Md. J. Maah, S.-W. Ng, R. Rahman, I. Caracelli and C.-H. Ng, Biometals, 2010, 23, 99–118 CrossRef CAS PubMed.
- X.-M. Mo, Z. F. Chen, X. Qi, Y. T. Li and J. Li, Bioinorg. Chem. Appl., 2012, 2012, 756374, DOI:10.1155/2012/756374.
- A. P. Neves, M. X. G. Pereira, E. J. Peterson, R. Kipping, M. D. Vargas, F. P. Silva-Jr., J. W. M. Carneiro and N. P. Farrell, J. Inorg. Biochem., 2013, 119, 54–64 CrossRef CAS PubMed.
- X.-W. Liu, S.-B. Zhang, L. Li, Y.-D. Chen and J.-L. Lu, J. Organomet. Chem., 2013, 729, 1–8 CrossRef CAS.
- B. M. Zeglis, V. Divilov and J. S. Lewis, J. Med. Chem., 2011, 54, 2391–2398 CrossRef CAS PubMed.
- D. Palanimuthu, S. V. Shinde, K. Somasundaram and A. G. Samuelson, J. Med. Chem., 2013, 56, 722–734 CrossRef CAS PubMed.
- K. W. Tan, H. L. Seng, F. S. Lim, S.-C. Cheah, C. H. Ng, K. S. Koo, M. R. Mustafa, S. W. Ng and M. J. Maah, Polyhedron, 2012, 38, 275–284 CrossRef CAS.
- L.-F. Chin, S.-M. Kong, H.-L. Seng, Y.-L. Tiong, K.-E. Neo, M. J. Maah, A. S.-B. Khoo, M. Ahmad, T.-S. A. Hor, H.-B. Lee, S.-L. San, S.-M. Chye and C.-H. Ng, JBIC, J. Biol. Inorg. Chem., 2012, 17, 1093–1105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3mt00099k |
| This journal is © The Royal Society of Chemistry 2014 |