Quantification of copper binding to amyloid precursor protein domain 2 and its Caenorhabditis elegans ortholog. Implications for biological function†‡§
Su Ling
Leong
ab,
Tessa R.
Young
ac,
Kevin J.
Barnham
ade,
Anthony G.
Wedd
ac,
Mark G.
Hinds
*ac,
Zhiguang
Xiao
*ac and
Roberto
Cappai
*ab
aThe Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Victoria 3010, Australia
bDepartment of Pathology, The University of Melbourne, Victoria 3010, Australia. E-mail: r.cappai@unimelb.edu.au
cSchool of Chemistry, The University of Melbourne, Victoria 3010, Australia. E-mail: mhinds@unimelb.edu.au; z.xiao@unimelb.edu.au
dDepartment of Pharmacology, The University of Melbourne, Victoria 3010, Australia
eFlorey Institute for Neuroscience and Mental Health, Victoria, Australia
First published on 19th November 2013
Abstract
Aberrant regulation of transition metals and the resultant disregulation of neuronal reactive oxygen species (ROS) are considered significant in the etiology of Alzheimer's disease (AD). We determined the solution structure of the D2 domain of APL-1 (APL1-D2), the Caenorhabditis elegans ortholog of the amyloid precursor protein domain 2 (APP-D2). The copper binding affinities of APL1-D2 and APP-D2 were estimated and the ability of their copper complexes to promote formation of ROS was tested. The two protein domains are isostructural, consisting of a three-stranded β-sheet packed against a short α-helix in a βαββ fold. A six-residue insert in APL1-D2, absent in APP-D2, forms an extended loop. The putative copper binding ligands in APP-D2 are not conserved in APL1-D2 and this delineates a clear difference between them. APL1-D2 and APP-D2 bind one equivalent of Cu(I) weakly, with dissociation constants KD ∼10−8.6 M and ∼10−10 M, respectively, and up to two equivalents of Cu(II) with KD values in the range 10−6 –10−8 M. The relative abilities of APL1-D2, APP-D2 and amyloid-β (Aβ) copper complexes to generate H2O2 correspond to their copper binding affinities. Copper affinities for Aβ (KD ∼ 10−10 M for both Cu(I) and Cu(II)) and APP-D2 are in a range allowing redox cycling to occur, albeit less efficiently for APP-D2. However, APL1-D2 binds Cu(I) and Cu(II) too weakly to sustain catalysis which further suggests that it plays no significant role in copper handling in C. elegans. Overall, the data are consistent with a possible role in copper homeostasis for APP-D2, but not APL1-D2.
Introduction
The amyloid β (Aβ) peptide is generated from the amyloid precursor protein (APP) by proteolysis. APP is a single pass transmembrane glycoprotein that belongs to a strongly conserved multi-gene family containing paralogs such as the amyloid precursor-like proteins APLP1 and APLP2 in mammals.1,2 The exact biological function of APP and its homologs remains unknown. However, APP and Aβ are able to bind copper and are thought to play a role in copper homeostasis.3In vitro experiments have implicated APP and Aβ in the Cu(I)–Cu(II) redox cycle,4 an ability thought to contribute to the cellular oxidative stress and neuronal cell loss that define Alzheimer's disease (AD) pathogenesis.5,6 Orthologs of APP have been identified in many species7,8 and sequence conservation suggests a conserved function.9 Copper binding has not been demonstrated for all orthologs.APP features an ectodomain consisting of a growth factor-like domain (D1), a copper binding domain (D2), an acidic region (D3), a carbohydrate domain (D6) that can be divided further into an E2 domain (D6a) and a juxtamembrane domain (D6b).10,11 APP is a modulator of copper homeostasis in vitro and in vivo. Compared to wild type mice, APP knockout mice have elevated copper levels in the liver and cerebral cortex12 while transgenic mice overexpressing APP have significantly reduced copper levels.13,14 Neurons expressing APP in culture are more susceptible to Cu toxicity compared to those deficient in APP.15In vitro, specific Cu toxicity appears to be mediated by residues 135 to 166 of human APP.16 In particular histidine residues H147, H149 and H151 were identified as pivotal to APP-mediated Cu toxicity.
The 65-residue copper binding sub-domain (CuBD) within D2 is defined by conserved histidine residues and is located in the N-terminal cysteine-rich region of APP termed E1.4,17 Solution and crystal structures of APP-D2 revealed a Cu(II) binding site consisting of residues H147, H151, Y168 and possibly M170.18 The dissociation constant, KD, for APP Cu(II) binding was estimated to be 10 μM.4 It has been proposed that APP-D2 binds to Cu(II) and that reduction to Cu(I) ensues.18
Another metal binding site was identified recently within the E2 domain from three high-resolution X-ray structures.19 Metal binding at this site appears to function as a molecular switch that regulates the conformation, flexibility and hence the physiological function of APP-E2 and full-length APP.
Unlike mammals that have multiple APP-like genes, the worm Caenorhabditis elegans bears only a single gene apl-1 that is orthologous to APP. Consequently, this organism is an ideal genetic model to delineate APL-1 function and its connections to APP.20,21 Several roles have been ascribed to APL-1 in the worm, including cholesterol-dependent regulation of neurotransmission,20 moulting22 and regulation of metabolic and developmental pathways.23 The domain structure of APL-1 is similar to its mammalian homologues. It consists of two highly structured extracellular regions, E1 and E2, followed by a transmembrane region and a C-terminal cytoplasmic domain. The E1 domain contains 6 conserved cystine bridges that delineate the putative growth factor-like heparin-binding domain (D1) and the putative metal binding domain (D2). These folded modules of E1 are followed by a low complexity acidic region connecting the E1 and E2 domains. The APL-1 E2 domain is an all-helical protein with an N-terminal coiled coil domain and C-terminal 4-helix bundle. It is similar structurally to the human APP E2 domain.24 It bears a potential heparin-binding site that, unusually, uses histidine residues to bind heparin analogues in a pH-dependent manner.24,25 Like its mammalian counterpart, APL-1 is expressed in multiple tissues and there is a high degree of sequence similarity with the extracellular domains of APP (approximately 47%).26 The most striking differences between the C. elegans and human sequences in this domain are the absence in the region equivalent to the CuBD of histidine residues as potential metal ligands plus the presence of a six-residue insertion in a loop region (Fig. 1).
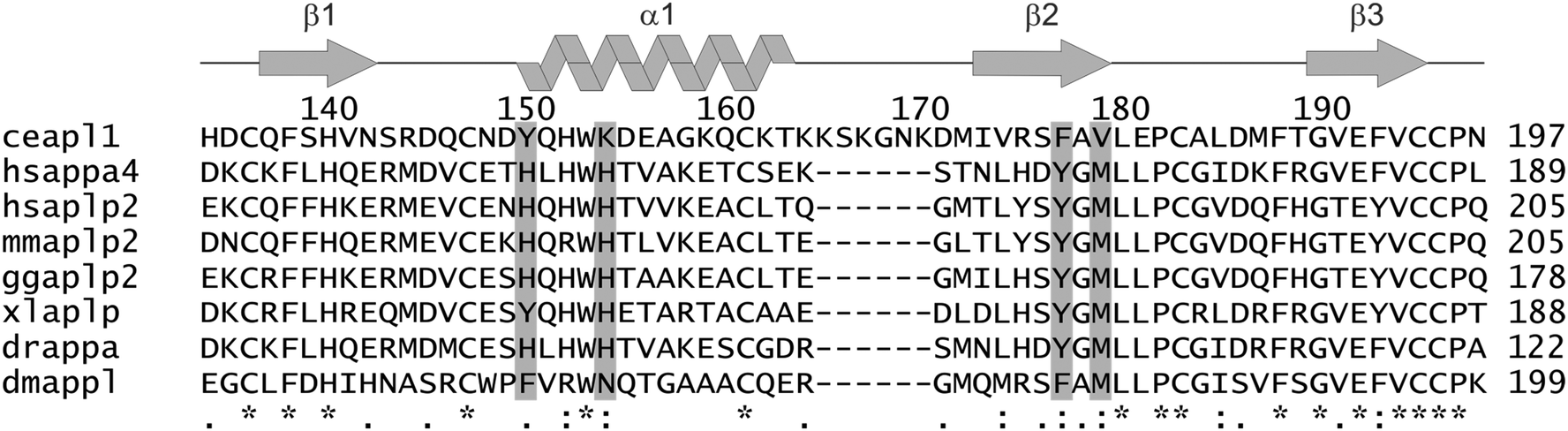 | ||
| Fig. 1 Sequence comparison of APL1-D2 with orthologous proteins. The position of the sequence insertion and the key copper ligand residues are indicated. The secondary structure elements and sequence numbers for C. elegans Apl1-D2 are depicted across the top of the sequence. The putative copper binding residues chosen on the basis of human APP-D2 are depicted in grey. Symbols: ‘*’ At the bottom, symbols indicate: conserved residue; ‘:’ conserved among residues of strongly similar properties; ‘.’ conservation among residues of weakly similar properties. ce, C. elegans; hs, Homo sapiens; mm, Mus musculus; gg, Gallus gallus; xl, Xenopus laevis; dr, Danio rerio; dm, Drosophila melanogaster. | ||
Loss of function or overexpression of APL-1 results in larval lethality and suggests a critical role in development for the apl-1 gene. This lethality can be rescued by neuronal expression of the extracellular domain of APL-1, highlighting the significance of this region for APL-1 function.22,27 Like mammalian APP and drosophila APPL, APL-1 is cleaved proteolytically by a secretase enzyme to release soluble extracellular APL-1. In contrast to the presence of multiple proteolysis sites in mammalian APP, only a single cleavage site exists in APL-1 and an equivalent of the copper-binding Aβ peptide is not formed.27
We have investigated the structure and function of C. elegans APL1-D2, the region homologous to APP-D2, the copper-binding domain of human APP. The solution structure of the APL1-D2 was solved and its copper binding properties investigated. APL1-D2 is closely related in structure to APP-D2. However, its intrinsic affinity for Cu is low and therefore of uncertain physiological relevance when compared to human APP. Cu–APP-D2 is less efficient than Cu–Aβ in catalysing aerobic oxidation of ascorbate (Asc) with production of H2O2. The lower affinity of APP-D2 for Cu(II) shifts its redox properties unfavourably with respect to H2O2 production so it is no longer catalytically efficient. On the other hand, APL1-D2 has only a marginal effect on the catalysis of either free Cu2+ or the Cu–Aβ complex, consistent with its weak affinity for both Cu(I) and Cu(II) and therefore its inability to compete for metal ions with Aβ. The conclusion from these studies is that APL1-D2 is not an effective copper trafficking domain and would not play a direct role in C. elegans copper homeostasis.
Experimental procedures
Materials and general methods
Ligand Ferene S (as its sodium salt Na2Fs), reductants NH2OH (as its H2SO4 salt) and ascorbic acid, copper standard (as atomic absorption standard solution), Amplex Red (also called Ampliflu Red) and horseradish peroxidase (HRP) were all purchased from Sigma and were used as received. Peptides Aβ16 (sequence: DAEFRHDSGYEVHHQK) and Ac-Aβ16 (sequence: Ac-DAEFRHDSGYEVHHQK) spanning the Cu binding site of the Aβ peptide were purchased from GL Biochem (Shanghai). Determination of Cu(I) binding affinities were conducted anaerobically in deoxygenated buffers containing reductants NH2OH or Asc in a glovebox ([O2] < 1 ppm). The samples were transferred in sealed containers for characterization.Protein expression and purification
Recombinant secreted APL-1 domain equivalent to APP-D2, APL1-D2 (Uniprot Accession: Q10651) residues 133–197, was expressed in Pichia pastoris. The expression plasmid was constructed and transformed into the P. pastoris strain GS115 as previously described.28 Expression of APL1-D2 produced a protein with two N-terminal residues (Glu, Ala) identical to those of APP-D2 that was obtained from the same expression vector. The identity of the isolated protein was confirmed using silver stain SDS-PAGE analysis and mass spectrometry. Uniformly labeled proteins (15N single labeled or 15N- and 13C-double labeled) were expressed using previously described methods.2915NH4Cl, and 13C glucose or 13C methanol were used as the isotope sources. The APL1-D2 protein was purified to homogeneity using a Superdex 75 10/300 size exclusion chromatography column (GE Healthsciences) followed by a C8 reverse phase column (Grace) with 0.1% TFA in acetonitrile as the mobile phase. Purified labeled proteins were confirmed using mass spectrometry and SDS-PAGE analysis. Circular dichroism (CD) spectroscopy (Jasco) confirmed that the protein was folded. Recombinant APP-D2 was expressed and purified as described previously.18 After purification, APP-D2 and the equivalent APL-1 proteins were lyophilized and stored at −20 °C prior to use. Recombinant human APP-D2 (Uniprot P05067, residues 133–189) with two additional N-terminal vector-derived residues (Glu, Ala) was prepared in a similar manner to APL1-D2.28Sequence alignment and analysis
Protein sequences were found through a BLAST30 search of the NCBI sequence database and aligned using Clustal-W31 in MEGA5.32 Sequence accession codes (Uniprot) and sequence positions for D2 are: Caenorhabditis elegans, Q10651 (133–197); Homo sapiens A4, P05067 (131–189); APLP2, Q06481 (147–205); Mus musculus APLP2, Q06335 (147–205); Gallus gallus APLP2, F1P0A9 (120–178); Xenopus laevis APLP1, Q52KN8 (130–188); Danio rerio APPA, Q919E7 (64–122); Drosophila melanogaster APPL, P14599 (141–199). The sequence numbers of the D2 domain are given in parentheses.NMR spectroscopy
NMR spectra were acquired at 30 °C on a Bruker-Biospin Avance 800 MHz spectrometer equipped with a cryogenically cooled triple-resonance pulsed field gradient probe. Sequential resonance assignments were made using a series of triple-resonance spectra acquired on either the double labeled 13C, 15N-enriched or the single labeled 15N-APL-1 domain. Spectra were obtained on samples that were typically 0.5 mM protein in 10 mM phosphate buffer at pH 6.9 in 1H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2H2O (9:1). Spectra were processed with TopSpin (Bruker BioSpin) and analyzed using XEASY.33 Backbone and side chain resonance assignment was completed using standard multidimensional heteronuclear NMR techniques. Experiments performed on uniformly 13C, 15N-labelled APL1-D2 included; HNCA, HNCO, HCCH-TOCSY, HNCACB, CBCA(CO)NH, HBHACONH, CCCONH-TOCSY, HCCH-TOCSY, CBCACO, 15N NOESY-HSQC, 13C NOESY-HSQC.34 Apart from the amide resonance of G161, a complete set of backbone resonance assignments was determined and the only side chain resonances not assigned were those of H145 where resonances of the imidazole ring were not observed. Assignments have been deposited in BioMagResBank with accession number 18794.
:2H2O (9:1). Spectra were processed with TopSpin (Bruker BioSpin) and analyzed using XEASY.33 Backbone and side chain resonance assignment was completed using standard multidimensional heteronuclear NMR techniques. Experiments performed on uniformly 13C, 15N-labelled APL1-D2 included; HNCA, HNCO, HCCH-TOCSY, HNCACB, CBCA(CO)NH, HBHACONH, CCCONH-TOCSY, HCCH-TOCSY, CBCACO, 15N NOESY-HSQC, 13C NOESY-HSQC.34 Apart from the amide resonance of G161, a complete set of backbone resonance assignments was determined and the only side chain resonances not assigned were those of H145 where resonances of the imidazole ring were not observed. Assignments have been deposited in BioMagResBank with accession number 18794.
Structure constraints and calculation
Approximate inter-proton distances were derived from 13C-and 15N-edited 3D-NOESY and 2D NOESY spectra with 150 ms and 75 ms mixing times recorded on unlabelled protein. Peak intensities were integrated in XEASY and converted into upper distance bounds. ϕ and ψ backbone torsion angles were derived using TALOS.35 Hydrogen bond restraints were employed at a late stage in the calculation based on the calculated structures. Initial structures were generated in CYANA36 and several rounds of structure calculation were performed to resolve violated distance constraints and determine possible assignments for ambiguous NOE cross-peaks. The structure was calculated with and without the presence of disulfide connections to check the pairings of the three disulfide bonds were correct. Once the final set of restraints had been obtained, a new family of structures was generated and refined using XPLOR-NIH using the disulfide bonds, hydrogen bonding and Ramachandran terms37 The final 20 structures were selected on the basis of their stereochemical energies having no distance violations >0.2 Å and angle violations >5°. Structures were analyzed using MOLMOL38 and PROCHECK_NMR.39 Surface pockets were determined using LigSite40 and POCASA.41 The constraint and structure statistics are summarized in Table S1 (ESI§). Structure figures were generated using MOLMOL. The coordinates of the final 20 structures and the experimental constraints have been deposited with the protein data bank (accession code 2M05).Quantification of Cu(II) binding
To minimise potential interference of binding of Cu(II) by ligands such as buffers KPi and Tris-HCl, purified protein samples were exchanged into Mops buffer (50 mM, pH 7.3) that exhibits a minimal affinity for Cu(II).42 Protein concentrations were estimated from solution spectra using molar absorbance at 280 nm calculated from the amino acid compositions (ε280 = 7360 M−1 cm−1 for both APL1-D2 and APP-D2). Two experimental approaches were used to define Cu(II) stoichiometry and binding: direct metal ion titration to determine Cu(II) stoichiometry and ligand competition to define Cu(II) binding affinity.Titration of Cu2+ into a solution of protein P may induce reaction 1 where the fraction of Cu(II) bound to protein P (i.e., [CuII–P]/[Cu(II)]tot) may be described by eqn (2):
| Cuaq2+ + P + L ⇌ CuII–P + ‘CuII–L’ | (1) |
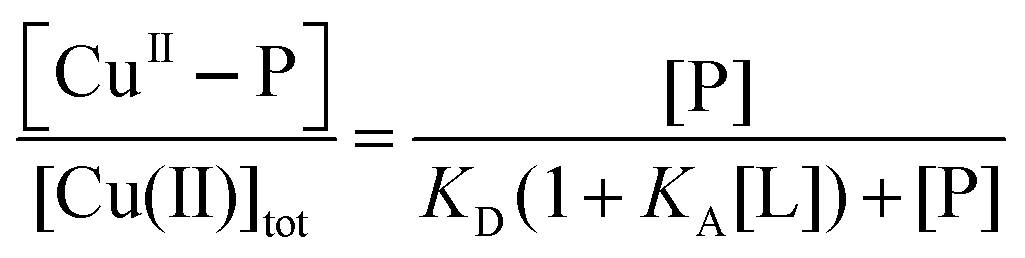 | (2) |
APP-D2 and APL1-D2 contain single Trp and Tyr residues that fluoresce when excited at ∼280 nm: the former emits intensively at λmax ∼ 330 nm and the latter more weakly at λmax ∼ 310 nm. Binding of paramagnetic Cu(II) quenches the fluorescence intensity permitting binding events to be monitored and analysed according to eqn (1) and (2). To avoid significant dilution, titration of aqueous CuSO4 at concentrations of 200, 200 and 2000 μM was performed by addition of small aliquots to apo-protein at respective concentrations of 2.0, 20 and 200 μM in Mops buffer (50 mM; pH 7.3). For the binding analysis, the fluorescence intensity was normalised to that of the apo form.
Estimation of Cu(II) affinities by direct metal ion titration is often subject to large uncertainties associated with possible contributions from other weak Cu(II) binding ligands such the proton buffer (i.e., the ‘CuII–L’ and KA[L] terms in eqn (1) and (2)). To suppress such effects, the Cu(II)-binding ligand glycine (Gly), which binds Cu2+ to yield 1:1 and 1:2 complexes with formation constants KA1 = 8.5 × 105 M−1 and KA2 = 3.4 × 104 M−1 at pH 7.3,43,44 was used to buffer the Cu2+ concentrations according to eqn (3)–(5):
| Cuaq2+ + ‘CuII–L’ + [CuII(Gly)+] + [CuII(Gly)2] + P ⇌ CuII–P + L + 3Gly | (3) |
| [CuII]tot = [Cuaq2+] + [‘CuII–L’] + [CuII(Gly)+] + [CuII(Gly)2] + [CuII–P] | (4) |
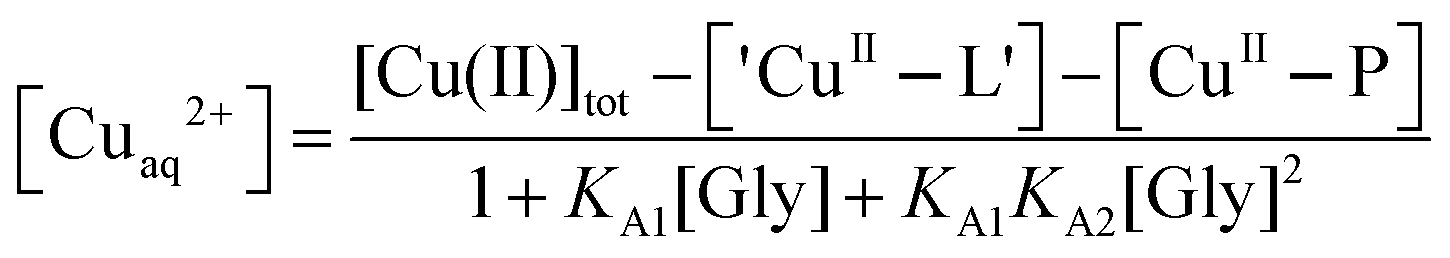 | (5) |
Eqn (5) is derived from the total Cu(II) speciation relationship of eqn (4) and allows, in principle, estimation of free [Cuaq2+] at various experimental points. In particular, at the point where a Cu(II) site is 50% occupied, [Cuaq2+] corresponds to the KD for that site. The term ‘CuII–L’ in eqn (3)–(5) is negligible in Mops buffer (50 mM, pH 7.3) for this system and may be ignored when the total Cu(II) content is distributed between competing ligands Gly and protein P.
Direct metal ion titrations suggested that APP-D2 and APL1-D2 at concentrations ≥20 μM bind two equivalents of Cu(II). Consequently, ligand competition experiments were conducted by preparation of a series of solutions containing identical total concentrations of Cu(II) (40 μM) and protein (20 μM) with increasing concentration of Gly (0–2.0 mM). Each solution was incubated at room temperature for at least 1 h to ensure reaction equilibrium and the fluorescence emission spectra for the set of solutions were recorded under identical conditions. Transfer of Cu(II) from CuII–P to Gly was monitored by recovery of fluorescence intensity and expressed quantitatively by ΔF/ΔFmax where F is the observed fluorescence, ΔF = F − Fmin and ΔFmax = Fmax − Fmin (Fmin and Fmax are the fluorescence intensities with 0 and 2.0 equivalents of Cu(II) being removed from the initial complex Cu2P, respectively).
When excited at 280 nm, APP-D2 emits a more intense fluorescence at 330 nm than does the Aβ16 peptide (single Tyr only). Consequently, it is possible to monitor the distribution of Cu(II) between Aβ16 and APP-D2 in competition experiments. Cu2+ (800 μM) was titrated into solution containing APP-D2 (20 μM) plus a Cu(II) competitor (Aβ16, Ac-Aβ16, EDTA; 40 μM) in Mops buffer (50 mM, pH 7.3).
Quantification of Cu(I) binding
This was conducted under anaerobic conditions ([O2] < 1 ppm) via competition for Cu(I) with probe ligand Ferene S (Fs) as detailed recently:45| CuI–P + 2Fs2− ⇌ [CuI(Fs)2]3− + P | (6) |
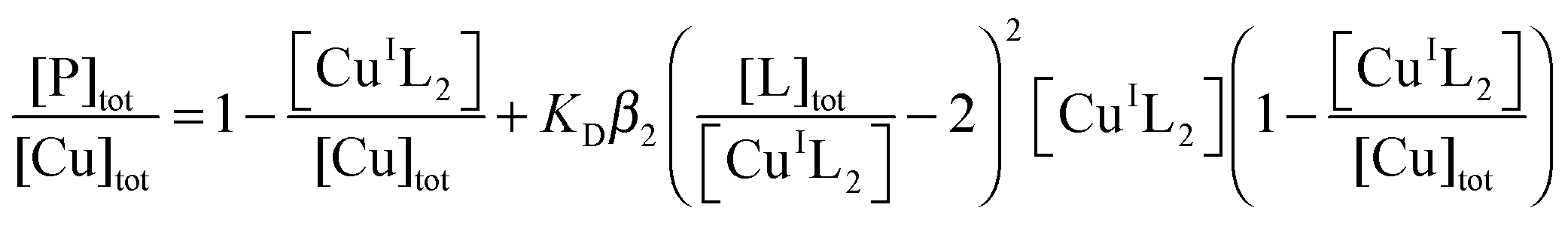 | (7) |
K
D is the dissociation constant of CuI–P. Anionic charges are omitted in eqn (7) for clarity. When in excess, Fs binds Cu(I) quantitatively to yield a well-defined 1:2 chromophoric complex anion [CuI(Fs)2]3− (ε484 = 6700 M−1 cm−1; β2 = 1013.7 M−2). Fs can probe Cu(I)-binding affinities in the range KD = 10−8.3–10−12.3 M, according to eqn (6) and (7).45 Concentrations of [CuI(Fs)2] may be estimated from A484 and KD determined by curve-fitting to eqn (7). Briefly, a series of de-oxygenated solutions were prepared containing increasing concentrations of protein but fixed concentrations of Fs and Cu(I) in Mops buffer (50 mM; pH 7.3) containing both reductants NH2OH (1.0 mM) and Asc (0.5 mM) (defined as buffer A hereafter). A suitable molar ratio of Cu to Fs was chosen to impose effective competition (eqn (6)). [CuI(Fs)2]3− was prepared by adding Cu2+ ions (supplied as CuSO4) into a solution of Fs in Mops buffer (50 mM, pH 7.3) containing NH2OH (1.0 mM). Most of the Cu2+ ions were rapidly trapped by excess Fs as [CuI(Fs)2]3−. Asc (final concentration 0.5 mM) was added to enforce complete reduction of Cu2+.45 The resultant [CuI(Fs)2]3− solution was aliquoted into a series of microtubes, followed by addition of increasing amounts of protein and dilution with Mops buffer to a fixed total volume. A second set of solutions with half the total concentration of Fs, Cu and P were prepared by 1:1 dilution with buffer A of each member of the first set of solutions. After incubation for 1–2 h under anaerobic conditions, spectra were recorded for each solution versus a buffer A background and the concentration of [CuI(Fs)2]3− calculated from its A484 value. The experimental data were fitted to eqn (7) to obtain Kex = KDβ2 and then KD from the known β2.45
Catalytic aerobic oxidation of ascorbate and generation of H2O2
Generation of H2O2 by catalytic aerobic oxidation of Asc was followed by UV-Vis spectroscopy using an Amplex Red assay.46 Consumption of Asc was monitored by decay of its absorbance at 265 nm. Production of H2O2 was monitored by formation of resorufin (λmax, 571 nm), the product of oxidation of Amplex Red by H2O2 in the presence of HRP. The Asc consumption rate was demonstrated to be ∼20% higher in the presence of the Amplex Red than that of the control in its absence. This is attributed to the accompanied extra oxidation of ascorbate by the Amplex Red radicals generated in situ in the former case.46 However, the rate of this side-reaction is proportional to the rate of the overall oxidation and has little impact on the quantitative evaluation of the relative rate of different catalysts in this work and thus is not considered. The solution conditions for a typical reaction were: [Asc]tot ∼ 80 μM, [Amplex Red]tot ∼ 100 μM; [HRP] ∼ 0.8 U mL−1; [Cu]tot = 10 μM (if added) and [ligand]tot = 15 μM. The ligands tested were APL1-D2, APP-D2, Aβ16, Ac-Aβ16 and EDTA. The reactions were started by addition of Asc into a solution containing all other components in air-saturated Mops buffer (20 mM, pH 7.2–7.3). Spectral changes were recorded at intervals of 50 s. A control solution without Asc served for baseline correction. The initial absorbance observed at 265 nm was sensitive to the reaction rate and solution composition. Use of Cuaq2+ as catalyst led to considerably lower initial absorbance than the other test solutions due to its high relative reaction rate. Other solutions differed marginally but were normalized to the initial spectrum to facilitate comparison.Results
Structure of APL1-D2
A protein spanning residues 133–197 of APL1-D2 corresponding to the CuBD of mammalian APP18 (Fig. 1) was expressed and purified in P. pastoris. Mass spectrometry confirmed the expected molar mass with six half-cystine residues involving in the formation of three intra-molecular disulfide bonds (expected 7411.4 Da; found 7411.7 Da), as expected for this extracellular domain.18 CD spectroscopy indicated that the purified protein was folded and featured some α-helix content (Fig. S1, ESI§). APL1-D2 provided a well-dispersed 1H, 15N HSQC solution spectrum (Fig. S2, ESI§) indicating that the protein was folded, corroborating the CD measurements. A near-complete set of chemical shift assignments was obtained by standard methods.34 Structures were calculated in CYANA and refined in XPLOR-NIH.36,37The resultant structures are well-ordered over the entire sequence and have good stereochemical properties with more than 98% of backbone angles falling in the allowed regions of the Ramachandran plot (Table S1, ESI§). As expected from the shared sequence identity with APP-D2 and related proteins, APL1-D2 consists of an α-helix (residues 149 to 162) packed against a triple stranded β-sheet with β-strands corresponding to residues 136–141 (β1), 172–178 (β2) and 189–195 (β3), respectively, forming a βαββ fold (Fig. 2A and B). Nuclear Overhauser effects (NOE) observed between the CiαH–CjβH and CiβH–CjβH of cysteines enabled the unambiguous assignment of the three disulfide pairings to C135–C195, C146–C182 and C160–C194, corresponding to the expected pairings for this domain (Fig. 1).18 The disulfide C135–C195 links β1 with β3, C160–C1194 connects the α-helix with β3 and C146–C182 fixes two loops at distal ends of the molecule (Fig. 2A and B). A search of the protein surface using either LigSite40 or Pocasa41 found 3 pockets, the most significant of which is formed between the C-terminus of α1 and the β-sheet and is encompassed by residues Q136, S138, C160, K163, M171 and C194 (Fig. 2).
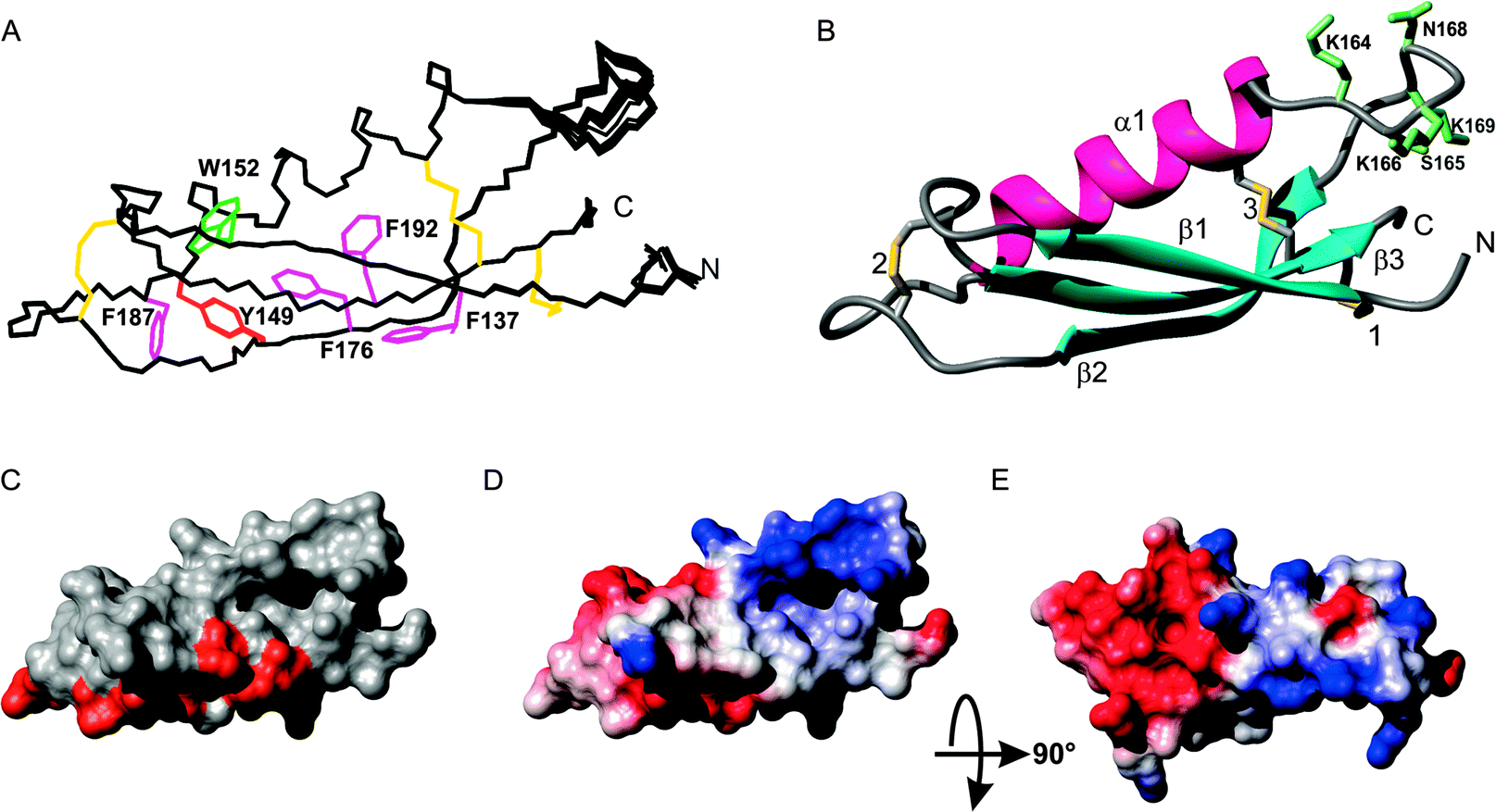 | ||
| Fig. 2 APL1-D2 structure. (A) Backbone (N, Cα, C) traces of the solution structure of C. elegans APl1-D2 showing the 20 lowest energy structures. Aromatic side chains are shown (Trp, green; Tyr, orange; Phe, pink) and the 3 disulfide bonds in yellow. (B) Ribbon diagram of the APL1-D2 structure closest to the mean in the same orientation as (A). Disulfide bonds are shown in yellow and side chains of residues in the α1–β2 loop are shown in pale green. The N- and C-termini are indicated and the disulfides indicated: 1, C135–C195; 2, C146–C182, 3 C160–C194. (C) Solvent accessible surface showing the conserved residues from alignment from worm APL-1 homologues in orange and the position of a pocket. (D) and (E) Electrostatic potential surface showing patches of basic (blue) and acidic (red) residues. (D) has the same orientation as in (A) while (E) has been rotated about the horizontal axis 90°. | ||
The C-terminal region of the βαββ motif of C. elegans APL1-D2 is highly conserved with related species (Fig. S3, ESI§). Plotting the conserved residues among the worm sequences for this domain onto the surface of APL1-D2 (Fig. 2C) shows they form a contiguous solvent accessible surface indicative of a potential protein binding site with a total exposed surface area of 750 Å2 located on strand β1 and the β2–β3 loop. The surface exposed (>25% accessible residue area) conserved residues in the C-terminal element encompassing the β2, β3 strands and intervening loop are E180 P181 and L184 and D185 and all reside in the β2–β3 loop, all the other residues are buried. A basic patch from residues at the C-terminal of the helix and the loop insert (residues K163, K164, K166) lies on one side of the cavity (Fig. 2D) while is an acidic patch (D144, D148, D154, E155, D185) resides on the opposite face of APL1-D2 (Fig. 2E).
The potential metal binding ligands of APP-D2 are not conserved in C. elegans APL1-D2 (Fig. 1). Furthermore, these residues are not conserved over all equivalent domains of APP ortholog sequences, suggesting that copper binding may not be the primary function or that differentiation in the function of this domain has occurred. The proposed metal ligands for human APP-D2 are H147, H151, Y168 and M170,18 and the equivalent residues in APL1-D2 (Y149, K153, F176, V178 respectively) are unlikely to form a metal binding site (Fig. 1 and 2). A further feature that differentiates the C. elegans APL1-D2 sequence from the mammalian APP-D2 is the presence of a six-residue insertion in the α1–β2 loop (Fig. 1). The addition of this loop has few structural consequences for the domain.
Cu(II) binding to APP-D2 and APL1-D2
Copper binding to APP-D2 and APL1-D2 may be conveniently monitored by fluorescence spectroscopy. Titration of Cu(II) into solutions of APP-D2 (Fig. 3b) or APL1-D2 (Fig. 3c) in Mops buffer quenched the fluorescence intensity progressively. Individual behaviour depended significantly upon protein concentration and turning points tended to be ill-defined in the concentration range 2–200 μM. These direct metal ion titration experiments allowed definition of the Cu(II) binding stoichiometry when [P]tot ≥ 20 μM and of the Cu(II) binding affinity with [P]tot = 2.0 μM. Detailed analysis (viaeqn (1) and (2); see Experimental procedures and ESI§) led to the conclusion that APP-D2 can bind two equivalents of Cu(II) at separated sites with comparable affinities in the range KD < 1.0 μM and that APL1-D2 binds two equivalents of Cu(II) but the affinity for at least one site is weaker (i.e., KD ≥ 1.0 μM).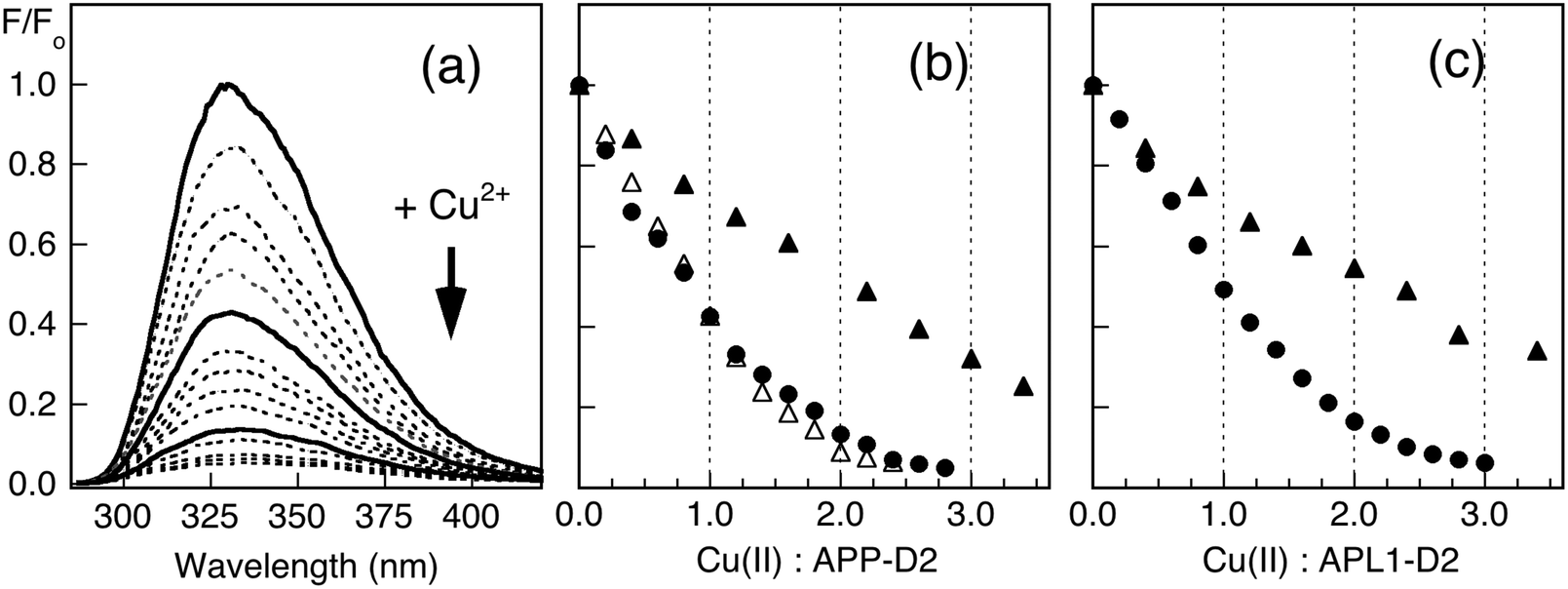 | ||
| Fig. 3 Quantification of Cu(II) binding by direct metal ion titration. (a) Change in fluorescence intensity of APP-D2 (20 μM) in Mops (50 mM, pH 7.3) upon titration with CuSO4 in H2O. The intensities were normalised to that at [Cu(II)] = 0 (i.e., F0) and the spectra after addition of 0, 1, 2 equiv. of Cu(II) are shown in solid lines. (b) Plots of F/F0 at ∼330 nm versus total molar ratio of Cu(II): APP-D2 for the titration of APP-D2 (2.0 μM, filled triangles; 20 μM, filled circles; 200 μM, empty triangles) with CuSO4. (c) Plots of F/F0 at 330 nm versus total molar ratio of Cu(II): APL1-D2 for the titrations of APL1-D2 (2.0 μM, triangles; 20 μM, circles) with CuSO4 in H2O. The concentration of CuSO4 in H2O used was 0.20 mM for titration of protein of concentration 2.0 and 20 μM and 2.0 mM for protein of concentration 200 μM. | ||
Addition of increasing amounts of Gly into a series of solutions containing equal concentrations of Cu2P (20 μM; P = APL1-D2 or APP-D2) recovered the protein fluorescence intensity partially (Fig. 4a), demonstrating that eqn (3) governs the metal ion equilibrium under the conditions. Detailed analysis (see Experimental procedures and ESI§) revealed that the experiments were not able to distinguish the difference in affinities of the two Cu(II) sites in these proteins under the conditions (Fig. 4b). The analysis provides an average Cu(II) KD value of 10−6.8 and 10−7.8 M for APL1-D2 and APP-D2, respectively, estimates consistent with the direct metal ion titration in Fig. 3.
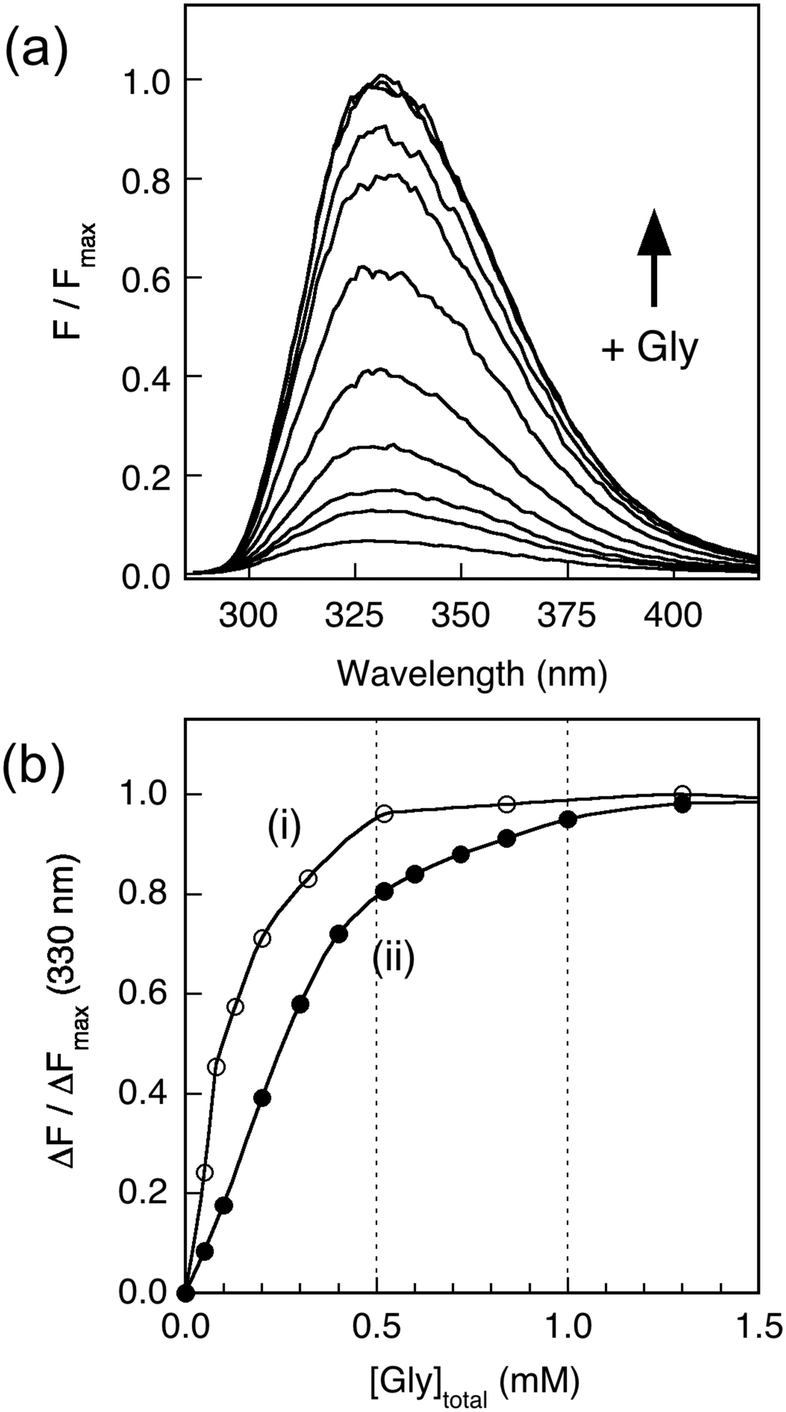 | ||
| Fig. 4 Estimation of Cu(II) affinity by ligand competition with Gly. (a) Change in fluorescence intensity of Cu2APP-D2 (20 μM) in Mops (50 mM, pH 7.3) with increasing concentration of glycine in solution. (b) Plots of ΔF/ΔFmax at 330 nm of Cu2P (20 μM; P = APP-D2 in filled circles; P = APL1-D2 in empty circles) versus total glycine concentration in solution. Each solution was prepared separately with different glycine concentrations and thus there is no dilution effect (see Experimental procedures for details). | ||
To better define the affinity of APP-D2 for Cu(II), an experiment was designed to directly compare its affinity with those of the two peptides Aβ16 and its N-terminally acetylated form Ac-Aβ16, that span the Cu binding site of highest affinity in amyloid-β (Aβ). The fluorescence intensity of APP-D2 at 330 nm is >50 times more intense than that of Aβ16 (Fig. 5a). Consequently, titration of Cu2+ into a solution containing both APP-D2 and Aβ16 allows monitoring of the Cu(II) distribution between the two competing molecules. The affinity of the APP-D2 domain was compared with those of Aβ16 (two sites with consensus values KD ∼ 10−10 and ∼10−8 M at pH 7.4),47–49 Ac-Aβ16 (single site with KD ∼ 10−8 M due to acetylated N-terminus)50 and EDTA (single high affinity site with KD = 10−15.8 M at pH 7.3).44 Detailed analysis of Fig. 5b (titrations i–iv; see ESI§) confirmed that APP-D2 features two binding sites of affinity KD ∼ 10−8 M, matching that (KD ∼ 10−7.8 M) estimated via competition with Gly (Fig. 4).
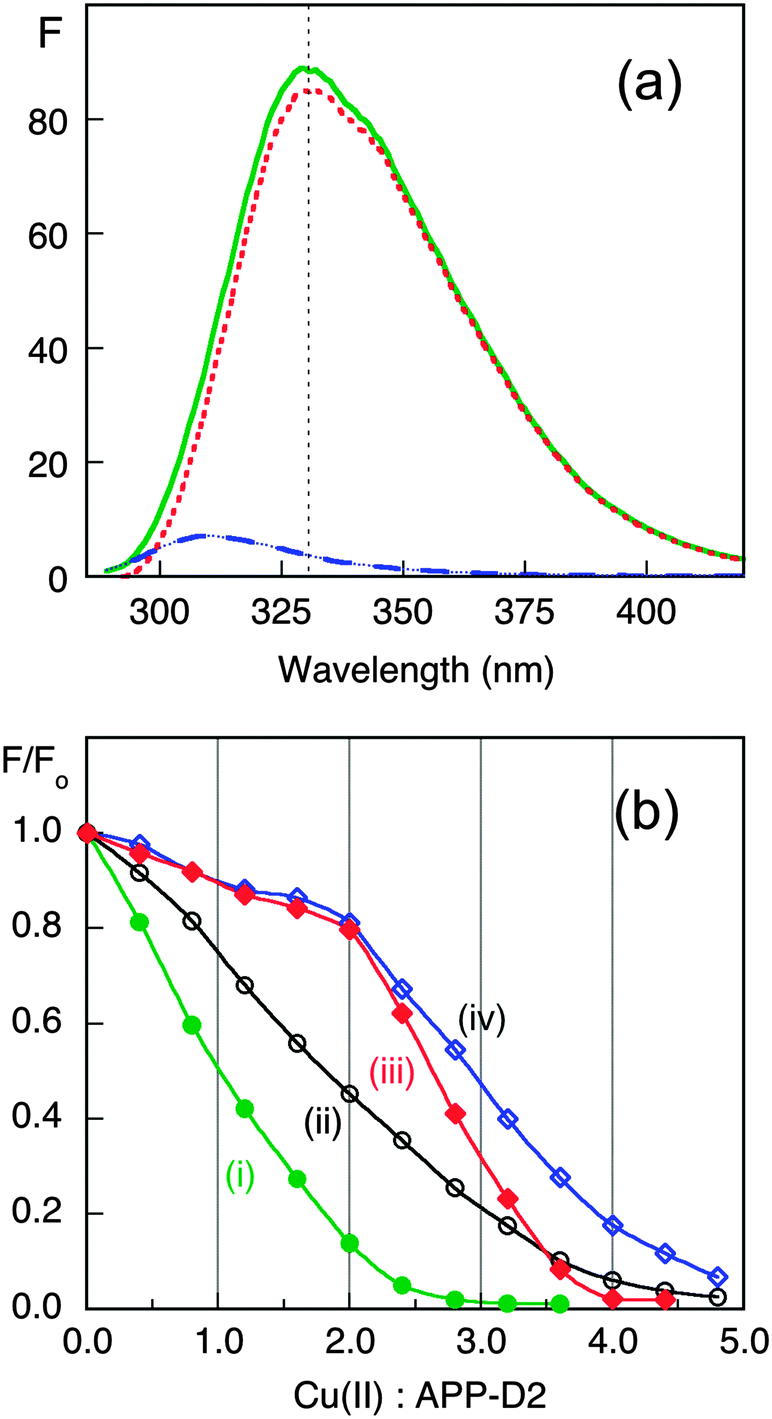 | ||
| Fig. 5 Estimation of Cu(II) affinity by ligand competition with Aβ16, Ac-Aβ16 and EDTA. (a) Fluorescence spectra in Mops buffer (50 mM, pH 7.3) of APP-D2 (20 μM; solid green trace), Aβ16 peptide (40 μM; dash blue trace) and their difference (dot red trace); (b) Plots of F/F0 at ∼330 nm versus total molar ratio of Cu(II): APP-D2 for the titrations with CuSO4 (800 μM): (i) APP-D2 (20 μM); (ii) a mixture of APP-D2 (20 μM) and Ac-Aβ16 (40 μM); (iii) a mixture of APP-D2 (20 μM) and EDTA (40 μM); (iv) a mixture of APP-D2 (20 μM) and Aβ16 (40 μM). Note that Cu(II) binding to EDTA and Aβ16 induces an inner filter effect that led to the minor declines in fluorescence intensity during the initial stages of titrations (iii) and (iv). | ||
Cu(I) binding to APP-D2 and APL1-D2
The Cu(I) binding ability of APP-D2 and APL1-D2 was investigated with Cu(I) ligand Ferene S (Fs). Fs may be used as a competitive Cu(I) ligand to probe proteins with Cu(I) affinities in the range KD = 10−8–10−12 M, according to eqn (6) and (7).45 Competition experiments using increasing amounts of APP-D2 induced a non-linear decrease in the absorbance of [CuI(Fs)2]3− (Fig. 6a, curve i) that documented a transfer of Cu(I) from the probe complex to the protein. A simple dilution of each solution caused further transfer of Cu(I) to APP-D2 (curve ii (solid trace) vs. prediction for simple dilution (dot trace)). These observations demonstrated that an effective competition described by eqn (6) was occurring under the experimental conditions. Fitting of each data set (i) and (ii) to eqn (7) (based on a single binding site model) provided the same estimate of the Cu(I) affinity as KD = 10−10.1 M.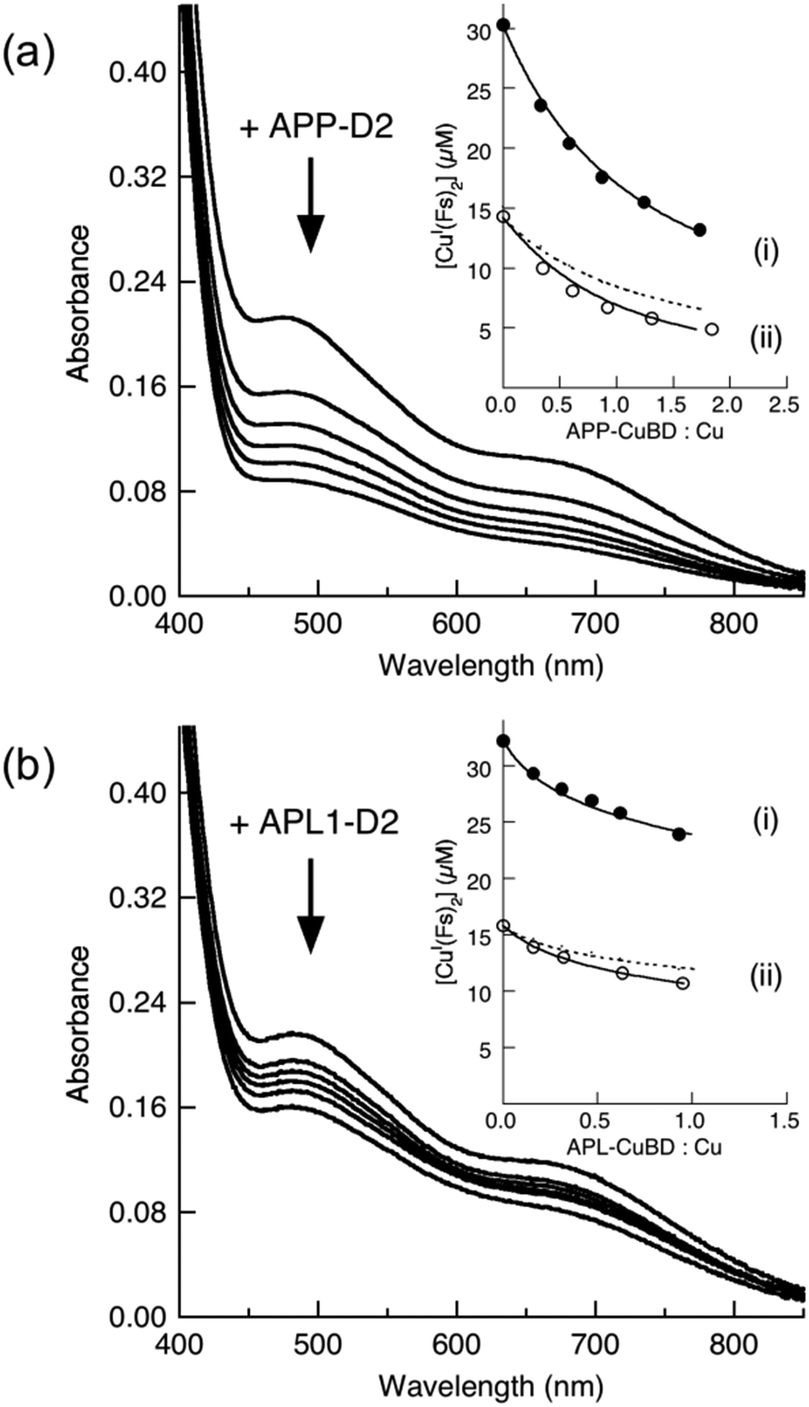 | ||
| Fig. 6 Estimation of Cu(I) binding affinity using the probe [CuI(Fs)2]3−. (a) Change in spectrum of [CuI(Fs)2]3− (prepared in situ from a mixture of CuSO4 (30 μM) and Fs (120 μM)) with increasing concentration of APP-D2; (b) change in spectrum of [CuI(Fs)2]3− (prepared in situ from a mixture of CuSO4 (32 μM) and Fs (70 μM)) with increasing concentration of APL1-D2. Shown in inset are the experimental data points for the originally prepared solutions (in solid circles) and for solutions obtained by a 1:1 dilution (in empty circles). The solid traces are the fitting curves of the experimental data to eqn (7), which yield Cu(I) dissociation constant KD = 10−10.1 M for APP-D2 in (a) and KD ∼ 10−8.6 M for APL-D2 in (b). The dash traces are the simple 1:1 dilution of data set (i). Experiments were performed in Mops buffer (50 mM, pH 7.0) containing NH2OH (2.0 mM) and Asc (1.0 mM) as reductants. | ||
A probe solution with a minimum molar ratio of Fs/Cu ∼ 2.3 (to ensure the dominance of [CuI(Fs)2]3−) was needed to observe transfer of Cu(I) to APL1-D2 (Fig. 6b). Curve-fitting to eqn (7) of the two data sets provided a KD ∼ 10−8.6 M. However, this value should be treated as an approximate value only, since it is at the detection limit for the Fs probe.45
Catalytic aerobic oxidation of ascorbate and generation of H2O2
The possibility that the Cu–APP-D2 complex can catalyse aerobic oxidation of Asc and generation of H2O2 was tested and compared with two Cu–Aβ peptide complexes and ‘free Cu2+ ion’ under the same conditions. The reaction was followed by UV-visible spectroscopy (Fig. 7a).46 The metal-free proteins and peptides (APL1-D2, APP-D2, Aβ peptides) were catalytically inactive. Aerobic oxidation of Asc was catalyzed efficiently by ‘free Cu2+ ion’ with a half-life of ∼2.5 min under the conditions. This activity was reduced three-fold in the presence of the Aβ16 peptide and about 18-fold in the presence of APP-D2 or Ac-Aβ16 (compare Fig. 7b and c, i–iv). The activity in the presence of APP-D2 or Ac-Aβ16 was about the same (Fig. 7b and c, iii and iv).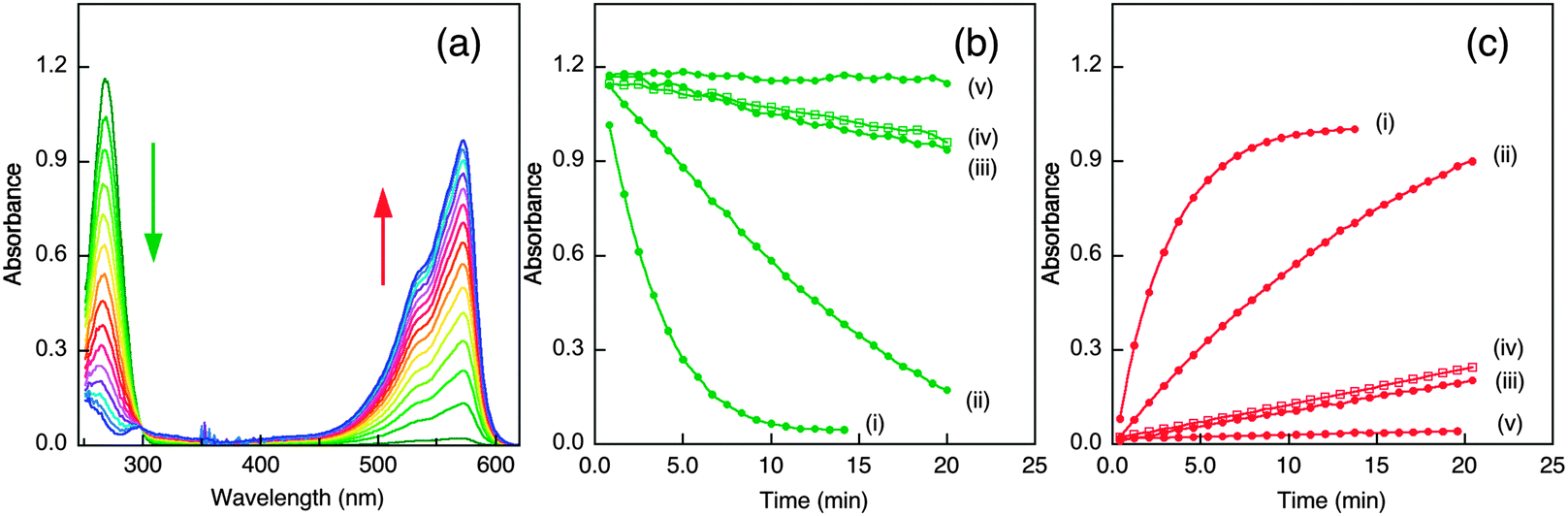 | ||
| Fig. 7 Catalytic aerobic oxidation of Asc and production of H2O2. (a) UV-Vis monitoring of Asc consumption and resorufin formation (thus H2O2 production; the spectrum of initial solution containing all components except Asc was subtracted from each recorded spectra); (b, c) Kinetic curves for Asc consumption (b) and resorufin formation (c) with following components as catalyst: (i) free Cu2+ (10 μM); (ii) Cu2+ (10 μM) plus Aβ16 (15 μM); (iii) Cu2+ (10 μM) plus APP-D2 (15 μM); (iv) Cu2+ (10 μM) plus Ac-Aβ16 (15 μM); (v) Aβ16 or APP-D2 without Cu (15 μM) or Cu2+ (10 μM) plus EDTA (15 μM). Other initial solution conditions: [Asc] = 80 μM, [Amplex Red] ∼ 100 μM; [HRP] ∼ 0.8 U mL−1. The reactions were conducted in air-saturated Mops buffer (20 mM, pH 7.2–7.3), started by introduction of catalyst and monitored by change in solution spectra. | ||
Addition of Cu2+ into an equimolar mixture of APP-D2 and Aβ16 produced an activity that was the average of controls containing APP-D2 or Aβ16 alone (Fig. S6b and c, iv–vi, ESI§). The copper ions appeared to be evenly distributed between the two competing molecules under the conditions, a result consistent with their similar affinities for Cu(I) that appeared to be the resting oxidation state of the metal ion in the presence of Asc. Consequently, the Cu(II) site located in the N-terminus of APP-D2 has little contribution to the catalysis. On the other hand, APL1-D2 had little effect on the catalytic activity of either ‘free Cu2+ ion’ or the Cu–Aβ16 complex (Fig. S6b and c; i–iv, ESI§), consistent with its weak binding affinity for either Cu(I) or Cu(II). The high affinity Cu(II) ligand EDTA (KD = 10−15.8 M at pH 7.3) quenched the catalytic activities of all copper complexes tested in this work completely (Fig. 7b and c; curve v). The catalytic activity is controlled by the metal coordination environment and is discussed below in more detail.
Discussion
The molecular structures of APP-D2 and APL1-D2 are similar
The βαββ structure determined for APL1-D2 is nearly identical to that of APP-D2 (the so-called APP copper binding domain, CuBD; Fig. 2), as expected for proteins with a shared sequence identity of 35.4% and similarity of 47.7%. A search of the protein database using secondary structure matching51 found that APP-D2 was the highest scoring match for the APL1-D2 domain, followed by proteins with related βαββ folds such as scorpion toxins. The extended α1–β2 loop of C. elegans APL1-D2 is conserved among the worm sequences but not in other species including Drosophila that also bear a single APP-like gene only (Fig. S3, ESI§). Although of undetermined function, the presence of surface-exposed lysines (Fig. 2) suggests a potential role in protein regulation through ubiquitylation pathways for these loop residues. The lack of conservation of any sets of residues in the APL1-D2 sequence and other worm sequences that could be considered as metal binding sites (Fig. 1) indicates that metal homeostasis is not the primary function that this domain plays in APL-1. This correlates with the absence of high affinity copper binding activity observed here for APL1-D2.Nature of Cu(I) and Cu(II) binding
Quantitative estimation of the relatively weak affinities of APL1-D2 and APP-D2 for Cu(II) was problematic due to possible contributions from other available weak Cu(II) binding sites such as those provided by buffer and other unidentified components present in experimental solutions (‘CuII–L’ in eqn (1)–(5)). Three separate approaches were applied: direct metal ion titration; competition with the Cu(II) buffer Gly; competition with the similar ligand Aβ16 of defined affinity (see Fig. 3–5 and ESI§). Each approach estimated that both APL1-D2 and APP-D2 bind two Cu(II) centres and that the average KD values for these centres are 10−7 and 10−8 M, respectively. The latter estimate for APP-D2 is about three orders of magnitude smaller (higher affinity for Cu(II)) than a previous estimate of KD ∼ 10−5 M.4Structural studies on APP-D2 have identified two Cu(II) binding sites. One features His147, His151, Tyr168 and possibly Met170 (HHY site).18 The other is poorly characterized but does involve the N-terminus. However, the latter site is an artefact of the protein construct used for structural studies and is not present at this position in full-length APP. An X-ray crystallographic analysis of crystals of apo-APP-D2 soaked in concentrated CuCl2 (100 mM) confirmed His147, His151 and Tyr168, but not Met170, as Cu(II) ligands (Fig. 8a, right).52 These findings are consistent with the present work that identifies two Cu(II) binding sites in APP-D2 with an average affinity KD ∼ 10−8 M. One of these may be assigned to the serendipitous site at the N-terminus and the second to the HHY site.
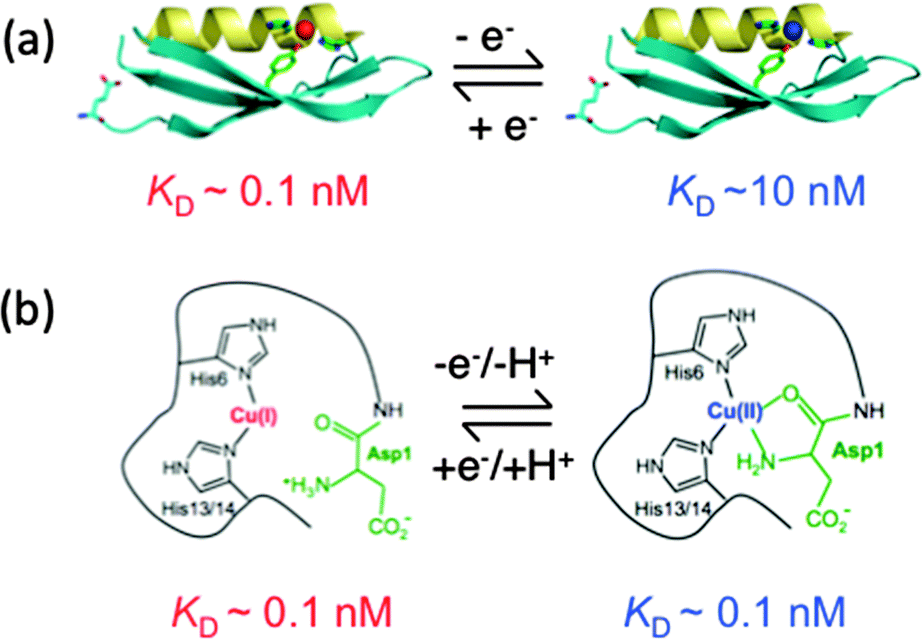 | ||
| Fig. 8 (a) Redox chemistry of the copper centre bound in APP-D2; (b) schematic of redox chemistry of the copper center bound in Aβ peptide. Red represents values for Cu(I) and blue those for Cu(II). | ||
Structural analysis has also detected Cu(I) binding to the HHY site (Fig. 8a, left)52 and the present work assigns a value of KD = 10−10.1 M for Cu(I) bound here (Fig. 6a). This affinity is about 100 times stronger than that of the same (or equivalent) site for Cu(II).
Comparison of the affinities for both Cu(I) and Cu(II) with APP-D2, APL1-D2 and the Aβ16 peptides corroborates the structural and competition equilibrium experiments. It has been proposed that only two of the three His side-chains available in Aβ16 are required to coordinate a single Cu(I) in a linear fashion (Fig. 8b, left).50,53–55 Such a binding mode suggests that the Cu(I) site in APP-D2 is likely to be dominated in solution by the side-chains of His147 and His151 and that an affinity for Cu(I) comparable to Aβ16 might be expected. Indeed, the present work confirms that the affinity of APP-D2 for Cu(I) (KD = 10−10.1 M at pH 7.0) is very close to that of Aβ16 (KD = 10−10.3 M), determined under identical conditions.45 Further support is provided by the observation that the catalytic activity of Cu for aerobic oxidation of Asc in a solution containing both APP-D2 and Aβ16 in a 1:1 molar ratio is about halfway between those of controls containing either APP-D2 or Aβ16 alone at the same individual concentrations (Fig. S6b and c, iv–vi, ESI§). It is apparent that, in the presence of reductant Asc, the resting copper forms are dominated by CuI–APP-D2 and CuI–Aβ16 and their KD values for Cu(I) are comparable.
In contrast, the APL1-D2 domain displays much weaker affinities: KD ∼ 10−8.6 M for Cu(I) (Fig. 6b) and KD ≥ 10−7 M for Cu(II) (Fig. 3 and 4). These values almost certainly reflect adventitious copper binding in APL1-D2 and are consistent with the absence of the equivalent metal binding site in its structure that is present in APP-D2 (Fig. 1 and 2). As a result, APL1-D2 has only minimum impact on copper redox chemistry of either ‘free Cu2+ ion’ or the Cu–Aβ16 complex (Fig. S6b and c; i–iv, ESI§). Consequently, APL1-D2 is unlikely to be involved in native copper handling.
The higher affinity Cu(II) site in Aβ16 at neutral pH involves two His side-chains and the N-terminal amine that collectively contribute to a square planar coordination site (Fig. 8b, right).56 However, other amino acid side-chains in APP-D2 appear to be unable to augment His147 and His151 to assemble such a site, nor is the N-terminus of APP-D2 able to contribute. This is the apparent source of the weaker affinity of APP-D2 for Cu(II) (KD ∼ 10−8 M) compared to the higher affinity site in Aβ16 (KD ∼ 10−10 M). In fact, it is comparable to that of the lower Cu(II) site in Aβ16 (KD ∼ 10−8 M; Fig. 5b, iv). As expected, blocking of the N-terminal amine of Aβ16 led to an affinity essentially identical to that of APP-D2 (Fig. 5b, ii).
Redox chemistry and implication for biological function
Ascorbic acid has a wide tissue distribution and is more concentrated in the CNS. It may function as a neuromodulator and/or neuroprotective agent in the brain.57,58 Its oxidation by dioxygen produces H2O2 that is liable to undergo further reduction via Fenton-type reactions to generate the hydroxyl radical ˙OH, and so lead to oxidative stress and inflammation.5Aerobic oxidation of ascorbate involves two redox half-reactions: the oxidation of ascorbic acid (AA) to dehydroascorbate (DA) and the reduction of O2 to H2O2 (Fig. 9). Their respective half reduction potentials at pH 7.0 are ∼+50 mV59 and ∼+300 mV60 and hence oxidation is a thermodynamically favored process. However, the reaction is very slow in healthy cells but, if copper homeostasis is perturbed, O2 reduction may be catalyzed by ‘free copper ions’ and certain Cu–protein/peptide complexes.61 The reduction potentials of favored catalysts must therefore fall within the range +50 and +300 mV. ‘Free Cu2+ ion’ is a robust catalyst for this reaction (Fig. 7b and c), consistent with its reduction potential of +164 mV,62 a value midway between the dehydroascorbate/ascorbate and O2–H2O2 couples (Fig. 9). Consequently, ‘free Cu2+ ion’ is highly toxic in an environment rich with Asc and dioxygen and so its concentration is tightly controlled in healthy cells.6
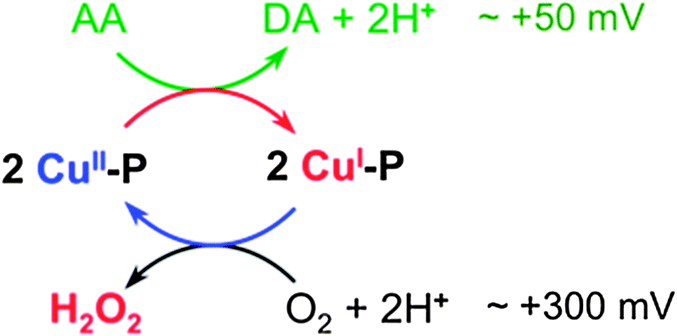 | ||
| Fig. 9 Scheme for catalytic aerobic oxidation of Asc and production of H2O2. | ||
The amyloid-β peptide Aβ possesses a flexible N-terminal amine that, together with the available His residues, is able to assemble a square planar Cu(II) site (Fig. 8b). Consequently, Aβ binds Cu(I) and Cu(II) with comparable affinities (KD ∼ 10−10 M).45,47,49 These affinities translate to a reduction potential of ∼170 mV for the CuII/CuI couple of the Aβ16 complex. This is close to that observed for ‘free Cu2+’, making Cu–Aβ16 an effective catalyst (Fig. 7b and c). The less efficient catalysis relative to ‘free copper ion’ probably arises from the significant structural modification and proton transfer necessary upon the change in oxidation state (Fig. 8b).
Aβ16 and APP-D2 sequester Cu(I) with essentially the same affinity. However, due to the absence of ligands able to provide a square planar binding site, the affinity of APP-D2 for Cu(II) is about 100 fold weaker (Fig. 8a). This imposes a positive shift in the reduction potential of the Cu–APP-D2 complex by ∼120 mV to ∼+300 mV, disfavoring its ability to reduce O2. This interpretation is consistent with the observation that the catalytic activity of ‘free copper’ ions for aerobic Asc oxidation are suppressed by ∼95% upon addition of APP-D2 (Fig. 7). The same argument applies to Ac-Aβ16 peptide that exhibits catalytic activity much weaker than that of Aβ16 but similar to that of APP-D2 (Fig. 7).
As well as domain D2, other domains of APP feature adjacent His residues that would be expected to bind Cu(I) with significant affinity (KD ∼ 10−10 M). Such sites may be neuroprotective in the sense that ROS generation in the CNS by the Cu–Aβ catalyst could be suppressed partially by competitive Cu(I) binding with APP-D2 and Ac-Aβ. Notably the soluble APPα fragment generated via a cleavage by α-secretase contains the Cu(I) sites in both APP-D2 and Ac-Aβ, but not in Aβ peptides and thus could be effective in neuroprotection. On the other hand, APL1-D2 from C. elegans lacks effective copper binding sites and therefore its functional role cannot directly involve copper binding.
Acknowledgements
This work was funded by grants from the Australian Research Council and the National Health and Medical Research Council (NHMRC). RC and KJB are NHMRC Senior Research Fellows. NMR spectra were acquired at the Bio21 Institute NMR Facility, University of Melbourne. SLL is currently at Wellcome Trust Centre for Cell Biology, University of Edinburgh, Edinburgh, U.K.Notes and references
- W. Wasco, K. Bupp, M. Magendantz, J. F. Gusella, R. E. Tanzi and F. Solomon, Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid beta protein precursor, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 10758–10762 CrossRef CAS.
- H. H. Slunt, G. Thinakaran, C. Von Koch, A. C. Lo, R. E. Tanzi and S. S. Sisodia, Expression of a ubiquitous, cross-reactive homologue of the mouse beta-amyloid precursor protein (APP), J. Biol. Chem., 1994, 269, 2637–2644 CAS.
- T. A. Bayer, S. Schafer, A. Simons, A. Kemmling, T. Kamer, R. Tepest, A. Eckert, K. Schussel, O. Eikenberg, C. Sturchler-Pierrat, D. Abramowski, M. Staufenbiel and G. Multhaup, Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Abeta production in APP23 transgenic mice, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14187–14192 CrossRef CAS PubMed.
- L. Hesse, D. Beher, C. L. Masters and G. Multhaup, The beta A4 amyloid precursor protein binding to copper, FEBS Lett., 1994, 349, 109–116 CrossRef CAS PubMed.
- X. Zhu, B. Su, X. Wang, M. A. Smith and G. Perry, Causes of oxidative stress in Alzheimer disease, Cell. Mol. Life Sci., 2007, 64, 2202–2210 CrossRef CAS PubMed.
- G. Eskici and P. H. Axelsen, Copper and oxidative stress in the pathogenesis of Alzheimer's disease, Biochemistry, 2012, 51, 6289–6311 CrossRef CAS PubMed.
- E. J. Coulson, K. Paliga, K. Beyreuther and C. L. Masters, What the evolution of the amyloid protein precursor supergene family tells us about its function, Neurochem. Int., 2000, 36, 175–184 CrossRef CAS PubMed.
- H. C. Huang and Z. F. Jiang, Amyloid-beta protein precursor family members: a review from homology to biological function, J. Alzheimer's Dis., 2011, 26, 607–626 CAS.
- D. M. Walsh, A. M. Minogue, C. Sala Frigerio, J. V. Fadeeva, W. Wasco and D. J. Selkoe, The APP family of proteins: similarities and differences, Biochem. Soc. Trans., 2007, 35, 416–420 CrossRef CAS PubMed.
- C. Reinhard, S. S. Hebert and B. De Strooper, The amyloid-beta precursor protein: integrating structure with biological function, EMBO J., 2005, 24, 3996–4006 CrossRef CAS PubMed.
- E. Storey and R. Cappai, The amyloid precursor protein of Alzheimer's disease and the Abeta peptide, Neuropathol. Appl. Neurobiol., 1999, 25, 81–97 CrossRef CAS PubMed.
- A. R. White, R. Reyes, J. F. Mercer, J. Camakaris, H. Zheng, A. I. Bush, G. Multhaup, K. Beyreuther, C. L. Masters and R. Cappai, Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knockout mice, Brain Res., 1999, 842, 439–444 CrossRef CAS PubMed.
- C. J. Maynard, R. Cappai, I. Volitakis, R. A. Cherny, A. R. White, K. Beyreuther, C. L. Masters, A. I. Bush and Q. X. Li, Overexpression of Alzheimer's disease amyloid-beta opposes the age-dependent elevations of brain copper and iron, J. Biol. Chem., 2002, 277, 44670–44676 CrossRef CAS PubMed.
- S. A. Bellingham, G. D. Ciccotosto, B. E. Needham, L. R. Fodero, A. R. White, C. L. Masters, R. Cappai and J. Camakaris, Gene knockout of amyloid precursor protein and amyloid precursor-like protein-2 increases cellular copper levels in primary mouse cortical neurons and embryonic fibroblasts, J. Neurochem., 2004, 91, 423–428 CrossRef CAS PubMed.
- A. R. White, G. Multhaup, F. Maher, S. Bellingham, J. Camakaris, H. Zheng, A. I. Bush, K. Beyreuther, C. L. Masters and R. Cappai, The Alzheimer's disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures, J. Neurosci., 1999, 19, 9170–9179 CAS.
- A. R. White, G. Multhaup, D. Galatis, W. J. McKinstry, M. W. Parker, R. Pipkorn, K. Beyreuther, C. L. Masters and R. Cappai, Contrasting, species-dependent modulation of copper-mediated neurotoxicity by the Alzheimer's disease amyloid precursor protein, J. Neurosci., 2002, 22, 365–376 CAS.
- J. Rossjohn, R. Cappai, S. C. Feil, A. Henry, W. J. McKinstry, D. Galatis, L. Hesse, G. Multhaup, K. Beyreuther, C. L. Masters and M. W. Parker, Crystal structure of the N-terminal, growth factor-like domain of Alzheimer amyloid precursor protein, Nat. Struct. Biol., 1999, 6, 327–331 CrossRef CAS PubMed.
- K. J. Barnham, W. J. McKinstry, G. Multhaup, D. Galatis, C. J. Morton, C. C. Curtain, N. A. Williamson, A. R. White, M. G. Hinds, R. S. Norton, K. Beyreuther, C. L. Masters, M. W. Parker and R. Cappai, Structure of the Alzheimer's disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis, J. Biol. Chem., 2003, 278, 17401–17407 CrossRef CAS PubMed.
- S. O. Dahms, I. Konnig, D. Roeser, K. H. Guhrs, M. C. Mayer, D. Kaden, G. Multhaup and M. E. Than, Metal binding dictates conformation and function of the amyloid precursor protein (APP) E2 domain, J. Mol. Biol., 2012, 416, 438–452 CrossRef CAS PubMed.
- M. Wiese, A. Antebi and H. Zheng, Regulation of neuronal APL-1 expression by cholesterol starvation, PLoS One, 2012, 7, e32038 CAS.
- C. Y. Ewald and C. Li, Caenorhabditis elegans as a model organism to study APP function, Exp. Brain Res., 2012, 217, 397–411 CrossRef CAS PubMed.
- M. Wiese, A. Antebi and H. Zheng, Intracellular trafficking and synaptic function of APL-1 in Caenorhabditis elegans, PLoS One, 2010, 5, e12790 Search PubMed.
- C. Y. Ewald, D. A. Raps and C. Li, APL-1, the Alzheimer's Amyloid precursor protein in Caenorhabditis elegans, modulates multiple metabolic pathways throughout development, Genetics, 2012, 191, 493–507 CrossRef CAS PubMed.
- J. T. Hoopes, X. Liu, X. Xu, B. Demeler, E. Folta-Stogniew, C. Li and Y. Ha, Structural characterization of the E2 domain of APL-1, a Caenorhabditis elegans homolog of human amyloid precursor protein, and its heparin binding site, J. Biol. Chem., 2010, 285, 2165–2173 CrossRef CAS PubMed.
- Y. Xue, S. Lee and Y. Ha, Crystal structure of amyloid precursor-like protein 1 and heparin complex suggests a dual role of heparin in E2 dimerization, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16229–16234 CrossRef CAS PubMed.
- I. Daigle and C. Li, apl-1, a Caenorhabditis elegans gene encoding a protein related to the human beta-amyloid protein precursor, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 12045–12049 CrossRef CAS.
- A. Hornsten, J. Lieberthal, S. Fadia, R. Malins, L. Ha, X. Xu, I. Daigle, M. Markowitz, G. O'Connor, R. Plasterk and C. Li, APL-1, a Caenorhabditis elegans protein related to the human beta-amyloid precursor protein, is essential for viability, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1971–1976 CrossRef CAS PubMed.
- A. Henry, C. L. Masters, K. Beyreuther and R. Cappai, Expression of human amyloid precursor protein ectodomains in Pichia pastoris: analysis of culture conditions, purification, and characterization, Protein Expression Purif., 1997, 10, 283–291 CrossRef CAS PubMed.
- Y. Laroche, V. Storme, J. De Meutter, J. Messens and M. Lauwereys, High-level secretion and very efficient isotopic labeling of tick anticoagulant peptide (TAP) expressed in the methylotrophic yeast, Pichia pastoris, Biotechnology, 1994, 12, 1119–1124 CrossRef CAS PubMed.
- S. F. Altschul, T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller and D. J. Lipman, Gapped BLAST and PSI-BLAST: a new generation of protein database search programs, Nucleic Acids Res., 1997, 25, 3389–3402 CrossRef CAS PubMed.
- R. Chenna, H. Sugawara, T. Koike, R. Lopez, T. J. Gibson, D. G. Higgins and J. D. Thompson, Multiple sequence alignment with the Clustal series of programs, Nucleic Acids Res., 2003, 31, 3497–3500 CrossRef CAS PubMed.
- K. Tamura, D. Peterson, N. Peterson, G. Stecher, M. Nei and S. Kumar, MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods, Mol. Biol. Evol., 2011, 28, 2731–2739 CrossRef CAS PubMed.
- C. Bartels, T. H. Xia, M. Billeter, P. Guntert and K. Wuthrich, The program XEASY for computer-supported NMR spectral analysis of biological macromolecules, J. Biomol. NMR, 1995, 6, 1–10 CrossRef CAS PubMed.
- M. Sattler, J. Schleucher and C. Griesinger, Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 34, 93–158 CrossRef CAS.
- G. Cornilescu, F. Delaglio and A. Bax, Protein backbone angle restraints from searching a database for chemical shift and sequence homology, J. Biomol. NMR, 1999, 13, 289–302 CrossRef CAS PubMed.
- P. Guntert, Automated NMR structure calculation with CYANA, Methods Mol. Biol., 2004, 278, 353–378 CAS.
- C. D. Schwieters, J. J. Kuszewski, N. Tjandra and G. M. Clore, The Xplor-NIH NMR molecular structure determination package, J. Magn. Reson., 2003, 160, 65–73 CrossRef CAS PubMed.
- R. Koradi, M. Billeter and K. Wuthrich, MOLMOL: a program for display and analysis of macromolecular structures, J. Mol. Graphics, 1996, 14, 51–55 CrossRef CAS PubMed , 29–32.
- R. A. Laskowski, J. A. Rullmannn, M. W. MacArthur, R. Kaptein and J. M. Thornton, AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR, J. Biomol. NMR, 1996, 8, 477–486 CrossRef CAS PubMed.
- B. Huang and M. Schroeder, LIGSITEcsc: predicting ligand binding sites using the Connolly surface and degree of conservation, BMC Struct. Biol., 2006, 6, 19 CrossRef PubMed.
- J. Yu, Y. Zhou, I. Tanaka and M. Yao, Roll: a new algorithm for the detection of protein pockets and cavities with a rolling probe sphere, Bioinformatics, 2010, 26, 46–52 CrossRef CAS PubMed.
- K. Y. Djoko, L. X. Chong, A. G. Wedd and Z. Xiao, Reaction mechanisms of the multicopper oxidase CueO from Escherichia coli support its functional role as a cuprous oxidase, J. Am. Chem. Soc., 2010, 132, 2005–2015 CrossRef CAS PubMed.
- A. Albert and E. P. Serjeant, The Determination of Ionisation Constants-A Laboratory Manual, Cambridge University Press, London, 1984 Search PubMed.
- Z. Xiao and A. G. Wedd, The challenges of determining metal-protein affinities, Nat. Prod. Rep., 2010, 27, 768–789 RSC.
- Z. Xiao, L. Gottschlich, R. V. D. Meulen, S. R. Udagedara and A. G. Wedd, Evaluation of quantitative probes for weaker Cu(I) binding sites completes a set of four capable of detecting Cu(I) affinities from nanomolar to attomolar, Metallomics, 2013, 5, 501–513 RSC.
- J. V. Rodrigues and C. M. Gomes, Enhanced superoxide and hydrogen peroxide detection in biological assays, Free Radicals Biol. Med., 2010, 49, 61–66 CrossRef CAS PubMed.
- C. J. Sarell, C. D. Syme, S. E. Rigby and J. H. Viles, Copper(II) binding to amyloid-beta fibrils of Alzheimer's disease reveals a picomolar affinity: stoichiometry and coordination geometry are independent of Abeta oligomeric form, Biochemistry, 2009, 48, 4388–4402 CrossRef CAS PubMed.
- C. Sacco, R. A. Skowronsky, S. Gade, J. M. Kenney and A. M. Spuches, Calorimetric investigation of copper(II) binding to Abeta peptides: thermodynamics of coordination plasticity, JBIC, J. Biol. Inorg. Chem., 2012, 17, 531–541 CrossRef CAS PubMed.
- B. Alies, E. Renaglia, M. Rozga, W. Bal, P. Faller and C. Hureau, Cu(II) Affinity for the Alzheimer's Peptide: Tyrosine Fluorescence Studies Revisited, Anal. Chem., 2013, 85, 1501–1508 CrossRef CAS PubMed.
- T. R. Young, A. G. Wedd and Z. Xiao, Unpublished observations.
- E. Krissinel and K. Henrick, Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2256–2268 CrossRef CAS PubMed.
- G. K. Kong, J. J. Adams, H. H. Harris, J. F. Boas, C. C. Curtain, D. Galatis, C. L. Masters, K. J. Barnham, W. J. McKinstry, R. Cappai and M. W. Parker, Structural studies of the Alzheimer's amyloid precursor protein copper-binding domain reveal how it binds copper ions, J. Mol. Biol., 2007, 367, 148–161 CrossRef CAS PubMed.
- S. Furlan, C. Hureau, P. Faller and G. La Penna, Modeling the Cu+ binding in the 1-16 region of the amyloid-beta peptide involved in Alzheimer's disease, J. Phys. Chem. B, 2010, 114, 15119–15133 CrossRef CAS PubMed.
- S. Furlan, C. Hureau, P. Faller and G. La Penna, Modeling copper binding to the amyloid-beta peptide at different pH: toward a molecular mechanism for Cu reduction, J. Phys. Chem. B, 2012, 116, 11899–11910 CrossRef CAS PubMed.
- J. Shearer, P. E. Callan, T. Tran and V. A. Szalai, Cu K-edge X-ray absorption spectroscopy reveals differential copper coordination within amyloid-beta oligomers compared to amyloid-beta monomers, Chem. Commun., 2010, 46, 9137–9139 RSC.
- C. Hureau and P. Dorlet, Coordination of redox active metal ions to the amyloid precursor protein and to amyloid-β peptides involved in Alzheimer disease. Part 2: Dependence of Cu(II) binding sites with Aβ sequences, Coord. Chem. Rev., 2012, 256, 2175–2187 CrossRef CAS.
- R. A. Grünewald, Ascorbic acid in the brain, Brain Res. Rev., 1993, 18, 123–133 CrossRef.
- M. E. Rice, Ascorbate regulation and its neuroprotective role in the brain, Trends Neurosci., 2000, 23, 209–216 CrossRef CAS PubMed.
- B. E. Conway, Electrochemical Data, Greenwood Press, New York, 1969 Search PubMed.
- D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, Freeman, W.H., New York, 2004 Search PubMed.
- F. Zhou and G. L. Millhauser, The rich electrochemistry and redox reactions of the copper sites in the cellular prion protein, Coord. Chem. Rev., 2012, 256, 2285–2296 CrossRef CAS PubMed.
- C. J. Hawkins and D. D. Perrin, Oxidation–reduction potentials of metal complexes in water. Part II. Copper complexes with 2,9-dimethyl- and 2-chloro-1,10-phenanthroline, J. Chem. Soc., 1963, 2996–3002 RSC.
Footnotes |
| † This paper is part of a themed issue from 4th International Symposium on Metallomics 2013. |
| ‡ The atomic co-ordinates and structural constraints (code 2M05) have been deposited in the protein data bank (http://www.pdb.org) and chemical shifts deposited in biological magnetic resonance data bank (http://www.bmrb.wisc.edu) with code 18794. |
| § Electronic supplementary information (ESI) available: Analysis of Cu(II) binding to APP-D2 and APL1-D2; Fig. S1–S6; Table S1. See DOI: 10.1039/c3mt00258f |
| This journal is © The Royal Society of Chemistry 2014 |