Combination studies of platinum(II)-based metallointercalators with buthionine-S,R-sulfoximine, 3-bromopyruvate, cisplatin or carboplatin†
K. Benjamin
Garbutcheon-Singh
a,
Benjamin W. J.
Harper
a,
Simon
Myers
bc and
Janice R.
Aldrich-Wright
*a
aNanoscale Organisation and Dynamics Group, School of Science and Health, University of Western Sydney, Locked Bag, 1797, Penrith South DC, 2751, NSW, Australia. E-mail: j.aldrich-wright@uws.edu.au; Fax: +61 (02) 4620 3025; Tel: +61 (02) 4620 3218
bMolecular Medicine Research Group, School of Medicine, University of Western Sydney, Locked Bag 1797, Penrith South DC, NSW. 2751, Australia
cNeuro-cell Biology Lab, University of Western Sydney, Locked Bag 1797, Penrith South DC, NSW. 2751, Australia
First published on 26th September 2013
Abstract
With current chemotherapeutic treatment regimes often limited by adverse side effects, the synergistic combination of complexes with anticancer activity appears to offer a promising strategy for effective cancer treatment. This work investigates the anti-proliferative activity using a combination therapy approach where metallointercalators of the type [Pt(IL)(AL)]2+ (where IL is the intercalating ligand and AL is the ancillary ligand) are used in combination with currently approved anticancer drugs cisplatin and carboplatin and organic molecules buthionine-S,R-sulfoximine and 3-bromopyruvate. Synergistic relationships were observed, indicating a potential to decrease dose-dependent toxicity and improve therapeutic efficacy.
Introduction
The 1960s saw the introduction of combination chemotherapy for the treatment of cancer.1,2 If two or more drugs used in combination increase the biological activity to a greater extent than the treatment of each drug alone, the effect is considered to be synergistic. If a decrease in activity is observed then an antagonistic effect is observed3 whereas if no change is observed the combination produces an additive effect. Anticancer chemotherapy utilises this treatment strategy so much so that combination therapy has become common place. The practice, of using already approved chemotherapeutic drugs, is seen as a more time efficient methodology which builds on existing research. A number of platinum–organic drug combinations have already been reported.4 Many platinum complexes covalently bind to DNA forming adducts, while organic drugs tend to interact with signaling pathways, inhibiting protein synthesis and potentially affect DNA repair mechanisms.5 Combining platinum- and organic-based drugs to increased therapeutic efficacy over single drug treatments, that is to produce a synergistic effect, has the potential to aid in overcoming resistance by targeting different mechanisms of action.6 This therapeutic approach has significant implications for metallodrugs.Combination therapy could be applied to other classes of platinum(II) complexes, such as metallointercalators in combination with clinical anticancer compounds in order to improve cytotoxic activity. There have been many metallointercalators produced in previous studies which utilise an intercalating ligand and a vicinal diamine as the ancillary ligand.7 Compounds of this type include [(5,6-dimethyl-1,10-phenanthroline)(1S,2S-diaminocyclohexane)platinum(II)] dichloride (56MESS), [(5,6-dimethyl-1,10-phenanthroline)(1R,2R-diaminocyclohexane)platinum(II)] dichloride (56MERR) and [(5,6-dimethyl-1,10-phenanthroline)(1,2-diaminoethane)platinum(II)] dichloride (56MEEN). These metallointercalators have a range of activity against the L1210 cell line, with cytotoxicities for 56MESS,8 56MERR9 and 56MEEN as shown in Table 1.10
| Complex | t 1/2 (h) | IC50 (μM) |
|---|---|---|
| Cisplatin | 3.312 | 0.5 |
| 56MESS | 68 | 0.009 ± 0.002 |
| 56MERR | 31 | 0.46 ± 0.08 |
| 56MEEN | 20 | 1.50 ± 0.3 |
Combination chemotherapy can increase the efficacy of anticancer drugs like cisplatin through minimising problems of cellular resistance, reducing adverse side-effects and decreasing the dosage required to produce the same cytotoxic activity.11 This work endeavours to observe the effect metallointercalators in combination with cisplatin, carboplatin, 3-bromopyruvate (BrPy) or buthionine-S,R-sulfoximine (BSO) to determine the potential of these complexes for use in combination therapy.
Methods and materials
Reagents
Metallointercalators were synthesised by previously published methods.7 The ovarian carcinoma cell line (A2780) and its resistant cell line (A2780cisR) were obtained from the Peter MacCallum Cancer Centre Melbourne. The Madine Darbey canine kidney (MDCK) cell line was provided by Dr Simon Myers from the University of Western Sydney. Human ovarian carcinoma (A2780 and A2780cisR) cells were cultured with Roswell Park Memorial Institute (RPMI) medium supplemented with 10% fetal calf serum (FCS), L-glutamine and penicillin/streptomycin. MDCK cells were cultured in Dulbecco's Modified Eagle medium (DMEM) supplemented with 10% FCS, L-glutamine (20 mM) and penicillin (100 units mL−1)/streptomycin (100 μg mL−1). All cell lines were cultured in an atmosphere of 5% CO2 at 37 °C. Sulforhodamine B (SRB), cisplatin, carboplatin, BrPy, BSO, dimethyl sulfoxide (DMSO) trichloroacetic acid, acetic acid and Tris base were purchased from Sigma-Aldrich chemicals. DMEM, FCS, phosphate buffered saline (PBS), L-glutamine (200 mM), penicillin (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units mL−1), streptomycin (10000 μg mL−1) and 0.25% trypsin-EDTA were purchased from Gibco® Invitrogen™. PBS tablets were purchased from Astral scientific. Sodium chloride was purchased from Fronine.
000 units mL−1), streptomycin (10000 μg mL−1) and 0.25% trypsin-EDTA were purchased from Gibco® Invitrogen™. PBS tablets were purchased from Astral scientific. Sodium chloride was purchased from Fronine.
SRB cytotoxicity assay
The cytotoxicity was assessed by observing the protein content using the SRB assay. Cells were seeded at a concentration of 2 × 104 cells per well in 96 well plate and left to adhere for 24 h before treatment. Cells were incubated with drugs over a range of concentrations for 24 h. The metallointercalators, BrPy and BSO were dissolved in media to the desired concentration, while cisplatin and carboplatin were dissolved in a minimum volume of DMSO and then diluted with media due to their limited water solubility (DMSO final concentration less than 0.5%). After which the media was removed and trichloroacetic acid (100 μL, 12% w/v) was added and the cells were allowed to incubate at 4 °C for an hour. The trichloroacetic acid was then removed and the wells washed with water (3 × 200 μL). The cells were stained with SRB (0.4% w/v in 1% acetic acid, 100 μL) for 20 min followed by washing with acetic acid (1% v/v, 3 × 200 μL). To each well Tris base is then added (200 μL, 10 mM). The absorbance was measured on a Microplate reader 680 (Bio-Rad) at 490 nm, using 660 nm as a reference wavelength.13 Cytotoxicity studies were performed in three independent experiments with three duplicates.GSH depletion experiments were performed in MDCK and A2780cisR cell lines. Cells were pretreated with media containing BSO (400 μM for MDCK and 200 μM for A2780cisR) for 24 h. After this time period the medium containing BSO was removed and media containing the platinum complexes was applied to each well and allowed to incubate for 24 h. Then the growth inhibition of the complexes after 24 h was then determined using the SRB cytotoxicity assay.14 GraphPad Prism 5 was used for all statistical data analysis. For comparison of the differences between the groups, a two-tailed unpaired, students t-test was used. A p value of less than 0.05 was considered statistically significant.
Results and discussion
Experimental design for combination studies
Loewe and Muischnek were the first to introduce a quantitative method for evaluating drug combinations utilising the process of isobolographic analysis.15,16 This analysis allows for the determination of additivity as well as the degree of synergism or antagonism. The interaction of the drugs can be established by the relationship between the dose and the effect on cells. Eqn (1) is utilised to calculate the interaction of an infinite set of drug combinations by analysing the sum of the dose of n drugs that inhibits or kills a percentage of cells (Fig. 1).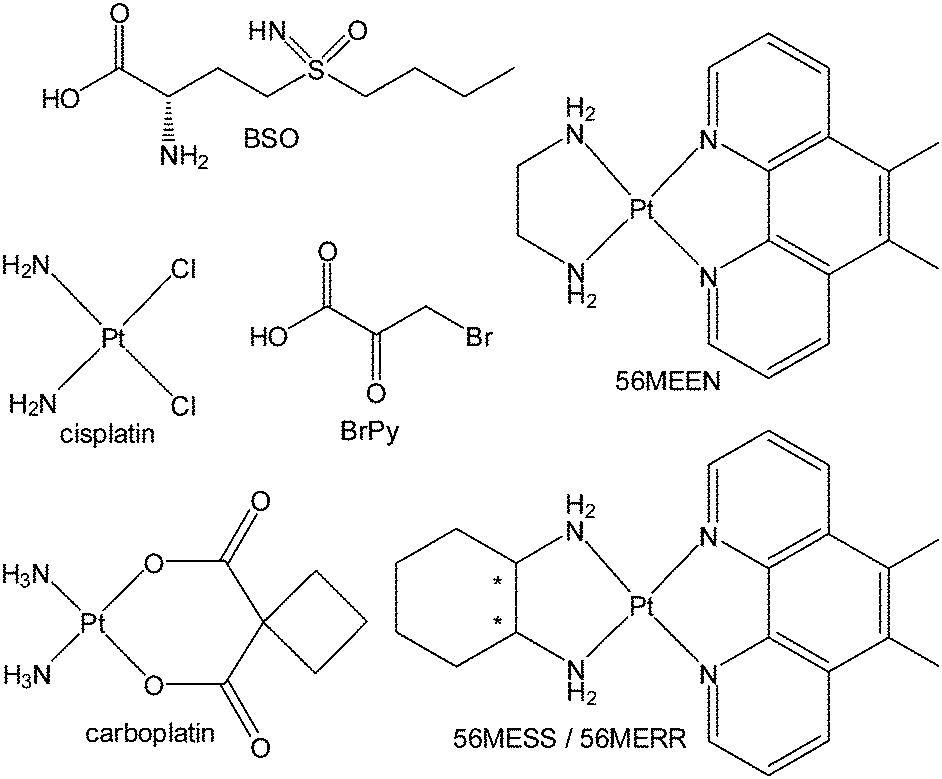 | ||
| Fig. 1 The chemical structure of compounds tested in combination: BSO; cisplatin; BrPy; 56MEEN; carboplatin; 56MESS and 56MERR. | ||
Where di is the concentration of individual drug that produced the IC50 values and Di is the concentration of the combination of drugs used to produce the IC50. From the line of zero interaction, synergism is interpreted as a greater than expected additive effect while antagonism is determined as a less than expected effect.
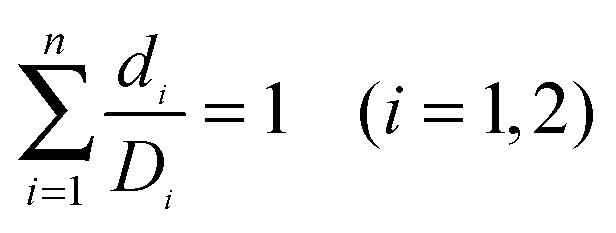 | (1) |
The Combination Index (CI) gives the line of zero interaction for two drugs. Zero interaction is represented by the concentration of the drugs used in combination that produce the same IC50 as they do individually. The CI is calculated using eqn (2) to determine the zero interaction line which represents the additive effect of two drugs.17
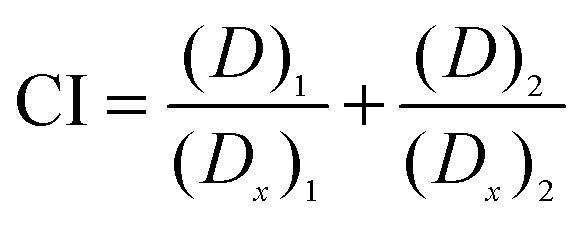 | (2) |
| CI values | Agonistic effect |
|---|---|
| <0.10 | Very strong synergism |
| ≈0.10–0.30 | Strong synergism |
| ≈0.30–0.70 | Synergism |
| ≈0.70–0.90 | Moderate synergism |
| ≈0.90–1.10 | Additive |
| ≈1.10–1.45 | Moderate antagonism |
| ≈1.45–3.30 | Antagonism |
| >3.30 | Strong antagonism |
There are many mathematical models that could be used for predicting and interpreting combination therapy data. The CI value was used here to evaluate the combinations in this preliminary investigation primarily due to its simplicity. Once we have established which combinations are synergistic we will be able to undertake further analysis with more robust methods in a wider range of cell lines. Isobolographic analysis was used to determine a range of cytotoxic properties for a combination of metallointercalators with cisplatin, carboplatin, BSO and BrPy, within this study.
With consideration that the IC50 values produced by compounds are proportional to the number of cells they are tested against, combination studies were undertaken as follows. For every microtiter plate that was used to conduct a combination study, both complexes were used on their own as well as in combination. This was considered important, as even small variations to cell number between experiments may produce significant differences in IC50 values. Cell numbers were determined by haemocytometer counting which allowed for a more accurate determination of the combined complexes cellular effects to be observed.
Combination of platinum anticancer complexes
SRB cytotoxicity assay was used to determine the agonistic affect (as described in Table 2) as a result of combination treatment between metallointercalators (56MESS, 56MERR or 56MEEN) with cisplatin or carboplatin. The results of this preliminary combination therapy are reported in Table 3. 56MESS produced a synergistic effect in all the cell lines tested, whether in combination with cisplatin or carboplatin. 56MERR and 56MEEN, which are less cytotoxic than 56MESS (Table 1), also produced a synergistic effect in most of the experiments.| Complex | Complex | Cell line | CI | Agonistic effect |
|---|---|---|---|---|
| a Denotes statistical significance set at p < 0.05. | ||||
| 56MESS | Cisplatin | MDCK | 0.65 ± 0.07 | Synergism |
| A2780 | 0.66 ± 0.12 | Synergism | ||
| A2780cisR | 0.28 ± 0.18a | Synergism | ||
| Carboplatin | MDCK | 0.59 ± 0.09 | Synergism | |
| A2780 | 0.85 ± 0.13 | Moderate synergism | ||
| A2780cisR | 0.36 ± 0.20a | Synergism | ||
| BrPy | MDCK | 0.39 ± 0.03a | Synergism | |
| A2780 | 0.85 ± 0.11 | Moderate synergism | ||
| A2780cisR | 0.30 ± 0.11 | Synergism | ||
| 56MERR | Cisplatin | MDCK | 0.87 ± 0.11 | Moderate synergism |
| A2780 | 0.74 ± 0.06 | Moderate synergism | ||
| A2780cisR | 0.39 ± 0.08a | Synergism | ||
| Carboplatin | MDCK | 1.24 ± 0.35 | Moderate antagonism | |
| A2780 | 0.87 ± 0.13 | Moderate synergism | ||
| A2780cisR | 0.69 ± 0.12 | Synergism | ||
| BrPy | MDCK | 1.13 ± 0.16 | Moderate antagonism | |
| A2780 | 0.49 ± 0.12 | Synergism | ||
| A2780cisR | 0.39 ± 0.08a | Synergism | ||
| 56MEEN | Cisplatin | MDCK | 0.84 ± 0.08 | Moderate synergism |
| A2780 | 0.64 ± 0.06 | Synergism | ||
| A2780cisR | 0.71 ± 0.04 | Synergism | ||
| Carboplatin | MDCK | 1.31 ± 0.23 | Moderate antagonism | |
| A2780 | 0.92 ± 0.07 | Additive | ||
| A2780cisR | 0.58 ± 0.12 | Synergism | ||
| BrPy | MDCK | 1.21 ± 0.08 | Moderate antagonism | |
| A2780 | 0.39 ± 0.15a | Synergism | ||
| A2780cisR | 0.97 ± 0.07 | Additive | ||
Metallointercalators in combination with cisplatin or carboplatin in the resistant cell line, A2780cisR, showed the greatest synergism. The use of these platinum complexes together may provide an approach to overcome acquired cellular resistance. The synergistic response produced by the less active metallointercalator 56MEEN, suggests that complexes with unremarkable individual cytotoxicities may also have potential in combination therapies and necessitates their investigation. Combinations of cisplatin and carboplatin together were not undertaken here as previous studies have shown that their combination is moderately synergistic in platinum sensitive cell lines.18,19
Signalling pathways and combination therapy
Many organic molecules have different effects on various signalling pathways within cells. Cyclin-dependant kinases, aerobic glycolysis, telomerase, vascular endothelial and epidermal growth factors are a few molecules and signalling pathways that currently under investigation as alternative targets for cancer therapy.21 Two pathways that will be investigated here are the glycolysis and glutathione metabolism pathways. Organic compounds BrPy and BSO influence glycolysis and glutathione metabolism respectively and were used in combination with metallointercalators 56MESS, 56MERR and 56MEEN in order to determine any changes in their cytotoxic effect. There are a number of physiological differences between normal and cancerous tissues. Understanding these differences, particularly in terms of cellular processes, can aid in the development of anticancer compounds with increased selectivity for cancer cells over typical somatic cells.22 The usual process for screening and selecting anticancer compounds tends to be based on the different rates of proliferation between cancerous and normal tissues. The tumour microenvironment is now being considered a distinguishing feature from normal tissues, with contributing characteristics like hypoxia, reduced growth rate and intrinsic drug resistance.23 This targeted cellular approach has potential, using combinational therapy, but a wider range of compounds that interact with DNA (both organic and inorganic), need to be investigated.Glycolysis inhibiting agents in combination with metallointercalators
Cancer cells are heavily reliant on glycolysis as their primary source of energy for the generation of ATP.24 To eliminate cancerous cells, a high level of ATP depletion is needed. This can be achieved by using ATP depleting agents that act on different enzymes in the glycolytic pathway in combination with clinical anticancer agents.25–27Numerous organic compounds that interact with different enzymes in the glycolysis pathway, such as BrPy are currently under investigation.22,26,27 BrPy has been shown to severely reduce intracellular ATP levels by inhibition of hexokinase in the glycolytic pathway.22 Here BrPy has been used in combination with 56MESS, 56MERR or 56MEEN against MDCK, A2780 or A2780cisR cell lines (Table 3).26,28 Whereas the combination of BrPy with 56MESS produced synergistic activity in both the MDCK and A2780cisR cell lines it was only moderately synergistic in the A2780 cell line. The combination of BrPy with 56MERR produced synergistic activity in both the ovarian carcinoma cell lines. The combination of BrPy with 56MEEN only produced a synergistic result in the A2780 cell line. These results demonstrated an overall positive synergistic outcome when metallointercalators are used in combination with BrPy. The results also reveal that complexes that may have been over looked in previous studies due to their unremarkable IC50 concentrations may be utilised more effectively in combinational treatments. This is exemplified by 56MEEN in the A2780 cell line where it demonstrates a far more synergistic CI value than 56MESS or 56MERR. Synergy was also produced with BrPy in combination with 56MESS or 56MERR in the resistant ovarian cell line A2780cisR.
Glutathione metabolism
Glutathione (GSH) is known to deactivate covalent DNA binding anticancer complexes therefore inhibiting their interactions with molecular targets.9 There are a number of compounds that are able to inhibit GSH synthesis. In this study BSO was used to inhibit GSH synthesis. BSO is an irreversible inhibitor of γ-glutamylcysteine synthetase responsible for GSH synthesis (Fig. 2).29,30 GSH is important within cells due to its high concentrations and ability to react readily with many exogenous compounds, such as cisplatin, causing them to become deactivated before they exert their toxic cellular effects.31 BSO has been shown to suppress intracellular glutathione in vitro, therefore increasing the cells sensitivity to cisplatin as well as reversing resistance to a number of other anticancer compounds.32 This therapeutic principle has also been shown to work inversely, whereby elevated glutathione production by the addition of N-acetyl cysteine increased cisplatin resistance.33 Degradation of metallointercalators by glutathione has been shown to be inversely proportional to cytotoxicity (Table 1).9 Therefore it is of interest to see if reducing the intracellular glutathione levels in cells produces a proportional increase in cytotoxicity for these complexes.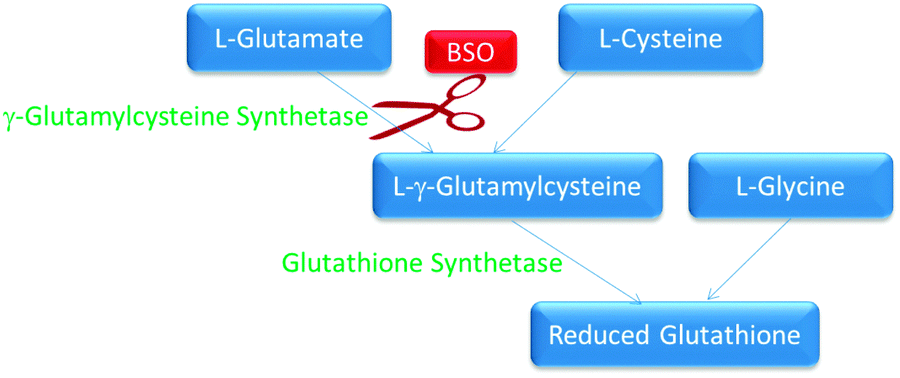 | ||
| Fig. 2 The biological pathway for the synthesis of glutathione. The enzymes are in green and the substrates are in blue. BSO inhibition is indicated in red. | ||
The mechanism by which glutathione is involved in the cytotoxicity of 56MESS is far more complex than the mere binding of 56MESS to glutathione which has already been described by other experiments.34 Studies have shown that 56MESS down regulated 45 genes in Saccharomyces cerevisiae which are involved in metabolic processes regarding amino acids and cellular stress response. Cellular respiration related to the biosynthesis of sulphur-containing amino acids (such as cysteine and methionine) and other amino acids like arginine are affected by 56MESS and may implicate mitochondrial processes in the mechanism of action.34
Previous combination studies have been conducted in A2780cisR cell lines with BSO and platinum(II) covalent binding complexes of bi- and tridentate pyrazolyl.14 This previous study utilised BSO to reduce intracellular GSH levels in order to overcome the acquired resistance observed in the A2780cisR cell line. This method was adapted and used to determine if similar results would be observed for 56MESS, 56MERR and 56MEEN. The effect of depleting GSH levels using BSO was investigated in the A2780cisR and MDCK cell lines, where the amount of BSO used to pre-treat each cell line was determined from cytotoxicity assays at the highest concentration for a 24 h period that resulted in no cell death. Metal complexes were added to cells, pre-treated with BSO, and not incubated on the cells together, as carried out in previous studies. Pre-incubation with BSO was intended to effectively deplete GSH concentrations and subsequently minimise platinum(II) drug deactivation.14 The results demonstrated that for all cells pre-treated with BSO, a decreased IC50 was observed for all complexes. The intrinsic resistance of A2780cisR cells was also decreased for all compounds (Table 4). The modulation factor is measured as a ratio of the IC50 values in the cells without pre-treatment to IC50 values in cells with BSO pre-treatment. The modulation factor has been used in previous studies to indicate the influence of reducing intracellular GSH levels on complex cytotoxicity. The cisplatin resistant cell line A2780cisR is known to have increased glutathione levels as well as decreased drug influx.35,36
| Cell line | Complex | IC50 without BSO pre-treatment | IC50 with BSO pre-treatment | Reduction in IC50 (% of concentration) | Modulation factora |
|---|---|---|---|---|---|
| a Modulation factor is defined as the ratio of the IC50 in the cell line: with and without BSO pre-treatment. b Error given as SEM. c 400 μM BSO pre-treatment. d 200 μM BSO pre-treatment. | |||||
| MDCKc | 56MESS | 0.25 ± 0.03b | 0.22 ± 0.03 | 11.5 | 1.13 ± 0.03 |
| 56MERR | 1.25 ± 0.14 | 0.74 ± 0.08 | 44.5 | 2.23 ± 0.23 | |
| 56MEEN | 2.50 ± 0.17 | 1.80 ± 0.11 | 28.0 | 1.46 ± 0.14 | |
| A2780cisRd | 56MESS | 0.22 ± 0.09 | 0.19 ± 0.04 | 25 | 1.33 ± 0.11 |
| 56MERR | 2.80 ± 0.13 | 2.30 ± 0.15 | 17 | 1.2 ± 0.09 | |
| 56MEEN | 3.50 ± 0.12 | 2.75 ± 0.11 | 31 | 1.4 ± 0.15 | |
As the experiment was conducted over 48 h, the greatest effect would be on those complexes that have short GSH half-lives. Compounds that react with GSH rapidly are more readily deactivated, rendering them unable to react with cellular components to produce an effect. 56MERR and 56MEEN have relatively short half-lives when exposed to GSH in compared to 56MESS, with half-lives shown to be 31, 20 and 68 hours respectively (Table 1).9 Pre-treatment with BSO influences the effectiveness of 56MERR and 56MEEN, producing the greatest reduction in IC50 compared to 56MESS in MDCK cells.
Cisplatin in combination with BSO has shown the greatest change in modulation factor in previous studies, with A2780cisR cells producing a range of 2.6–4.5.14,35 This significant improvement may be because the half-life of cisplatin is only 3.3 hours. In addition to the platinum bi- and tridentate pyrazolyl complexes, a series of BBR3464 derivatives were also studied using BSO pre-treatment; they produced a range of modulation factors between 1.5–2.9 and 1.6–1.9, respectively.14,35 Both of these studies showed that the more cytotoxic metal complex had the smallest reduction in IC50, perhaps due to their resistance to GSH degradation.
Conclusions
The combination study of platinum(II) metallointercalators with cisplatin or carboplatin demonstrated a reduced dose required to achieve the same cytotoxic effect. This result could mean that the nephrotoxicity observed in the preclinical trials for metallointercalators with syngeneic mice could be reduced.8 This study shows that the metallointercalators tested in combination with cisplatin or carboplatin exhibited synergy in most cases. Results indicate that less active metallointercalators may also be useful in combination with for example, cisplatin.The metallointercalators evaluated here (56MESS, 56MERR and 56MEEN) also showed that when used in combination with molecules that act on the glycolysis pathway, were able to produce a synergistic interaction therefore allowing dose reduction to produce the same cytotoxic effect. Reduction in GSH levels in cells by pre-treatment with BSO shows that the metallointercalators which have the shortest GSH half-life, are the most significantly affected and their IC50 values improved. This would mean that previously over looked complexes could be again explored as combination therapy agents.
The results warrant further investigation of metallointercalators with a wider range of anticancer compounds to illicit any synergistic combinations. This study also indicates that complexes that have low activity against cancer cell lines when administered individually can demonstrate increased activity when used in combination. The influence of the sequence of administration on effectiveness should also be investigated.
Acknowledgements
K. Benjamin Garbutcheon-Singh and Benjamin W. J. Harper were supported by an Australian Postgraduate Award from the University of Western Sydney. We wish to acknowledge UWS for funding through internal research grants and International Research Initiative Scheme as well as the School of Science and Health for support through a New Research Project Development Grant. Aspects of this research were supported by Australian Government, through the International Science Linkages program. Neville S. Ng is acknowledged for helpful discussion and editorial comments on this manuscript.References
- V. T. DeVita Jr., G. P. George, P. Canellos and J. H. Moxley, Cancer, 1972, 30, 1495–1504 CrossRef CAS.
- A. Goldin, J. M. Venditti, N. Mantel, I. Kline and M. Gang, Cancer Res., 1968, 28, 950–960 CAS.
- L. Zhao, M. G. Wientjes and J. L. S. Au, Clin. Cancer Res., 2004, 10, 7994–8004 CrossRef CAS PubMed.
- K. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef PubMed.
- T. Boulikas and M. Vougiouka, Oncol. Rep., 2003, 10, 1663–1682 CAS.
- F. Greco and M. J. Vicent, Adv. Drug Delivery Rev., 2009, 61, 1203–1213 CrossRef CAS PubMed.
- N. J. Wheate, R. Taleb, A. Krause-Heuer, R. Cook, S. Wang, V. Higgins and J. Aldrich-Wright, Dalton Trans., 2007, 5055–5064 RSC.
- D. M. Fisher, R. R. Fenton and J. R. Aldrich-Wright, Chem. Commun., 2008, 5613–5615 RSC.
- S. Kemp, N. Wheate, M. Pisani and J. Aldrich-Wright, J. Med. Chem., 2008, 51, 2787–2794 CrossRef CAS PubMed.
- C. R. Brodie, J. Collins and J. Aldrich-Wright, Dalton Trans., 2004, 1145–1152 RSC.
- J. C. Boik and R. A. Newman, BMC Pharmacol., 2008, 8, 1–12 Search PubMed.
- B. J. Corden, Inorg. Chim. Acta, 1987, 137, 125–130 CrossRef CAS.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
- C. Francisco, S. Gama, F. Mendes, F. Marques, I. Cordeiro dos Santos, A. Paulo, I. Santos, J. Coimbra, E. Gabanoc and M. Ravera, Dalton Trans., 2011, 40, 5781–5792 RSC.
- S. Loewe and H. Muischnek, Arch. Exp. Pathol. Pharmakol., 1926, 114, 313–326 CrossRef CAS.
- R. J. Tallarida, Pharmacol. Ther., 2007, 113, 197–209 CrossRef CAS PubMed.
- T.-C. Chou, Cancer Res., 2010, 70, 440–446 CrossRef CAS PubMed.
- R. P. Perez, K. M. Perez, L. M. Handel and T. C. Hamilton, Cancer Chemother. Pharmacol., 1992, 29, 430–434 CrossRef CAS PubMed.
- K. Kobayashi, S. Kudoh, T. Takemoto, M. Hino, K. Hayashihara, K. Nakahiro, M. Ando and H. Niitani, J. Cancer Res. Clin. Oncol., 1995, 121, 715–720 CrossRef CAS PubMed.
- C. P. Reynolds and B. J. Maurer, Chemosensitivity: Volume 1: In Vitro Assays, Humana Press, Totowa, NJ, vol. 110, 2005 Search PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- H. Pelicano, D. S. Martin, R. H. Xu and P. Huang, Oncogene, 2006, 25, 4633–4646 CrossRef CAS PubMed.
- G. Maschek, N. Savaraj, W. Priebe, P. Braunschweiger, K. Hamilton, G. F. Tidmarsh, L. R. De Young and T. J. Lampidis, Cancer Res., 2004, 64, 31–34 CrossRef CAS PubMed.
- O. Warburg, Science, 1956, 123, 309–314 CAS.
- D. S. Martin, D. Spriggs and J. A. Koutcher, Apoptosis, 2001, 6, 125–131 CrossRef CAS PubMed.
- P. E. Porporato, S. Dhup, R. K. Dadhich, T. Copetti and P. Sonveaux, Front. Pharmacol., 2011, 2, 1–18 Search PubMed.
- P. Dell'Antone, Med. Chem., 2009, 5, 491–496 CrossRef.
- S. Ganapathy-Kanniappan, M. Vali, R. Kunjithapatham, M. Buijs, L. H. Syed, P. P. Rao, S. Ota, B. K. Kwak, R. Loffroy and J. F. Geschwind, Curr. Pharm. Biotechnol., 2010, 11, 510–517 CAS.
- A. Meister, J. Biol. Chem., 1988, 263, 17205–17208 CAS.
- O. W. Griffith and A. Meister, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4668–4672 CrossRef CAS.
- A. Meister, J. Biol. Chem., 1988, 263, 17205–17208 CAS.
- C. P. Anderson, J. M. Tsai, W. E. Meek, R. M. Liu, Y. Tang, H. J. Forman and C. P. Reynolds, Exp. Cell Res., 1999, 246, 183–192 CrossRef CAS PubMed.
- C. M. Rudin, Z. Yang, L. M. Schumaker, D. J. VanderWeele, K. Newkirk, M. J. Egorin, E. G. Zuhowski and K. J. Cullen, Cancer Res., 2003, 63, 312–318 CAS.
- S. Wang, M. J. Wu, V. J. Higgins and J. R. Aldrich-Wright, Metallomics, 2012, 4, 950–959 RSC.
- B. A. J. Jansen, J. Brouwer and J. Reedijk, J. Inorg. Biochem., 2002, 89, 197–202 CrossRef CAS PubMed.
- J. Holford, P. J. Beale, F. E. Boxall, S. Y. Sharp and L. R. Kelland, Eur. J. Cancer, 2000, 36, 1984–1990 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3mt00191a |
| This journal is © The Royal Society of Chemistry 2014 |