 Open Access Article
Open Access ArticleZinc-selective inhibition of the promiscuous bacterial amide-hydrolase DapE: implications of metal heterogeneity for evolution and antibioticdrug design†‡
Narasimha Rao
Uda
a,
Grégory
Upert
a,
Gaetano
Angelici
a,
Stefan
Nicolet
a,
Tobias
Schmidt
b,
Torsten
Schwede
b and
Marc
Creus
*a
aUniversity of Basel, Department of Chemistry, Spitalstrasse 51, Basel 4056, Switzerland. E-mail: marc.creus@unibas.ch; Fax: +41 (0)61 267 10 05; Tel: +41 (0)61 267 10 63
bUniversity of Basel, Biozentrum, Klingelbergstrasse 50/70, Basel 4056, Switzerland
First published on 8th August 2013
Abstract
The development of resistance to virtually all current antibiotics makes the discovery of new antimicrobial compounds with novel protein targets an urgent challenge. The dapE-encoded N-succinyl-L,L-diaminopimelic acid desuccinylase (DapE) is an essential metallo-enzyme for growth and proliferation in many bacteria, acting in the desuccinylation of N-succinyl-L,L-diaminopimelic acid (SDAP) in a late stage of the anabolic pathway towards both lysine and a crucial building block of the peptidoglycan cell wall. L-Captopril, which has been shown to exhibit very promising inhibitory activity in vitro against DapE and has attractive drug-like properties, nevertheless does not target DapE in bacteria effectively. Here we show that L-captopril targets only the Zn2+-metallo-isoform of the enzyme, whereas the Mn2+-enzyme, which is also a physiologically relevant isoform in bacteria, is not inhibited. Our finding provides a rationale for the failure of this promising lead-compound to exhibit any significant antibiotic activity in bacteria and underlines the importance of addressing metallo-isoform heterogeneity in future drug design. Moreover, to our knowledge, this is the first example of metallo-isoform heterogeneity in vivo that provides an evolutionary advantage to bacteria upon drug-challenge.
Introduction
It has been estimated that about 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people die in Europe alone every year from an infection due to common antibiotic-resistant bacteria, causing an economic burden in excess of 1.5 billion euro per year.1 Similar alarming statistics are reported around the world, making the current inevitable rise of antibiotic-resistance to our limited subset of clinically useful drugs an issue of great concern.2 It is therefore crucially important and urgent that new protein targets are identified and that their specific inhibitors are developed.
000 people die in Europe alone every year from an infection due to common antibiotic-resistant bacteria, causing an economic burden in excess of 1.5 billion euro per year.1 Similar alarming statistics are reported around the world, making the current inevitable rise of antibiotic-resistance to our limited subset of clinically useful drugs an issue of great concern.2 It is therefore crucially important and urgent that new protein targets are identified and that their specific inhibitors are developed.
DapE, N-succinyl-L,L-diaminopimelic acid desuccinylase of the meso-diaminopimelate (mDAP)/lysine biosynthetic pathway of bacteria, has been identified as an attractive potential antibiotic target for two main reasons:3 (i) the growth of many bacteria is highly dependent on the biosynthesis of lysine and diaminopimelic (DAP) acid, which are essential for protein synthesis and elaboration of their peptidoglycan cell wall, respectively,4 and (ii) both DAP and lysine are synthesized by the dap operon bacterial enzymes and no similar pathways take place in humans.
L-Captopril was identified as a low-micromolar inhibitor of the H. influenzae DapE enzyme in vitro.4a Considering that L-captopril is a marketed drug against hypertension5 one may suppose that the compound already exhibits attractive pharmacokinetic properties. Moreover, considering the structural and functional conservation among DapE from various bacteria, one would expect that L-captopril may inhibit various bacterial homologues in a similar manner, thus potentially proving to be a broad-selectivity antibiotic. Disappointingly, however, L-captopril exhibited only very modest antimicrobial activity when tested in bacteria (including strains of S. enterica and E. coli). Moreover, this weak activity was revealed to be totally DapE-independent.6
The failure of a promising drug-like compound to exhibit any significant antimicrobial effect in vivo or to target the enzyme target identified in vitro is perplexing and acts as a sobering reminder of the difficulty of translating in vitro data to effects in vivo. In an attempt to overcome this initial setback, we aimed at understanding why L-captopril should fail to have any significant antimicrobial potency in bacteria. Here, we present a simple answer to this conundrum, based on the surprising metal-selectivity of L-captopril and its inability to inhibit all microbiologically relevant metallo-isoforms. Further, our findings imply that heterogeneity in metal content of metallo-enzymes may confer selective advantages.
Materials and methods
Construction of C-terminus polyhistidine-tagged DapE (DapE-C6His)
A pET28b-based plasmid (Novagen) for DapE overexpression was constructed according to published methods.7 Briefly, the 1.2 kb dapE gene was PCR amplified from the plasmid pCM655dapE by using the primers from Microsynth, F-SEQpCM655 (5′ GCGTACGTCGCGACCGCGGACATGT 3′) & R-pCM655XhoI (5′ AGCCAAGCCTCGAGGGCGACGAGCTGTTCC 3′), with Pfu turbo polymerase (Stratagene) and sub-cloned into pET28b plasmid (Novagen) at the restriction sites HindIII and XhoI (using restriction enzymes from Biolabs).Expression and purification of recombinant DapE-C6His
The expression plasmid, pET28b-DapE, was transformed into strain BL21(DE3) (Stratagene); the pre-culture was made in LB-medium with 30 μg ml−1kanamycin and 2% glucose at 37 °C and the over-expression was induced with 1 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG) when the OD600 was between 0.5 and 0.8, in LB-medium containing 30 μg ml−1kanamycin at 20 °C throughout. The cells were harvested after 6 hours of IPTG induction and the cell pellet was frozen at −20 °C, which was then thawed on ice and re-suspended in lysis buffer (50 mM tricine pH 7.8, 0.5 M NaCl, 1 mM PMSF, 3 μg ml−1 DNase I and 5 μg ml−1 lysozyme) and the cell-lysis was carried out at 37 °C and 200 rpm for 1 hour. The soluble protein fraction was transferred into a fresh bottle at 4 °C after the centrifugation at high speed (9000g) for 30 minutes in a pre-cooled (4 °C) centrifuge.The soluble protein fraction was filtered using a 0.45 μm filter and the loading buffer (50 mM tricine pH 7.8 & 0.5 M NaCl) and elutionbuffer (50 mM tricine, 0.5 M NaCl & 0.5 M imidazole and the final pH was adjusted to 7.8) were filtered using a 0.2 μm filter. Filtered DapE-C6His protein was purified on HiTrap 5 ml chelating HP columns (GE Healthcare) loaded with 0.1 M ZnCl2 in 50 mM tricine pH 7.8 with an ÅKTA Purifier-10 from GE Life Sciences.
Removal of the labile metal from the purified protein solution was achieved by 3 step sequential dialysis: first dialysis against 10 l of 50 mM tricine pH 7.8 with 1 mM 1,10-phenanthroline, second dialysis against 10 l of 50 mM tricine pH 7.8 with 5 mM ethylenediaminetetraacetic acid (EDTA) and third dialysis with 50 mM de-metallated tricine pH 7.8. Each dialysis step was for 8 to 12 hours at 4 °C using the dialysis bags with an approximate molecular weight cut-off of 6 to 8 kDa (Spectrum Labs). Chelex-100 (Bio-Rad) chelating ion exchange resins were used for the de-metallation of the buffer, i.e. 50 mM tricine pH 7.8 used for dialysis.
Purified DapE protein from S. enterica exhibited a single band on SDS-PAGE, indicating a molecular weight of approximately 42 kDa. Protein concentrations were determined by using the Pierce BCA protein assay kit (Thermo Scientific). The yield of the purified DapE protein was about 1 mg L−1 of bacterial culture.
Synthesis of SDAP
N-succinyl-L,L-diaminopimelic acid (SDAP) was synthesized in liquid phase through a standard coupling procedure between mono-tert-butyl succinate and enantiopure mono-tert-butyl protected L,L-diaminopimelic dimethyl ester (protected DAP), using O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU) as a coupling agent. The product of the coupling was then conveniently deprotected by standard cleavage of the protecting groups. DAP was previously synthesized according to published protocols viaolefincross-metathesis between enantiopure Boc-L-allylglycine-OMe and Cbz-L-vinylglycine-OMe, in yields above 70%.8DapE kinetic assays
The enzymatic assays were carried out in triplicate in 50 mM tricine pH 7.8 at 37 °C, using the purified DapE-C6His to a final concentration of 1 ng μl−1. The final substrate concentration of the native substrate (SDAP) was 20 mM in a reaction volume of 50 μl. Initially the DapE enzyme was pre-incubated with metals (0.2 mM ZnCl2 & 0.5 mM MnCl2 for the native substrate and 1 mM MnCl2 for the promiscuous substrate) at 37 °C for an hour and then with the inhibitor for another 10 minutes. L-Captopril was purchased from Sigma-Aldrich. In all cases the reactions were initiated by adding the substrate, either SDAP or Asp-Leu (Shanghai FWD Chemicals). The enzymatic reaction was followed by measuring the hydrolysis of the peptide bond of the substrates.9 Readings were carried out every 2 minutes for about 3 hours at UV230nm in UV transparent 96-well plates (Greiner) using a TECAN Safire plate reader.Growth assays
Bacterial growth assays were carried out with strain TN5911, a knockout strain for DapE and for several dipeptidases; therefore, mDAP (Bachem) has to be supplemented in both minimal and enriched media. An appropriate source of leucine and proline is also required in minimal medium and supplementation with lysine provides better growth (Prof. Charles G. Miller, personal communication). For DapE expression and selection assays, strain TN5911 was transformed with the plasmid pCM655/DapE to form strain TN5935.6,7The metal-dependent anti-microbial activity of L-captopril was monitored by measuring the growth curves of the TN5935 in LB-medium containing L-captopril (0, 1 mM, 5 mM, 25 mM, 100 mM & 300 mM) under native activity selection (no mDAP supplementation) and under no selection (supplemented with 1 mM mDAP) and with and without 0.35 mM metal (MnCl2 or ZnCl2) supplementation. All measurements were carried out at least in duplicate. The main cultures were grown and the OD600 was measured in flat bottom 96-well plates (200 μl culture per well) in a plate reader (TECAN Safire) at 37 °C for every 16 minutes. The main cultures were started by adding the pre-culture of OD600 ∼ 1 to an initial OD600 between 0.1 and 0.2.
The effect of the metals on DapE enzyme activities in bacteria was monitored by measuring the growth curves of the TN5935 under no selection and under selection for native & promiscuous activities with and without 0.35 mM metal salt (MnCl2 or ZnCl2) supplementation. These growth curves were measured from the cultures of minimal medium (1% MgSO4·7H2O, 10% citric acid·H2O, 50% K2HPO4, 17.5% NaHNH4PO4·4H2O, 0.4% glucose, 0.4 mM proline, 0.4 mM lysine, 0.4 mM leucine or Asp-Leu, 1 mM IPTG and, if required, 1 mM mDAP) by following the OD600 every 4 hours at 37 °C at 200 rpm. The cultures were started by adding the pre-culture of LB-medium of OD600 ∼ 1 to an initial OD600 between 0.1 and 0.2, which corresponded in this case to 0.4 ml of overnight LB-medium culture in 100 ml of minimal medium in a 1 l Erlenmeyer flask. We carried out a control culture to confirm that at this dilution of rich LB-medium in minimal medium (250-fold dilution), there is insufficient carry-over of leucine to allow for any detectable growth of the dapE knockout strain (TN5911) when supplementing with mDAP.
Structural modelling
A homology model of the Salmonella enterica DapE model was built with SWISS-MODEL.10 The di-zinc Haemophilus influenzae X-ray structure [PDB: 3IC1] sharing 61% identity with S. enterica without water molecules was used as a modelling template.The resulting model was energy minimized using the Yasara energy minimization server.11 The conformation of residue Lys174 was replaced by an alternative rotamer in Maestro12 to enlarge the ligand binding site. For the mono-zinc model, ZN2 was removed. The resulting structures were energy minimized using MacroModel.13 Subsequently, the L-captopril structure [PDBe: X8Z] was docked into the active site of the mono- and di-zinc structures using SwissDock.14Ligand conformations satisfying the constraints of metal-coordination that were imposed based on mechanistic considerations (see Results section) were found within the 25 and the 10 best ranked poses for the di-zinc and the mono-zinc models, respectively. These best-ranked models were submitted to the model archive (www.modelarchive.org) and are freely available for download (DOI: 10.5452/ma-a1nb6). All model figures were drawn with OpenStructure.15
Results
Metal substitution changes L-captopril inhibition in vitro
DapE was active as an SDAP desuccinylase in vitro both in its Zn2+ mono-metallated form (Zn/-) and as a Zn2+ binuclear (Zn–Zn) or mixed (Zn–Mn) form, with comparable Vmax (Scheme 1 and Table 1). As reported previously,7 no activity was detected for Asp-Leuhydrolysis with the Zn/- and Zn–Zn isoforms of the enzyme, whereas only the Zn–Mn isoform was active with this promiscuous substrate (in our hands, with kcat 383 s−1). These activity profiles correspond to the metallation status ascribed to the various enzyme preparations.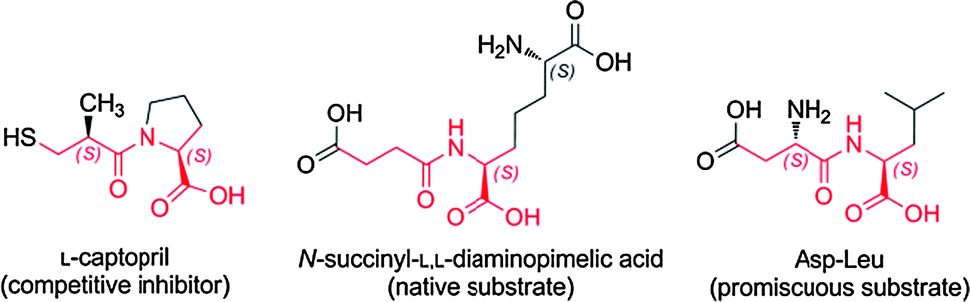 | ||
| Scheme 1 L-Captopril inhibitor and substrates of DapE used in this study. Elements common to the three molecules are highlighted in red. | ||
| DapE metallo-isoform | V max (μmol L−1 s−1) | k cat (s−1) |
|---|---|---|
| a V max and kcat values of desuccinylase activity of different metallo-isoforms of recombinant DapE from Salmonella enterica typhimurium. | ||
| Zn/- | 18.23 | 309 |
| Zn–Zn | 39.81 | 677 |
| Zn–Mn | 64.99 | 1104 |
Addition of L-captoprilin vitro inhibited the catalytic activity of the (Zn/-) or (Zn–Zn) metallated isoforms of DapE, with an IC50 of 10 μM and 28 μM at 20 mM substrate, respectively (Fig. 1). However, no significant inhibition of the Zn–Mn isoform was found even at 1 mM L-captopril.
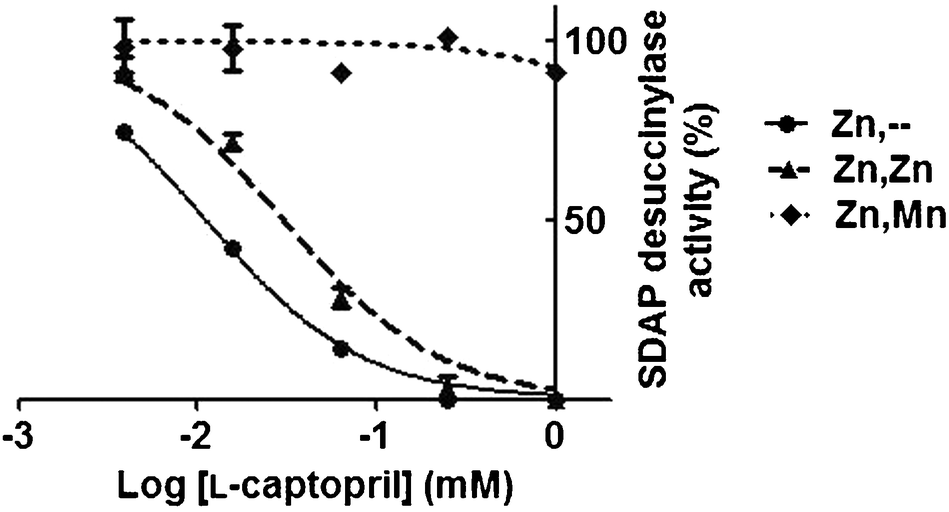 | ||
| Fig. 1 Inhibition of desuccinylase activity of different DapE metallo-isoforms by L-captopril. Measurements were carried out in triplicate and error bars represent the standard deviation from the mean. | ||
The substrate concentration at which half the maximum velocity was measured for SDAP hydrolysis (K50) was similar for the Zn–Zn and Zn–Mn metalloisoforms (12 mM and 12.5 mM respectively). DapE from Haemophilus influenzae was reported in two independent studies9,16 to have comparable Km for the Zn/-, Zn–Zn and Zn–Co metalloisoforms, which also supports the notion that the second metal only marginally affects the binding of the native substrate. Together, these results suggest that the metallation state at the second position influences the binding of both the inhibitorL-captopril and the promiscuous substrate Asp-Leu, but does not affect much the binding of the native substrate SDAP.
Manganese isoform is present in vivo
Traditionally, it has been considered that Zn2+ is the native and physiologically relevant metal of DapE. However, the enzyme presents also a promiscuous dipeptidase activity that is dependent on the presence of manganese ions (Mn2+), which can also be selected in vivo in an leucine auxotroph strain using Asp-Leu as a substrate.7 Since this non-native dipeptidase activity occurs in vitro only when the labile, co-catalytic Zn2+ is replaced by Mn2+ to form a mixed Zn–Mn binuclear centre, it is surmised that the DapE-dependent promiscuous activity in bacterial cultures also arises from Mn2+-metallo-isoforms of DapE in vivo.7When selecting for the native desuccinylase activity in bacteria, the conditions in which no metal was added and where Zn2+ was supplemented showed similar growth; in contrast Zn2+ slowed down the growth of cell cultures when selecting for dipeptidase activity, which suggests that Zn2+ is deleterious for the promiscuous activity of DapE, but not for its native activity (Fig. 2). DapE was able to desuccinylate SDAP in its Zn/-, Zn–Zn and Zn–Mn metallo-isoforms in vitro, albeit to different extents, with the following trends for kcat: Zn–Mn > Zn–Zn > Zn/- (Table 1). S. enterica grew appreciably better in Mn2+-supplemented media when selected for either native or promiscuous activity (Fig. 2). Therefore, it appears that addition of Mn2+ promotes dipeptidase activity of DapE both in vivo and in vitro, whereas Zn2+ inhibits the dipeptidase activity, presumably by competing with Mn2+ for the labile metal-binding site of DapE.
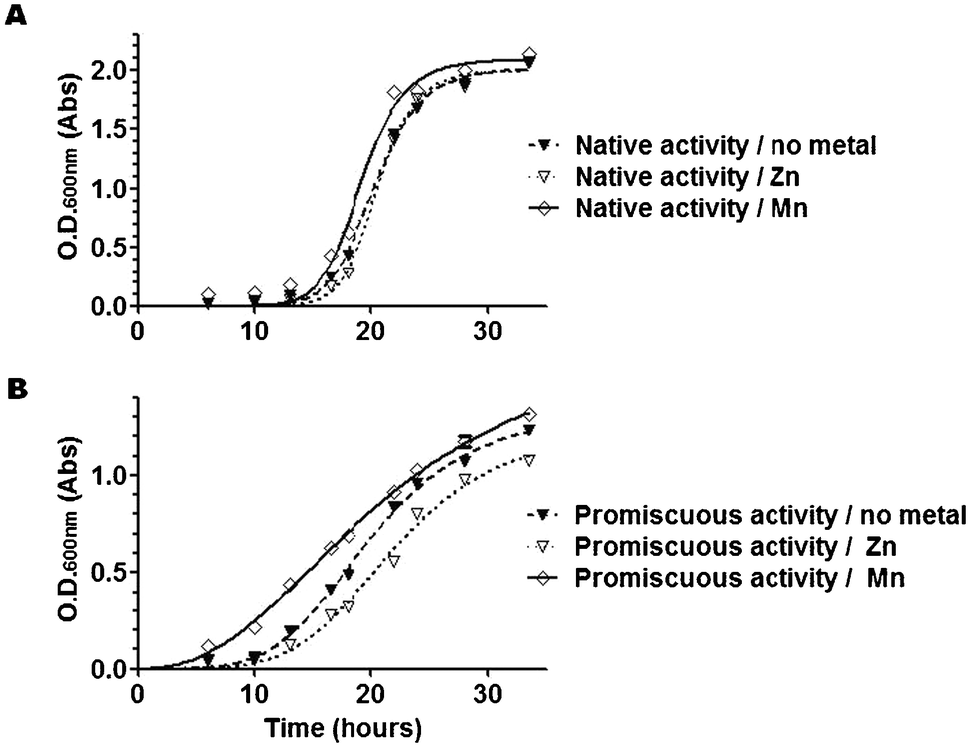 | ||
| Fig. 2 Growth curves of bacterial cultures selected for native (desuccinylase) DapE activity (A) or promiscuous (dipeptidase) DapE activity (B) in the absence or presence of Zn2+ or Mn2+ supplementation. Note that Mn2+ accelerates the growth of the culture appreciably when selecting for both native and promiscuous activity. In contrast, Zn2+ slows down the growth of the culture appreciably when selecting for promiscuous activity but has little effect on proliferation when selecting for native activity. Plotted are fitted curves from means of duplicate cultures, with individual values comprised in the symbols. The values plotted are representative of two independent experiments with similar findings. | ||
We conclude from these observations that (i) metallation of DapE can be influenced by metal-supplementation with either Zn2+ or Mn2+; (ii) Mn2+-metallation is growth-limiting in non-supplemented medium, particularly when selecting for the dipeptidase activity, and (iii) the Mn2+ isoform may be normally present in vivo but is not the only metallated species of DapE that occurs.
Zn2+ supplementation makes bacteria more susceptible to L-captopril inhibition compared to Mn2+
Having concluded from growth profiles that metal supplementation of bacterial cultures can influence the state of metallation of DapE, we next investigated whether metal-supplementation also affected susceptibility to L-captopril in bacterial cultures. We reasoned, since L-captopril targets only the Zn2+-isoform of DapE in vitro and since metal supplementation can influence the metallation of DapE in vivo, that we may be able to influence the sensitivity of bacterial cultures to L-captopril through metallation of DapE.First, we confirmed that L-captopril was toxic in a dose-dependent manner, shown by bacterial growth-curves in rich medium at different inhibitor concentrations (Fig. S1, ESI‡). Second, in the growth conditions tested, in rich medium and 25 mM L-captopril, metal supplementation with either divalent Zn or Mn lowered L-captopril toxicity. The influence of metal supplementation on bacterial growth under these conditions was much larger in the presence of 25 mM L-captopril than in the absence of the drug (Fig. 3): Mn2+-supplemented cultures clearly showed better growth as compared to Zn2+ supplemented or especially non-metal-supplemented cultures, which were more susceptible to L-captopril inhibition. The growth-profile in the presence of L-captopril was Mn2+ > Zn2+ > no metal. Intriguingly, L-captopril inhibition of DapE in vitro has exactly the complementary activity profile: L-captopril inhibition Mn2+ < Zn2+ < no metal.
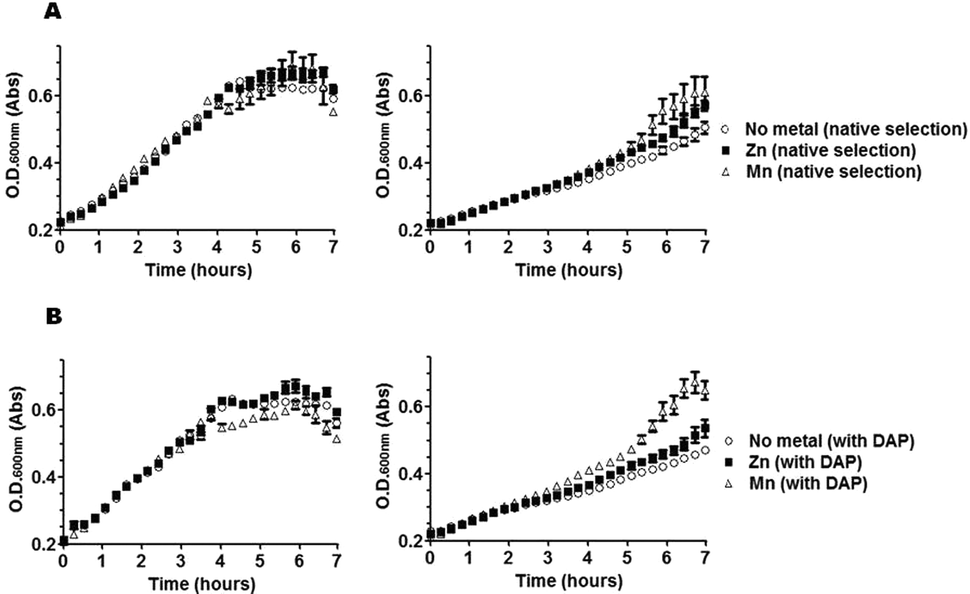 | ||
| Fig. 3 Effect of metal supplementation on growth curves of bacterial cultures selected for DapE activity (A) or non-selected (B), in the absence of L-captopril (left) or in the presence of 25 mM L-captopril (right). Cultures were grown in LB broth either in the presence or absence of mDAP for selection of native (desuccinylase) activity of DapE. The effect of divalent Zn or Mn was tested and compared to the absence of metal supplementation (no metal). Note that L-captopril toxicity is dependent on metal-supplementation and that Mn2+ is more protective than Zn2+, although the toxic effect of the drug is largely independent of DapE activity. Means of measurements from quadruplicate cultures are plotted with standard deviations shown as error bars. | ||
Despite the metal-dependent effect of L-captopril, bacterial growth in the absence of selection for DapE activity (i.e. with mDAP supplementation) showed that the toxicity of this drug is largely independent of DapE activity (Fig. 3B), confirming our previous finding that this essential enzyme is not the main target of inhibition of L-captoprilin vivo and suggesting that other metalloproteins may also be the main targets of L-captopril.6
We conclude that at least some of the targets of L-captopril are metal-dependent, i.e. metal-supplementation decreases sensitivity to L-captopril. The finding that Mn2+ supplementation is more protective than Zn2+ supplementation is consistent with the notion that L-captopril has a preference for Zn2+-containing targets, at least some of which may also be functionally active with Mn2+. We also surmise that the failure of L-captopril to exhibit any significant DapE-selective antibiotic activity in bacteria is due to the existence of sufficient amount of Zn–Mn isoform, which is not susceptible to L-captopril inhibition.
Model of L-captopril inhibition supports that thiol is the Zn2+-binding group
We created a model of S. enterica DapE (Fig. 4) based on the high-resolution crystal structure of the homologous enzyme from Haemophilus influenzae reported by Nocek et al.17 The DapE enzyme is a dimer, each monomer of which contains two domains: a dimerization domain and a catalytic domain. The active site of the enzyme can contain either two or one zinc ions. The strongly bound Zn2+ is referred to as Zn1, whereas the Zn2+ occupying the site of the labile metal (M2, which can be either Zn2+ or Mn2+) is referred to as Zn2.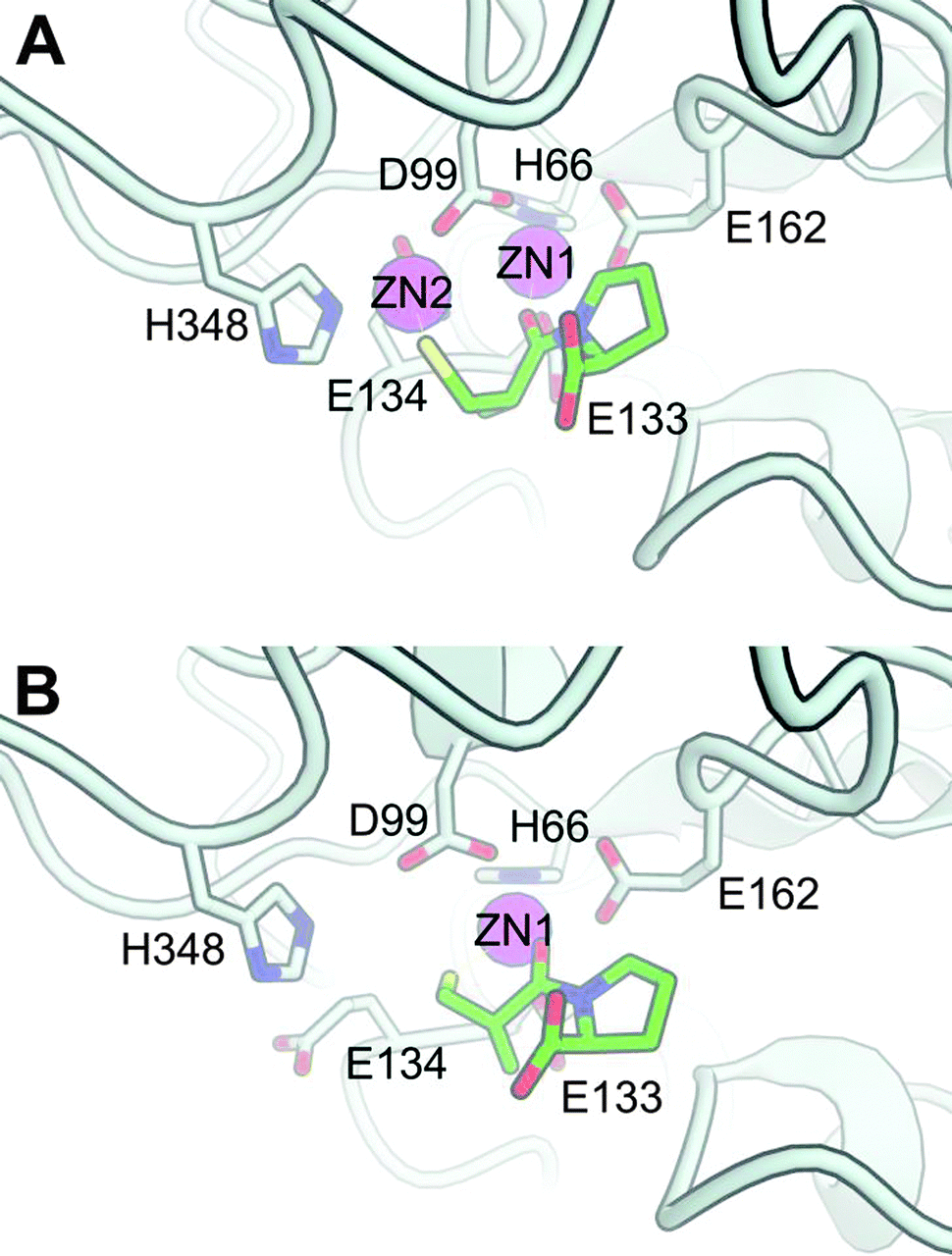 | ||
| Fig. 4 Model of S. enterica DapE based on the crystal structure of H. influenzae DapE (PDB: 3IC1), with L-captopril docked to the (A) di-zinc and (B) mono-zinc catalytic metal center. Only the side-chains of important residues for catalysis are drawn and labelled, i.e. coordinating amino acids and the catalytic base (E134). L-Captopril is drawn in the stick-form with each type of atom colored differently: green carbon; blue nitrogen; red oxygen and yellow sulfur. Although coordination bonds with the ligand are not explicitly drawn, in both the mono- and di-zinc form the L-captopril carbonyl group of the amide coordinates with the Zn1, whereas the L-captopril thiol group can additionally coordinate either with Zn2 in the di-zinc structure (A) or with Zn1 in the mono-zinc structure, to form a 6-membered ring (B). These two models are freely available for download with DOI: 10.5452/ma-a1nb6 (www.modelarchive.org). | ||
To generate the model of L-captopril binding to the active site, we imposed some constraints based on prior knowledge of DapE and L-captopril as a substrate analogue (Scheme 1). First, as in the suggested catalytic mechanism of DapE,17 we propose that the carbonyl group of the amide coordinates with the catalytic metal that acts as a Lewis acid (Zn1). Second, since captopril binds to the catalytic zinc of Angiotensin Converting Enzyme (ACE) through coordination by the sulfhydryl group5 and since we found experimentally that the nature of the co-catalytic metal (M2, either Zn2+ or Mn2+) influences inhibition by L-captopril, we expect the sulfhydryl group to be oriented toward this second metal. Satisfyingly, docking-poses that fulfil these constraints were found within the 25 highest ranked poses (Fig. 4).
Divalent manganese behaves similar to magnesium in having a preference for oxygen ligands, although Mn2+ is more receptive of nitrogenligands; in contrast, zinc prefers nitrogen and sulfur as ligands and, in general, a lower coordination number.18 Consequently, our model provides a reasonable and straightforward qualitative explanation for the metal-dependent selectivity of L-captopril inhibition of DapE: the presence of Mn2+ does not allow strong coordination by the sulfhydryl group, whereas either the absence of a co-catalytic metal or the presence of a second Zn2+ allows coordination with the sulfur liganding either Zn2 or Zn1, respectively.
Discussion
Implications for drug development
L-Captopril was the first marketed anti-hypertension drug, targeting the Zn-metalloprotease Angiotensin I Converting Enzyme (ACE) through coordination of the catalytic metal by a sulfhydryl group, with low nM affinity.5 However, L-captopril has also shown inhibitory activity towards a variety of other zinc metalloproteases, although this is typically several orders of magnitude weaker than with ACE.19 Recently, L-captopril was identified also as a low-micromolar inhibitor of H. influenzae DapE (IC50 = 3.3 μM). We found that L-captopril inhibited the Zn–Zn isoforms of S. enterica DapE with IC50 values comparable to those of the H. influenzae enzyme (i.e. IC50 of 10 μM vs. 3.3 μM, respectively), providing further evidence for the similarities in the structure and function of the active sites of DapE of different species.17,20 The low-micromolar affinity of L-captopril for the Zn/- and Zn–Zn isoforms of DapE could be seen as a reasonable starting point for further optimisation of this lead compound,3,4 akin to what succinyl-proline was for ACE.5,19 However, note that L-captopril at the concentrations at which it inhibits DapE might cause hypotension in normo-tensive individuals, which would warrant further development of its antibiotic properties. Moreover, at this low-micromolar level of affinity some off-targets are likely to occur, as indeed suggested by the poor activity of L-captopril6 and by the general protective effect of Zn2+ and especially Mn2+ supplementation (Fig. 3). We were nevertheless initially very surprised that we could detect no evidence of L-captopril inhibiting DapE activity in vivo and that the compound had such a modest antimicrobial activity, despite low-micromolar affinity of this drugin vitro for an essential enzyme. We now forward the proposal that a crucial contribution to this low antibiotic activity is that L-captopril does not target one of the physiologically relevant isoforms of DapE, i.e. the Zn–Mn form of the enzyme.It is generally acknowledged that any screen (or selection) should reflect the desired result as closely as possible, because ‘you get what you screen for’.21 Becker and colleagues4a identified L-captopril as an inhibitor of DapE in a screen biased toward compounds containing zinc-binding groups, which included thiols, carboxylic acids, boronic acids, phosphonates and hydroxamates, and using the Zn-form of DapE of H. influenzae as a target in vitro. Consequently, a Zn-selective compound was identified with low micromolar affinity in vitro. Our discovery offers a plausible explanation for the failure of an apparently promising compound to show any DapE-targeted antimicrobial activity:6 the Zn–Mn mixed-metal isoform, which is not inhibited by L-captopril, is normally present in bacteria, as originally suggested by Broder and Miller7 judging by the presence of a selectable Mn2+-dependent promiscuous activity, which we confirmed here also by exploring bacterial growth upon metal-supplementation of cultures.
Exact knowledge of the metallation state of proteinsin vivo is often thwarted by experimental difficulties.22 Moreover, metallation may exhibit strong dependence on the cellular context, differing for example in different media, cell-locations or in different species.23 Several other examples have been reported where incomplete knowledge of the physiologically relevant metal has hindered drug development. For example, potent inhibitors of methionine aminopeptidase discovered in vitro, which were active against either Co2+ or Mn2+-isoforms of the enzyme, failed to have any of the expected antibacterial activity in vivo.24 It was later suggested that Fe2+ is the native cofactor in E. coli, prompting a move either toward Fe2+-selective inhibitors that exhibited antimicrobial activity25 or, in what may be called a “bet-hedging move” in the absence of a clear candidate, toward exploring selectivity against a plethora of different metallo-isoforms.26 Similarly, peptide deformylase is another bacterial metalloenzyme and a promising antibiotic target27 that was originally thought to be a Zn-metalloenzyme in E. coli28 but was later shown instead to contain Fe2+, a metal that due to its tendency to oxidize in air leads to facile loss of enzyme activity.29 However, the precise identity of the metal in peptide deformylase may be species-specific, since another bacterial pathogen, Borrelia burgdorferi, does use Zn2+ in this enzyme.30 Despite considerable interest in peptide deformylase inhibitors in recent years27c it appears that little attention has been paid to the issue of metallo-selectivity of drugs targeting this essential enzyme.
Implications for evolution
In general, the presence of different metal ions in metalloenzymes can maintain the overall structure of the active site, but provide alternative coordination interactions with substrates and inhibitors. It has already been pointed out that non-strict metal incorporation can be used as a facile way to create catalytic diversity.31 For example, metal-heterogeneity has been proposed to have evolved in the carbonic anhydrase of marine diatoms that are challenged with low Zn-availability, thereby acquiring the capability of using either Zn2+ or Cd2+ in the active site of this essential enzyme.32 Indeed, we suggest that such metal-ambiguity and the resulting promiscuous behaviour in vivo, which may be considered a further example of “messiness in biology”,33 is in fact a more general survival strategy, for example to avoid facile drug inhibition of an essential enzyme like DapE. To our knowledge, this may be the first example of metal-promiscuity providing a source of (non-genetic) diversity that confers an intrinsic survival advantage in defending against potential inhibitors, namely to the synthetic drugL-captopril, but also likely against other naturally occurring dipeptide-like compounds. Considering the importance of diversity as a driver of Darwinian evolution, we speculate that such metal-promiscuity may be much more common than hitherto thought, not only to provide substrate promiscuity in the development of new catalytic function, but also promiscuity for inhibitor-escape.Conclusions
In summary, DapE in bacteria is heterogeneous, containing various metallated forms – mono-metallated with Zn2+ or binuclear with Zn2+ or Mn2+ – all of which are active as a desuccinylase, the essential and native function of the enzyme. We also conclude that the Zn–Mn binuclear isoform, which is very likely to be present in vivo as judged by bacterial selection of the promiscuous dipeptidase activity of DapE, is not inhibited by L-captopril, thus providing a mechanism of escape against this potential antibiotic. Such metal heterogeneity and resulting promiscuous properties may confer important selective advantages, including escape of inhibition, and therefore may have been selected for in evolution. Finally, the discovery of metal-selective inhibition of DapE that we describe here paves the way toward the discovery of compounds with improved antimicrobial properties. Our efforts are now directed toward finding novel inhibitors that target also the physiologically relevant Mn2+ isoform of DapE.Abbreviations
| mDAP | meso-Diaminopimelic acid |
| DAP | Diaminopimelic acid |
| SDAP | N-Succinyl-L,L-diaminopimelic acid |
| DapE | N-Succinyl-L,L-diaminopimelic acid desuccinylase |
| ACE | Angiotensin I converting enzyme |
Acknowledgements
The authors would like to thank the financial support from the Swiss National Science Foundation (Grants 200021-116344 and CR22I3_143770), the State Secretariat for Education and Research (Grant C11.0054) and COST Actions TD0905 and CM0902. MC is particularly grateful for the support from the Holcim Foundation and GA from the Novartis Foundation. The authors would also like to acknowledge Prof. Thomas R. Ward for general encouragement and for allowing the use of laboratory space and facilities, as well as thank Prof. Charles G. Miller (University of Illinois) for his generous donation of valuable research materials and insights.Notes and references
- ECDC/EMEA The bacterial challenge: time to react Joint Technical Report: 2009.
- (a) WHO Antimicrobial Resistance; Fact sheet No. 194; World Health Organisation: 2002; (b) S. B. Levy, The challenge of antibiotic resistance, Sci. Am., 1998, 278, 46–53 CrossRef CAS PubMed.
- D. M. Gillner, D. P. Becker and R. C. Holz, Lysine biosynthesis in bacteria: a metallodesuccinylase as a potential antimicrobial target, JBIC, J. Biol. Inorg. Chem., 2013, 18, 155–163 CrossRef CAS PubMed.
- (a) D. Gillner, N. Armoush, R. C. Holz and D. P. Becker, Inhibitors of bacterial N-succinyl-L,L-diaminopimelic acid desuccinylase (DapE) and demonstration of in vitro antimicrobial activity, Bioorg. Med. Chem. Lett., 2009, 19, 6350–6352 CrossRef CAS PubMed; (b) G. Scapin and J. S. Blanchard, Enzymology of bacterial lysine biosynthesis, Adv. Enzymol. Relat. Areas Mol. Biol., 1998, 72, 279–324 CAS; (c) C. Gilvarg, N-Succinyl-L-Diaminopimelic Acid, J. Biol. Chem., 1959, 234, 2955–2959 CAS.
- D. W. Cushman and M. A. Ondetti, Design of angiotensin converting enzyme inhibitors, Nat. Med., 1999, 5, 1110–1112 CrossRef CAS PubMed.
- N. Uda and M. Creus, Selectivity of inhibition of N-succinyl-L,L-diaminopimelic acid desuccinylase (DapE) in bacteria: DapE is not the target of L-captopril antimicrobial activity, Bioinorg. Chem. Appl., 2011, 2011, 306465 Search PubMed.
- D. H. Broder and C. G. Miller, DapE can function as an aspartyl peptidase in the presence of Mn2+, J. Bacteriol., 2003, 185, 4748–4754 CrossRef CAS PubMed.
- A. R. Chowdhury and G. J. Boons, The synthesis of diaminopimelic acid containing peptidoglycan fragments using metathesis cross coupling, Tetrahedron Lett., 2005, 46, 1675–1678 CrossRef CAS.
- T. L. Born, R. J. Zheng and J. S. Blanchard, Hydrolysis of N-succinyl-L,L-diaminopimelic acid by the Haemophilus influenzae dapE-encoded desuccinylase: metal activation, solvent isotope effects, and kinetic mechanism, Biochemistry, 1998, 37, 10478–10487 CrossRef CAS PubMed.
- K. Arnold, L. Bordoli, J. Kopp and T. Schwede, The SWISS-MODEL workspace: a web-based environment for protein structure homology modeling, Bioinformatics, 2006, 22, 195–201 CrossRef CAS PubMed.
- E. Krieger, K. Joo, J. Lee, J. Lee, S. Raman, J. Thompson, M. Tyka, D. Baker and K. Karplus, Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8, Proteins: Struct., Funct., Bioinf., 2009, 77, 114–122 CrossRef CAS PubMed.
- Suite 2012: Maestro, version 9.3 edn., Schrödinger, LLC, New York, NY, 2012 Search PubMed.
- MacroModel, version 9.9 edn., Schrödinger, LLC, New York, NY, 2012 Search PubMed.
- (a) A. Grosdidier, V. Zoete and O. Michielin, SwissDock, a protein-small molecule docking web service based on EADock DSS, Nucleic Acids Res., 2011, 39, W270–W277 CrossRef CAS PubMed; (b) A. Grosdidier, V. Zoete and O. Michielin, Fast Docking Using the CHARMM Force Field with EADock DSS, J. Comput. Chem., 2011, 32, 2149–2159 CrossRef CAS PubMed.
- (a) M. Biasini, V. Mariani, J. Haas, S. Scheuber, A. D. Schenk, T. Schwede and A. Philippsen, OpenStructure: a flexible software framework for computational structural biology, Bioinformatics, 2010, 26, 2626–2628 CrossRef CAS PubMed; (b) M. Biasini, T. Schmidt, S. Bienert, V. Mariani, G. Studer, J. Haas, N. Johner, A. D. Schenk, A. Philippsen and T. Schwede, OpenStructure: an integrated software framework for computational structural biology, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 701–709 CAS.
- D. L. Bienvenue, D. M. Gilner, R. S. Davis, B. Bennett and R. C. Holz, Substrate specificity, metal binding properties, and spectroscopic characterization of the DapE-encoded N-succinyl-L,L-diaminopimelic acid desuccinylase from Haemophilus influenzae, Biochemistry, 2003, 42, 10756–10763 CrossRef CAS PubMed.
- B. P. Nocek, D. M. Gillner, Y. Fan, R. C. Holz and A. Joachimiak, Structural Basis for Catalysis by the Mono- and Dimetalated Forms of the dapE-Encoded N-succinyl-L,L-Diaminopimelic Acid Desuccinylase, J. Mol. Biol., 2010, 397, 617–626 CrossRef CAS PubMed.
- C. W. Bock, A. K. Katz, G. D. Markham and J. P. Glusker, Manganese as a replacement for magnesium and zinc: Functional comparison of the divalent ions, J. Am. Chem. Soc., 1999, 121, 7360–7372 CrossRef CAS.
- M. A. Ondetti and D. W. Cushman, Design of Specific Inhibitors of Angiotensin-Converting Enzyme - New Class of Orally Active Antihypertensive Agents, Science, 1977, 196, 441–444 CAS.
- (a) T. L. Born, R. Zheng and J. S. Blanchard, Hydrolysis of N-Succinyl-L,L-diaminopimelic Acid by the Haemophilus influenzae dapE-Encoded Desuccinylase: Metal Activation, Solvent Isotope Effects, and Kinetic Mechanism, Biochemistry, 1998, 37, 10478–10487 CrossRef CAS PubMed; (b) K. S. Makarova and N. V. Grishin, The Zn-peptidase superfamily: functional convergence after evolutionary divergence, J. Mol. Biol., 1999, 292, 11–17 CrossRef CAS PubMed; (c) J. Badger, J. M. Sauder, J. M. Adams, S. Antonysamy, K. Bain, M. G. Bergseid, S. G. Buchanan, M. D. Buchanan, Y. Batiyenko, J. A. Christopher, S. Emtage, A. Eroshkina, I. Feil, E. B. Furlong, K. S. Gajiwala, X. Gao, D. He, J. Hendle, A. Huber, K. Hoda, P. Kearins, C. Kissinger, B. Laubert, H. A. Lewis, J. Lin, K. Loomis, D. Lorimer, G. Louie, M. Maletic, C. D. Marsh, I. Miller, J. Molinari, H. J. Muller-Dieckmann, J. M. Newman, B. W. Noland, B. Pagarigan, F. Park, T. S. Peat, K. W. Post, S. Radojicic, A. Ramos, R. Romero, M. E. Rutter, W. E. Sanderson, K. D. Schwinn, J. Tresser, J. Winhoven, T. A. Wright, L. Wu, J. Xu and T. J. Harris, Structural analysis of a set of proteins resulting from a bacterial genomics project, Proteins, 2005, 60, 787–796 CrossRef CAS PubMed; (d) T. S. Girish and B. Gopal, Crystal Structure of Staphylococcus aureus Metallopeptidase (Sapep) Reveals Large Domain Motions between the Manganese-bound and Apo-states, J. Biol. Chem., 2010, 285, 29406–29415 CrossRef CAS PubMed.
- L. You and F. H. Arnold, Directed evolution of subtilisin E in Bacillus subtilis to enhance total activity in aqueous dimethylformamide, Protein Eng., 1996, 9, 78 Search PubMed; L. You and F. H. Arnold, Protein Eng., 1996, 9, 719 CrossRef CAS.
- (a) Z. G. Xiao and A. G. Wedd, The challenges of determining metal-protein affinities, Nat. Prod. Rep., 2010, 27, 768–789 RSC; (b) M. Babor, S. Gerzon, B. Raveh, V. Sobolev and M. Edelman, Prediction of transition metal-binding sites from apo protein structures, Proteins: Struct., Funct., Bioinf., 2008, 70, 208–217 CrossRef CAS PubMed; (c) H. Zheng, M. Chruszcz, P. Lasota, L. Lebioda and W. Minor, Data mining of metal ion environments present in protein structures, J. Inorg. Biochem., 2008, 102, 1765–1776 CrossRef CAS PubMed.
- (a) W. Maret, Metalloproteomics, metalloproteomes, and the annotation of metalloproteins, Metallomics, 2010, 2, 117–125 RSC; (b) K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Metalloproteins and metal sensing, Nature, 2009, 460, 823–830 CrossRef CAS PubMed.
- (a) R. Schiffmann, A. Heine, G. Klebe and C. D. P. Klein, Metal ions as cofactors for the binding of inhibitors to methionine aminopeptidase: a critical view of the relevance of in vitro metalloenzyme assays, Angew. Chem., Int. Ed., 2005, 44, 3620–3623 CrossRef CAS PubMed; (b) C. Oefner, A. Douangamath, A. D'Arcy, S. Hafeli, D. Mareque, A. Mac Sweeney, J. Padilla, S. Pierau, H. Schulz, M. Thormann, S. Wadman and G. E. Dale, The 1.15 angstrom crystal structure of the Staphylococcus aureus methionyl-aminopeptidase and complexes with triazole based inhibitors, J. Mol. Biol., 2003, 332, 13–21 CrossRef CAS PubMed; (c) Q. L. Luo, J. Y. Li, Z. Y. Liu, L. L. Chen, J. Li, Z. Qian, Q. Shen, Y. Li, G. H. Lushington, Q. Z. Ye and F. J. Nan, Discovery and structural modification of inhibitors of methionine aminopeptidases from Escherichia coli and Saccharomyces cerevisiae, J. Med. Chem., 2003, 46, 2631–2640 CrossRef CAS PubMed.
- (a) S. C. Chai, W. L. Wang and Q. Z. Ye, Fe(II) is the native cofactor for Escherichia coli methionine aminopeptidase, J. Biol. Chem., 2008, 283, 26879–26885 CrossRef CAS PubMed; (b) S. C. Chai, W. L. Wang, D. R. Ding and Q. Z. Ye, Growth inhibition of Escherichia coli and methicillin-resistant Staphylococcus aureus by targeting cellular methionine aminopeptidase, Eur. J. Med. Chem., 2011, 46, 3537–3540 CrossRef CAS PubMed.
- F. Huguet, A. Melet, R. A. de Sousa, A. Lieutaud, J. Chevalier, L. Maigre, P. Deschamps, A. Tomas, N. Leulliot, J. M. Pages and I. Artaud, Hydroxamic Acids as Potent Inhibitors of FeII and MnII E. coli Methionine Aminopeptidase: Biological Activities and X-ray Structures of Oxazole Hydroxamate-EcMetAP-Mn Complexes, ChemMedChem, 2012, 7, 1020–1030 CrossRef CAS PubMed.
- (a) J. A. Leeds and C. R. Dean, Peptide deformylase as an antibacterial target: a critical assessment, Curr. Opin. Pharmacol., 2006, 6, 445–452 CrossRef CAS PubMed; (b) C. Giglione, M. Pierre and T. Meinnel, Peptide deformylase as a target for new generation, broad spectrum antimicrobial agents, Mol. Microbiol., 2000, 36, 1197–1205 CrossRef CAS PubMed; (c) C. T. Supuran, F. Carta and A. Scozzafava, Metalloenzyme inhibitors for the treatment of Gram-negative bacterial infections: a patent review (2009–2012), Expert Opin. Ther. Pat., 2013, 1–12 Search PubMed.
- T. Meinnel, C. Lazennec and S. Blanquet, Mapping of the Active-Site Zinc Ligands of Peptide Deformylase, J. Mol. Biol., 1995, 254, 175–183 CrossRef CAS PubMed.
- P. T. R. Rajagopalan and D. Pei, Oxygen-mediated inactivation of peptide deformylase, J. Biol. Chem., 1998, 273, 22305–22310 CrossRef CAS PubMed.
- K. T. Nguyen, J. C. Wu, J. A. Boylan, F. C. Gherardini and D. Pei, Zinc is the metal cofactor of Borrelia burgdorferi peptide deformylase, Arch. Biochem. Biophys., 2007, 468, 217–225 CrossRef CAS PubMed.
- M. Hogbom, Metal use in ribonucleotide reductase R2, di-iron, di-manganese and heterodinuclear-an intricate bioinorganic workaround to use different metals for the same reaction, Metallomics, 2011, 3, 110–120 RSC.
- Y. Xu, L. Feng, P. D. Jeffrey, Y. G. Shi and F. M. M. Morel, Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms, Nature, 2008, 452, 56–62 CrossRef CAS PubMed.
- D. S. Tawfik, Messy biology and the origins of evolutionary innovations, Nat. Chem. Biol., 2010, 6, 692–696 Search PubMed.
Footnotes |
| † This work is dedicated to the memory of Dr Peter Rellos (1961–2011), friend and mentor. |
| ‡ Electronic supplementary information (ESI) available: Fig. S1. See DOI: 10.1039/c3mt00125c |
| This journal is © The Royal Society of Chemistry 2014 |