Influence of structure-selective fluorene-based polymer wrapping on optical transitions of single-wall carbon nanotubes†
Masayoshi
Tange
*,
Toshiya
Okazaki
* and
Sumio
Iijima
Nanotube Research Center, National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba 305-8565, Japan. E-mail: masa-tange@aist.go.jp; toshi.okazaki@aist.go.jp
First published on 14th October 2013
Abstract
To understand how fluorene-based polymers selectively extract specific semiconducting single-wall carbon nanotubes (SWCNTs), we compared the optical transitions of SWCNTs wrapped with poly(9,9-dioctylfluorene-alt-pyridine) (PFOPy), i.e., structure-selective polymers, with those wrapped with poly(9,9-di-n-dodecylfluorene) (PFD), i.e., non-selective polymers, in organic solvents by using photoluminescence (PL) excitation spectroscopy. Two (n,m) species of PFOPy-wrapped SWCNTs with intermediate chiral angles exhibited blue-shifted emissions compared with the PFD-wrapped SWCNTs. The shifts in the peaks of PL signals cannot be explained in terms of the dielectric screening effect, but can plausibly be explained in terms of the strains of specific SWCNTs due to the PFOPy wrapping. Moreover, the emissions of specific SWCNTs wrapped with PFOPy were not blue-shifted as much when the solvent was changed from toluene to p-xylene, and this result could be accounted for by a change in the rigidity of the fluorene backbone. Moreover, using p-xylene instead of toluene lowered the selectivity of the SWCNT extraction, thereby suggesting the importance of having a rigid fluorene backbone for selective extraction of SWCNTs.
Introduction
Single-wall carbon nanotubes (SWCNTs) are one-dimensional materials showing a variety of electronic states depending on their tube structure (i.e., chiral angle θ and tube diameter dt, as identified by chiral indices (n,m)). In particular, the optical transitions of SWCNTs are very sensitive to the surrounding environment. Not only local dielectric screening but also strain induced by the surrounding and encapsulated molecules changes the optical transition energies.1–7 Consequently, the shift in the transition energy, appearing as near-infrared (NIR) photoluminescence (PL),8 of semiconducting SWCNTs, in particular, provides a lot of information on the surrounding dielectric constant and mechanical strain, and this suggests that these SWCNTs can be used as molecular and strain sensors.9–11Polymers are frequently used to disperse SWCNTs in organic solvents.12–15 Recently, we found that poly(9,9-dioctylfluorene-alt-pyridine) (PFOPy) can selectively extract specific semiconducting SWCNTs with a narrow diameter distribution.16 By measuring the optical absorption and PL spectra, we revealed that the tube-structure preference of PFOPy can be interpreted as a characteristic “wavy” conformation of PFOPy on the tube wall that is distinctly different from other polymer dispersants.16 Even though PFOPy was found to have this remarkable chirality selectivity unlike other fluorene-based polymers, the observed PL spectra of the PFOPy-wrapped SWCNTs were not characterized in detail in the previous report.16
The influence of wrapped polymers on SWCNTs has normally been investigated by comparing their PL peak positions with those of SWCNTs dispersed in water by surfactants such as sodium dodecyl sulfate (SDS) or sodium dodecylbenzene sulphonate (SDBS).12–15 Although the emission intensities and spectral shifts in aqueous SWCNT solutions are related to environmental effects due to the surrounding molecules,17 comparisons with SDS- or SDBS-dispersed SWCNTs would not be a suitable way to evaluate the influence of wrapping polymers in organic solvents. Because environmental effects such as dielectric screening are significantly different between organic solvents and water, it is very difficult to distinguish between the effects induced by the solvent and the wrapping polymers in the measured energy shifts.15 Therefore, instead of aqueous solutions, SWCNT organic solutions showing sharp emissions from a variety of semiconducting SWCNTs should be used as reference samples for evaluating energy shifts in the optical transitions of molecule-wrapped SWCNTs in organic solvents.
On the other hand, poly(9,9-di-n-dodecylfluorene) (PFD) can extract semiconducting large-diameter SWCNTs from mixtures of metallic and semiconducting SWCNTs regardless of the tube structure, i.e., chiral indices.18 The observed emission and excitation wavelengths of PFD-wrapped SWCNTs are systematically red-shifted in comparison with SDBS-dispersed SWCNTs. The ability to extract a variety of semiconducting SWCNTs and the systematic variation of the optical transitions associated with the “2n + m” family of SWCNTs suggest that PFD-wrapped SWCNTs can be utilized as a reference material for assigning (n,m) chiral indices to the PL peaks of polymer-wrapped SWCNTs in organic solvents. In addition, the energy shifts in the PL peaks relative to the peak positions of PFD-wrapped SWCNTs could be used to investigate the influence of wrapping molecules in organic solvents on the optical transitions of SWCNTs.
In the current study, we investigated the influence of PFOPy wrapping on the optical transitions of SWCNTs in toluene by using PFD-wrapped SWCNTs as reference materials. We found that emissions from PFOPy-wrapped SWCNTs are blue-shifted in comparison with PFD-wrapped SWCNTs, suggesting that the PFOPy wrapping induces strain in the nanotubes. Moreover, we analyzed the change in the PL peak positions in the photoluminescence excitation (PLE) map as a result of adding different organic solvents to the PFOPy-wrapped SWCNTs and found that the remarkable selectivity of PFOPy to certain tube structures is correlated with the emission energy shifts, suggesting the importance of the rigidity of the fluorene backbone.
Experimental section
Sample preparation
PLV-SWCNTs19 with diameters ranging from 1.0 to 1.4 nm were used as the initial material for polymer-wrapped SWCNTs dispersed in organic solvents. In order to compare the SWCNTs wrapped by structure-selective and non-selective polymers, the PLV-SWCNTs (∼5 mg) were dissolved in toluene (30 mL) with each polymer (∼20 mg): poly[(9,9-dioctylfluorenyl-2,7-diyl)-alt-(2,6-pyridine)] (PFOPy) and poly(9,9-di-n-dodecylfluorenyl-2,7-diyl) (PFD). PFOPy having an average molecular weight (MW) of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and PFD having an MW of 8195 were purchased from Sigma-Aldrich and American Dye Source, respectively. Using the SWCNT-polymer dispersions, we prepared each polymer-wrapped PLV-SWCNT solution via ultracentrifugation of dispersions obtained according to the procedure described in the literature.18 We centrifuged the dispersions for 1 h at 147000 × g and collected the upper 80% of supernatants to remove the bundlings of SWCNTs and metallic SWCNTs or to selectively extract specific semiconducting SWCNTs. To investigate the influence of different solvents on the optical transitions of PFOPy-wrapped SWCNTs, p-xylene solutions of PFOPy- and PFD-wrapped SWCNTs were also prepared via the same ultracentrifugation process as the toluene solution, using the dispersions of PLV-SWCNTs (∼5 mg) in p-xylene (∼30 mL) with PFOPy or PFD (∼20 mg).
000 and PFD having an MW of 8195 were purchased from Sigma-Aldrich and American Dye Source, respectively. Using the SWCNT-polymer dispersions, we prepared each polymer-wrapped PLV-SWCNT solution via ultracentrifugation of dispersions obtained according to the procedure described in the literature.18 We centrifuged the dispersions for 1 h at 147000 × g and collected the upper 80% of supernatants to remove the bundlings of SWCNTs and metallic SWCNTs or to selectively extract specific semiconducting SWCNTs. To investigate the influence of different solvents on the optical transitions of PFOPy-wrapped SWCNTs, p-xylene solutions of PFOPy- and PFD-wrapped SWCNTs were also prepared via the same ultracentrifugation process as the toluene solution, using the dispersions of PLV-SWCNTs (∼5 mg) in p-xylene (∼30 mL) with PFOPy or PFD (∼20 mg).
Spectroscopy
The absorption spectra of the solution samples of polymer-extracted SWCNTs were recorded with a Hitachi U-4100 spectrometer using cells with a 10 mm light path length. PLE spectroscopy was performed with a Shimadzu NIR-PL system with an IR-enhanced InGaAs detector (Princeton instruments, OMA-V2.2) for detection and a tunable Ti:sapphire laser (Spectra Physics, 3900S) for excitation. The PLE data were measured at 5 and 2 nm intervals for excitation and emission, respectively.Simulation
To speculate about the conformations of polymer backbones in PFOPy and PFD adhering to large-diameter SWCNTs with intermediate chiral angles via the π–π interaction, we simulated the configurations of SWCNTs and each oligomer (a fluorene–pyridine sixmer with meta-linkages for PFOPy and a fluorene sixmer with dodecyl alkyl chains for PFD) by a molecular mechanics method with the Optimized Potentials for Liquid Simulations (OPLS) force field using HyperChem (Hypercube, Inc., version 8.0.10). For simulation, (15,4) and (13,5) SWCNTs of about 10 nm in length were used; the ends of the structure of each SWCNT were capped with bonds to hydrogen atoms. The energy of the hybrid system consisting of each oligomer and an SWCNT was minimized using conjugate gradient minimization to a root mean square (RMS) gradient of 0.01 kcal Å−1 mol−1. To obtain optimized final geometries, we simulated the hybrid system with various initial wrapping angles ϕ of each oligomer backbone with respect to the tube axis of each SWCNT: we changed the initial wrapping angle over the range of 0–90° in 1° intervals (for ϕ less than 45°) and 5° intervals (for ϕ more than 45°).Results and discussion
Fig. 1 shows the absorption spectra of PFOPy- and PFD-wrapped SWCNTs in toluene. To prepare polymer-wrapped SWCNT solutions, we used SWCNTs synthesized by pulsed-laser vaporization (PLV) as the initial material, the diameters of which were distributed over a range of 1.0–1.4 nm. Each polymer-wrapped SWCNT solution was extracted via ultracentrifugation of polymer-SWCNT dispersions at 147000 × g for 1 h. Similarly to SDS- and SDBS-dispersed SWCNT solutions,16,20 polymer-SWCNT dispersions before ultracentrifugation showed not only absorption bands of semiconducting SWCNTs but also an absorption band with two peaks centered at about 648 and 695 nm because of the presence of metallic SWCNTs (see Fig. S1 of ESI†). The absorption band (M1) due to the first optical transitions (the interband transitions between the first van Hove singularities) of metallic SWCNTs was slightly red-shifted compared with the surfactant-dispersed SWCNT solutions because of the different surrounding materials.21 In contrast, PFD- and PFOPy-wrapped SWCNT solutions after ultracentrifugation did not show M1 absorption (Fig. 1). Moreover, structured absorption bands (S2) due to the second optical transitions (the interband transitions between the second van Hove singularities) of semiconducting SWCNTs were clearly observed in the wavelength range of 800–1100 nm. These dispersions had a relatively low absorption background compared with the polymer-SWCNT dispersions before ultracentrifugation; the low background would be due to the removal of bundled SWCNTs and metallic SWCNTs.22,23 However, one should note that almost the same optical transition energies in large-diameter tubes inhibit deconvolution of the absorption bands into different semiconducting (n,m) species. This means PLE spectroscopy rather than absorption measurements would be a more effective way to quantify the influence of polymer wrapping on the optical transitions of the large-diameter SWCNTs, because PLE can better resolve each optical transition of the (n,m) SWCNTs.
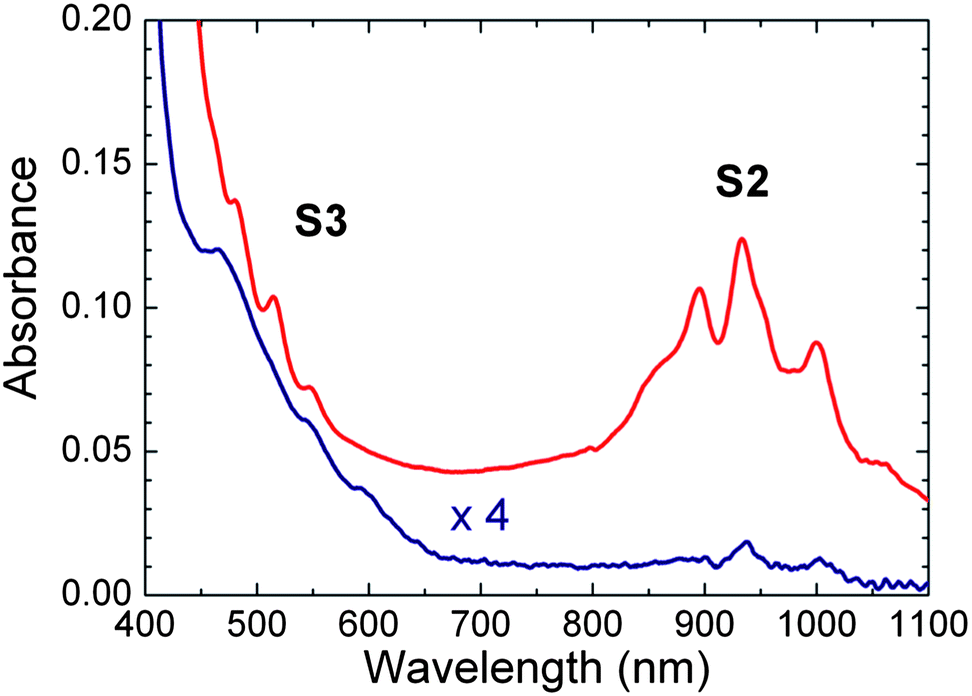 | ||
| Fig. 1 Absorption spectra of PFD- (red line) and PFOPy-extracted SWCNTs (blue line) in toluene. The absorption bands indicated by S3 and S2 are respectively attributed to the third and second optical transitions of semiconducting tubes. | ||
The PLE map of the PFD-extracted SWCNTs is shown in Fig. 2(a) (closed circles). The PL peaks associated with the tube structures of the semiconducting SWCNTs appeared as emissions at the first optical transitions caused by excitations of the second optical transitions. As a standard approach for characterizing PL peaks of SWCNTs in the PLE maps, each spectrum (in units of energy) was deconvoluted with Lorentzian line shapes to obtain the peak energies. The obtained PL peaks were assigned different chiral indices (n,m) by using the empirical functions of Weisman and Bachilo.24 Similarly to SDS-dispersed SWCNTs (crosses in Fig. 2(a)),16 the PFD-wrapped SWCNTs exhibited various emission peaks associated with a variety of tube structures. The red-shifted emissions relative to the PL peaks of the SDS-dispersed SWCNTs could be attributed to the change in the local dielectric constant around the SWCNTs due to the different dispersant and solvent (i.e., fluorene-based polymers and toluene). Furthermore, the emission peaks were made sharp by the removal of the metallic SWCNTs, and this allowed us to estimate the peak energies with a low experimental error (less than ± 1 meV).18 Therefore, instead of surfactant-dispersed SWCNTs, we used PFD-wrapped SWCNTs as a reference sample for characterizing the organic solutions of PFOPy-wrapped SWCNTs.
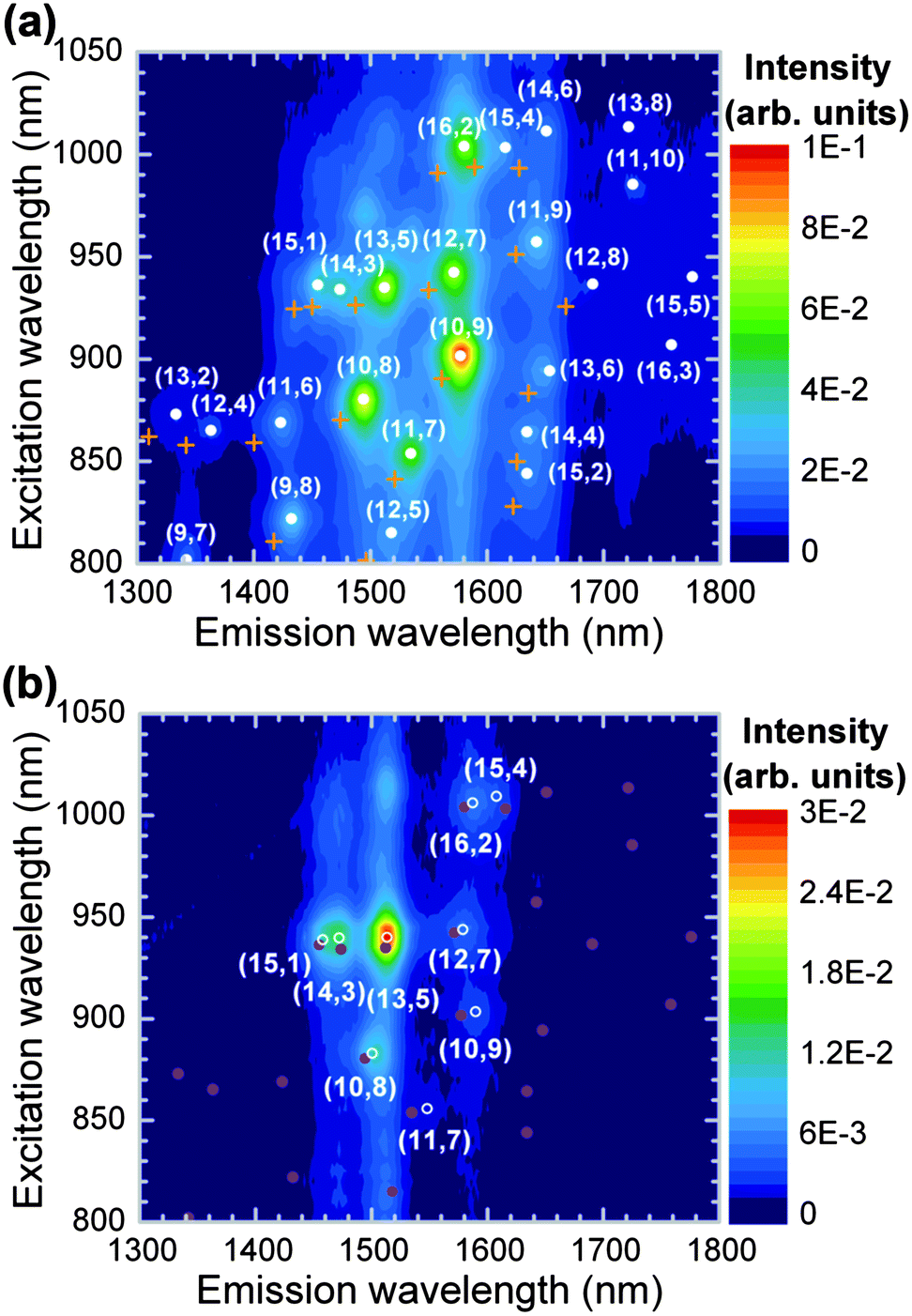 | ||
| Fig. 2 PLE maps of (a) PFD- and (b) PFOPy-extracted SWCNTs in toluene. In panel (a), the PL peak positions of SDS-dispersed PLV-SWCNTs16 are indicated by crosses. Closed and open symbols respectively represent the PL peak positions of PFD- and PFOPy-extracted SWCNTs. | ||
Fig. 2(b) shows the PLE map of the PFOPy-extracted SWCNTs in toluene. In contrast to the PLE map of the PFD-extracted SWCNTs showing various emission peaks from semiconducting SWCNTs with a variety of tube structures, sharp emission peaks from specific semiconducting SWCNTs, such as (13,5), (14,3), and (10,8) tubes, can be seen in this PLE map. The PFOPy-extracted SWCNTs have diameters in a range from 1.23 nm to 1.38 nm, showing a narrow diameter distribution: 0.15 nm.16 In particular, (13,5) tubes exhibit intense emissions; the θ and dt are 15.6° and 1.28 nm, respectively. Normally, chiral-angle selectivity of fluorene-based polymers depends on the rigid structure of aromatic rings in the polymers because relatively strong π–π interaction between polymers and specific SWCNTs is important for selective extraction. For example, the fluorene backbone of PFO prefers tube structures of near-armchair SWCNTs having large chiral angles close to θ = 30°,25 while the introduction of other aromatic rings into fluorene units leads to chiral-angle preference for SWCNTs with smaller chiral angles as reported in F8BT and other selective copolymers.18,26 The chiral-angle selectivity of PFOPy for SWCNTs with intermediate chiral angles such as (13,5) and (14,3) tubes may also be attributed to the introduction of a pyridine moiety into fluorene units. On the other hand, the remarkable tube-diameter selectivity of PFOPy would be explained with a wavy geometry of the polymer backbone. A molecular mechanical simulation indicated that the polymer backbone in PFOPy was like a wavy chain, different from the coil-like chain27,28 in PFD forming helical wrapping (see Fig. S2 of ESI† for the polymer conformations). The width of the wavy chain is expected to prefer a specific tube diameter when the wavy backbones align along the tube axis or form long-pitch helices. The simulation for SWCNTs with different diameters suggests that the amplitude of the wavy chain matches the diameter of a (13,5) tube rather than that of a (15,4) tube, which has a larger diameter: 1.38 nm. Moreover, smaller-diameter SWCNTs such as (9,7) tubes would deviate part of the PFOPy backbone from the tube surface because of a mismatch between the wavy backbone and the tube diameter, and weaken the interaction with the wrapping polymers.16 These results can be interpreted as that the wavy backbone of PFOPy selectively interacts with tube structures of SWCNTs with large diameters close to the diameter of (13,5) tubes: dt = 1.28 nm. For example, the diameter of (14,3) tubes extracted by PFOPy is 1.25 nm and similar to that of (13,5) tubes (see Fig. S3 of ESI† for the tube-diameter preference of PFOPy).
To investigate the influence of the PFOPy wrapping on the optical transitions of SWCNTs in detail, Fig. 3(a) plots the difference (ΔE11) in emission energy of the PL peak between PFOPy- and PFD-wrapped (n,m) SWCNTs as a function of the chiral angle of (n,m) SWCNTs. The shift in the emission energy of the PFOPy-wrapped SWCNTs relative to PFD-wrapped ones is negative for all tubes, except the (14,3) and (15,4) tubes. The ΔE11 values of (12,7) and (10,8) type-I tubes [(2n + m) mod 3 = 1] are slightly larger than those of (11,7) and (10,9) type-II tubes [(2n + m) mod 3 = 2]. This weak tube-type dependence is consistent with the results of an environmental effects' study by Ohno et al.,29 which suggest that the type dependence in the environmental effect on the optical transition energies is due to a difference in the effective mass. In addition, such a weak dependence was also found in a study on the influence of dielectric screening of excitons on optical transition energies.30
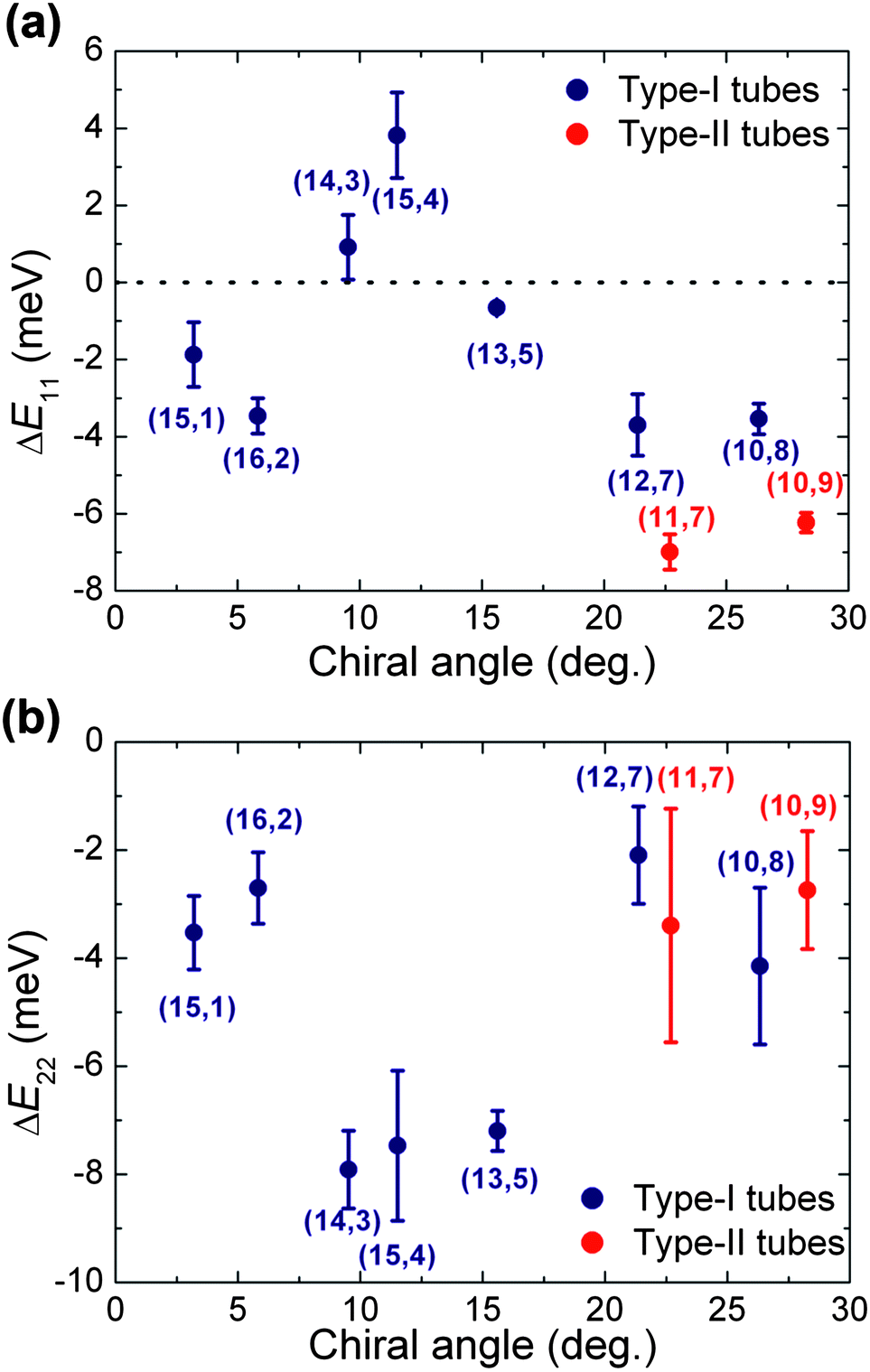 | ||
| Fig. 3 Energy shifts of the (a) first and (b) second optical transitions in PFOPy-wrapped SWCNTs relative to those of PFD-wrapped SWCNTs in toluene as a function of the chiral angle. | ||
Fig. 3(b) shows the energy shift (ΔE22) of the second optical transition of the PFOPy-wrapped SWCNTs relative to that of the PFD-wrapped SWCNTs. Almost all (n,m) species of PFOPy-wrapped SWCNTs exhibit the second optical transition energies that are lower by 2–4 meV, whereas the second optical transitions of specific PFOPy-wrapped SWCNTs with intermediate chiral angles, such as the (14,3) and (15,4) tubes, are shifted further toward the lower energy range; ΔE22 shifted down to about −8 meV. Obviously, the energy shifts in the (14,3) and (15,4) tubes are in the opposite direction to those of the first optical transition and are similar to the transition-dependent energy shifts due to strain.2
Adhesion of molecules with large π-planes to SWCNTs induces opposite energy shifts for the first and second optical transitions via strain of SWCNTs.4 Such opposite energy shifts were reported in near-armchair SWCNTs wrapped by other selective polymers including anthracene moieties by Mistry et al.31 They explained the energy shifts with a change in the local dielectric environment around SWCNTs. However, the explanation disagrees with the fact that a variation in dielectric constant around SWCNTs leads to energy shifts of the first and second optical transitions in the same directions.3,29,32 Not only the introduction of aromatic ring structures with relatively large π-planes into fluorene units, but also the wavy backbone revealed by the molecular mechanical simulation may strain SWCNTs with specific chiral angles in terms of the rigidity of polymer backbones. Interestingly, the (14,3) and (15,4) tubes showing the opposite energy shifts have similar chiral angles to each other (see Fig. 3(a) and Fig. S3 of ESI†). If we suppose that the PFOPy wrapping slightly contracts the SWCNTs along the tube axis, owing to its wavy backbone which may act as a spring, the energy shifts with opposite signs for excitations and emissions would be consistent with strain-induced changes in the energy of the SWCNT optical transitions.5
Similar shifts in the optical transition energies have been reported in chemically doped SWCNTs such as oxygen-doped SWCNTs33 and in polymer-wrapped SWCNTs, which form type-II heterojunctions between the polymers and SWCNTs on the basis of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of the polymers.15,4 However, these two kinds of energy shift can be ruled out. The emissions in oxygen-doped SWCNTs are shifted by a few hundred wavelengths owing to excitons becoming trapped at oxygen sites.33 The spectral shifts are significantly larger than those of PFOPy-wrapped SWCNTs. Moreover, the energy levels of PFOPy are such that they would form a type-I heterojunction with the SWCNTs.34 Except for the dielectric screening and strain effects, the first optical transitions of SWCNTs are insensitive to the type-I heterojunction.4 Therefore, the blue-shifted emissions in the PFOPy-wrapped (15,4) and (14,3) tubes cannot be explained in terms of oxygen doping or formation of a type-II heterojunction between polymers and SWCNTs. In addition, the SWCNTs in the above two situations did not show a significant change in the second optical transitions.33,4
If the PFOPy with the wave-like chain could be extended in the direction perpendicular to the tube axis (as shown in Fig. S2 of ESI†), the induced strain would be able to be estimated by using a theoretical equation. According to the study by Yang and Han,35 the strain along the tube axis can be expressed by the equation σ = ΔE11/[sgn(2p + 1)3t0(1 + ν)cos3θ], where t0 is the tight-binding overlap integral for the nearest-neighbor π-bonds, ν is the Poisson ratio, and the factor p is −1 for type-I tubes and 1 for type-II tubes. When the strain is estimated using the equation with t0 = 3 eV and ν = 0.2,36 uniaxial strains of about −0.04 and −0.01% are respectively deduced from the emission energy shifts of (15,4) and (14,3) SWCNTs. The estimated strain in the PFOPy-wrapped SWCNTs in solution is too small for resonant Raman spectroscopy to determine whether there are spectral shifts of the G-band or radial breathing modes due to strain;37 the strain is smaller by one order of magnitude than that of solution samples of SWCNTs encapsulating C60 fullerenes.2
Intriguingly, the change from toluene to p-xylene leads to noticeable differences in the chirality selectivity of PFOPy and the energy shifts of the optical transitions. Fig. 4(a) and (b) show PLE maps in PFD- and PFOPy-wrapped SWCNTs in p-xylene. The use of p-xylene suppressed the tube-structure selectivity in PFOPy, as can be seen from the emissions of other large-diameter (11,9), (13,6), and (14,4) tubes. The diameters of the extracted (11,9), (13,6), and (14,4) tubes were 1.38, 1.34, and 1.30 nm, respectively. Moreover, the diameter of the (10,8) tubes, which showed a stronger contribution to the PLE map in p-xylene, was 1.24 nm and similar to that of the (13,5) tubes which were the most prominent species in the PLE map of PFOPy-wrapped SWCNTs. The diameters of the PFOPy-wrapped SWCNTs in p-xylene were distributed over 1.23–1.38 nm. The toluene solution had a similar narrow diameter distribution.16 Therefore, changing the solvent had almost no effect on the diameter distribution of the PFOPy-extracted SWCNTs. The remarkable tube-diameter selectivity for SWCNTs with dt = 1.23–1.38 nm may thus be attributed to the wavelike chain of the polymer backbone formed by the meta-linkage of pyridine units;38,39 the extended π-plane of the backbone preferentially interacts with the side walls of the large-diameter SWCNTs via the π–π interaction. The width of the conformational modulation in the wavelike chain was estimated to be about 1 nm on the basis of a molecular mechanical simulation of a fluorene–pyridine oligomer. This wide wavelike chain would facilitate matching between PFOPy and specific large-diameter SWCNTs and lead to tube-diameter selectivity.
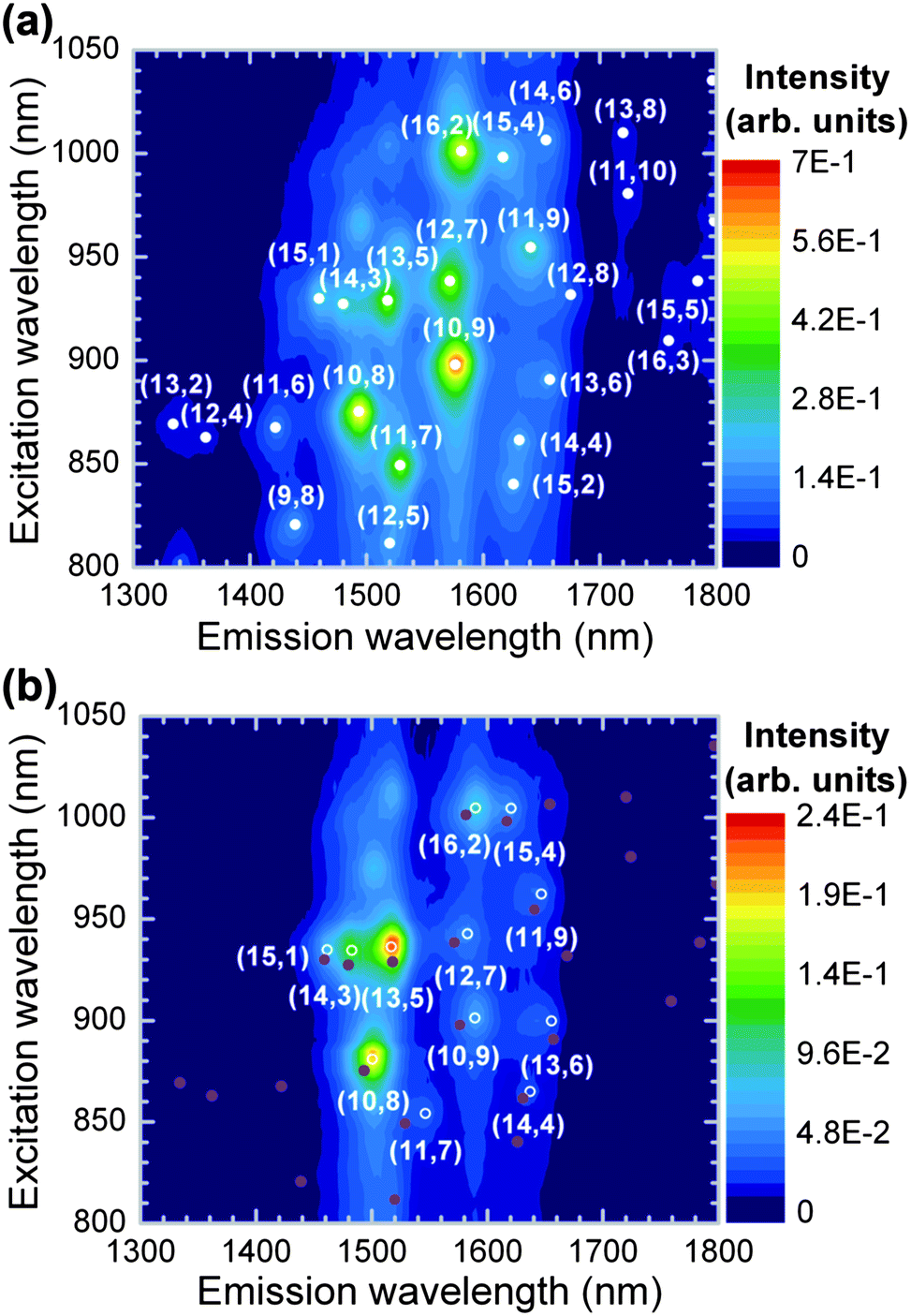 | ||
| Fig. 4 PLE maps of (a) PFD- and (b) PFOPy-extracted SWCNTs in p-xylene. For comparison, the PL peak positions of the PFD-extracted SWCNTs are indicated by closed circles in panel (b). | ||
Although the tube-diameter selectivity of PFOPy was observed in solutions with p-xylene as well as toluene, the chirality selectivity of PFOPy, in particular, the chiral-angle selectivity for SWCNTs with intermediate chiral angles, was obviously suppressed in p-xylene, and the suppression resulted not only in extracting (11,9) tubes but also in exhibiting intense emissions of (10,8) tubes. Such a suppression also occurs in PFO-wrapped SWCNTs when the solvent is changed from toluene to other solvents.27
The chain geometry of the polymer backbone in solution is dependent on the solvent used. In fluorene-based polymers, the β-phase associated with an extended chain conformation, i.e., the rigid backbone of the polymer is formed when the polymers are exposed to moderately poor solvent, while the phase is suppressed in good solvent.40 Therefore, in a solvent, such as p-xylene, in which polymers are more soluble, the polymer backbone may be weakened and be rather flexible in comparison with that in toluene solution. The reduced rigidity is expected to make the polymer less selective.27 In fact, the change from toluene to p-xylene led to a one-order-of-magnitude increase in the absorption intensity of PFOPy-extracted SWCNTs (see Fig. S4 of ESI† on the absorption spectra). Simultaneously, PFOPy in p-xylene additionally extracted not only SWCNTs with intermediate chiral angles ((13,6) and (14,4) tubes with θ = 18.0° and 12.2°), but also near-armchair SWCNTs ((11,9) tubes with θ = 26.7°).
A change in the rigidity of the polymer backbone presumably leads to a variation in the interactions between polymers and SWCNTs. If the rigidity of the PFOPy backbone is weakened by changing the solvent, the energy shifts of the optical transitions in PFOPy-wrapped SWCNTs may also change.
To quantitatively visualize the energy shifts of PFOPy-wrapped (14,3) and (15,4) SWCNTs in p-xylene, we plotted the energy shifts (ΔE11 and ΔE22) of the first and second optical transitions of PFOPy-wrapped SWCNTs relative to those of PFD-wrapped SWCNTs in p-xylene as a function of the chiral angle (Fig. 5(a) and (b)). Normally, the change from toluene to p-xylene affects the selectivity of the polymer for specific SWCNTs, but almost does not affect the PL peak positions of SWCNTs because the dielectric constants of the solvents are similar to each other: 2.38 for toluene and 2.27 for p-xylene.27,41 As shown in Fig. 5(a), the trend in emission energy shifts of other (n,m) SWCNTs besides (14,3) and (15,4) SWCNTs is similar to the one governed by the dielectric screening effect in the toluene solution.
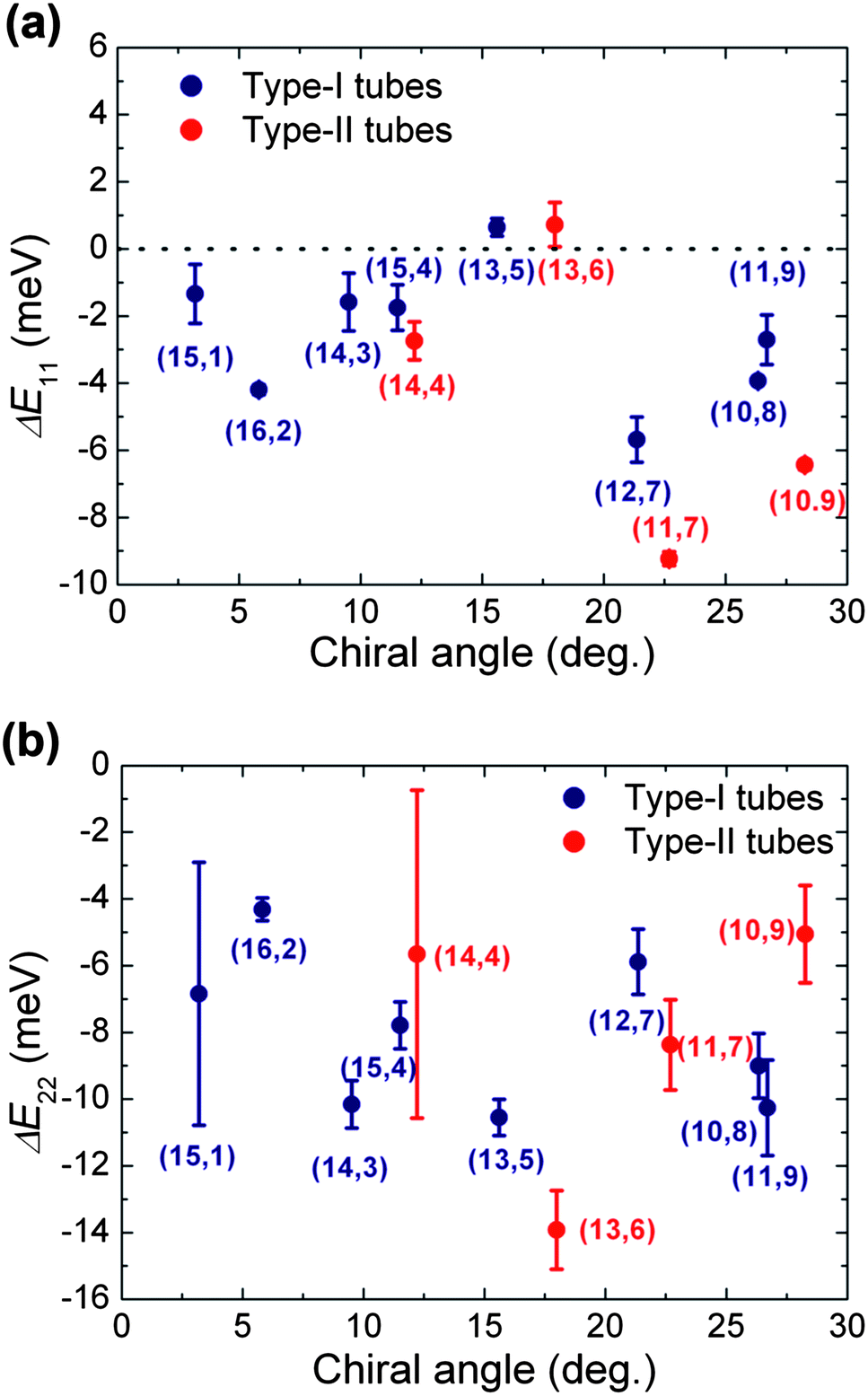 | ||
| Fig. 5 Energy shifts of the (a) first and (b) second optical transitions of PFOPy-wrapped SWCNTs in p-xylene relative to those of PFD-wrapped ones in p-xylene as a function of the chiral angle. | ||
Meanwhile, the change in solvent had a large effect on the energy shifts of PL peaks of the (14,3) and (15,4) SWCNTs in the solution. The emission energies of PFOPy-wrapped (14,3) and (15,4) SWCNTs shifted by about −2 meV in p-xylene, but from 1 to 4 meV in toluene (Fig. 3(a)). The emissions of the SWCNTs were not obviously blue-shifted in the p-xylene solution. Blue shifts in the emissions of PFOPy-wrapped SWCNTs were obviously suppressed in the p-xylene solution.
The trend in the excitation energy shifts (ΔE22) of PFOPy-wrapped (14,3) and (15,4) SWCNTs in p-xylene was also different from those in the toluene solution. The excitation energy shifted lower by about 8–10 meV, as shown in Fig. 5(b). Contrary to the characteristic energy shifts in the toluene solution, the excitation energy shifts of (14,3) and (15,4) SWCNTs in the p-xylene solution did not show striking deviations from those of other (n,m) species. The energy shifts of the first and second optical transitions in the same directions suggest that the energy shifts of optical transitions in the p-xylene solution are governed mainly by the variation in the environmental dielectric constant, i.e., the dielectric screening effect.
Conclusions
The emissions of selective PFOPy-wrapped SWCNTs were found to be blue-shifted in toluene in comparison with those of non-selective PFD-wrapped SWCNTs. Meanwhile, the blue shifts in the emissions were suppressed when the solvent was changed from toluene to p-xylene. Normally, in selective polymers such as PFO and F8BT,27 the rigidity of the polymer backbone is dependent on the solvent used and changes the selectivity for specific SWCNTs. In the case of PFOPy, changing the solvent from toluene to p-xylene made PFOPy less selective and increased the absorption intensity of PFOPy-extracted SWCNTs by one order of magnitude. The interpretation that the blue-shifted emissions result from the strain in SWCNTs caused by PFOPy wrapping leads us to conclude that the blue shifts in the emissions are suppressed as a result of p-xylene weakening the polymer backbone.Acknowledgements
This work was partly supported by a Grant-in-Aid for Young Scientists (B) (#24760607).Notes and references
- T. Hertel, A. Hagen, V. Talalaev, K. Arnold, F. Hennrich, M. Kappes, S. Rosenthal, J. McBride, H. Ulbricht and E. Flahaut, Nano Lett., 2005, 5, 511 CrossRef CAS PubMed.
- S. Okubo, T. Okazaki, N. Kishi, S.-K. Joung, T. Nakanishi, S. Okada and S. Iijima, J. Phys. Chem. C, 2009, 113, 571 CAS.
- Y. Ohno, S. Iwasaki, Y. Murakami, S. Kishimoto, S. Maruyama and T. Mizutani, Phys. Status Solidi B, 2007, 244, 4002 CrossRef CAS.
- S. D. Stranks, J. K. Sprafke, H. L. Anderson and R. J. Nicholas, ACS Nano, 2011, 5, 2307 CrossRef CAS PubMed.
- T. K. Leeuw, D. A. Tsyboulski, P. N. Nikolaev, S. M. Bachilo, S. Arepalli and R. B. Weisman, Nano Lett., 2008, 8, 826 CrossRef CAS PubMed.
- S. Cambré, S. M. Santos, W. Wenseleers, A. R. T. Nugraha, R. Saito, L. Cognet and B. Lounis, ACS Nano, 2012, 6, 2649 CrossRef PubMed.
- S. Chiashi, S. Watanabe, T. Hanashima and Y. Homma, Nano Lett., 2008, 8, 3097 CrossRef CAS PubMed.
- M. J. O'Connell, S. M. Bachilo, C. B. Huffman, V. C. Moore, M. S. Strano, E. H. Haroz, K. L. Rialon, P. J. Boul, W. H. Noon, C. Kittrell, J. Ma, R. H. Hauge, R. B. Weisman and R. E. Smalley, Science, 2002, 297, 593 CrossRef CAS PubMed.
- D. A. Heller, E. S. Jeng, T.-K. Yeung, B. M. Martinez, A. E. Moll, J. B. Gastala and M. S. Strano, Science, 2006, 311, 508 CrossRef CAS PubMed.
- L.-J. Li, R. J. Nicholas, R. S. Deacon and P. A. Shields, Phys. Rev. Lett., 2004, 93, 156104 CrossRef.
- Z. Li, P. Dharap, S. Nagarajaiah, E. V. Barrera and J. D. Kim, Adv. Mater., 2004, 16, 640 CrossRef CAS.
- V. C. Moore, M. S. Strano, E. H. Haroz, R. H. Hauge, R. E. Smalley, J. Schmidt and Y. Talmon, Nano Lett., 2003, 3, 1379 CrossRef CAS.
- W. Yi, A. Malkovskiy, Q. Chu, A. P. Sokolov, M. L. Colon, M. Meador and Y. Pang, J. Phys. Chem. B, 2008, 112, 12263 CrossRef CAS PubMed.
- S. G. Chou, H. B. Ribeiro, E. B. Barros, A. P. Santos, D. Nezich, G. G. Samsonidze, C. Fantini, M. A. Pimenta, A. Jorio, F. P. Filho, M. S. Dresselhaus, G. Dresselhaus, R. Saito, M. Zheng, G. B. Onoa, E. D. Semke, A. K. Swan, M. S. Ünlü and B. B. Goldberg, Chem. Phys. Lett., 2004, 397, 296 CrossRef CAS PubMed.
- T. Schuettfort, A. Nish and R. J. Nicholas, Nano Lett., 2009, 9, 3871 CrossRef CAS PubMed.
- M. Tange, T. Okazaki and S. Iijima, ACS Appl. Mater. Interfaces, 2012, 4, 6458 CAS.
- R. K. Wang, W.-C. Chen, D. K. Campos and K. J. Ziegler, J. Am. Chem. Soc., 2008, 130, 16330 CrossRef CAS PubMed.
- M. Tange, T. Okazaki and S. Iijima, J. Am. Chem. Soc., 2011, 133, 11908 CrossRef CAS PubMed.
- T. Okazaki, T. Saito, K. Matsuura, S. Ohshima, M. Yumura, Y. Oyama, R. Saito and S. Iijima, Chem. Phys. Lett., 2006, 420, 286 CrossRef CAS PubMed.
- T. Okazaki, S. Okubo, T. Nakanishi, S.-K. Joung, T. Saito, M. Otani, S. Okada, S. Bandow and S. Iijima, J. Am. Chem. Soc., 2008, 130, 4122 CrossRef CAS PubMed.
- C. A. Silvera-Batista, R. K. Wang, P. Weinberg and K. J. Ziegler, Phys. Chem. Chem. Phys., 2010, 12, 6990 RSC.
- Y. Miyauchi, H. Hirori, K. Matsuda and Y. Kanemitsu, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 081410(R) CrossRef.
- A. V. Naumov, S. Ghosh, D. A. Tsyboulski, S. M. Bachilo and R. B. Weisman, ACS Nano, 2011, 5, 1639 CrossRef CAS PubMed.
- R. B. Weisman and S. M. Bachilo, Nano Lett., 2003, 3, 1235 CrossRef CAS.
- A. Nish, J.-Y. Hwang, J. Doig and R. J. Nicholas, Nat. Nanotechnol., 2007, 2, 640 CrossRef CAS PubMed; F. Chen, B. Wang, Y. Chen and L.-J. Li, Nano Lett., 2007, 7, 3013 CrossRef PubMed.
- H. Ozawa, T. Fujigaya, Y. Niidome, N. Hotta, M. Fujiki and N. Nakashima, J. Am. Chem. Soc., 2011, 133, 2651 CrossRef CAS PubMed.
- J.-Y. Hwang, A. Nish, J. Doig, S. Douven, C.-W. Chen, L.-C. Chen and R. J. Nicholas, J. Am. Chem. Soc., 2008, 130, 3543 CrossRef CAS PubMed.
- H. Wang, J. Mei, P. Liu, K. Schmidt, G. Jiménez-Osés, S. Osuna, L. Fang, C. J. Tassone, A. P. Zoombelt, A. N. Sokolov, K. N. Houk, M. F. Toney and Z. Bao, ACS Nano, 2013, 7, 2659 CrossRef CAS PubMed.
- Y. Ohno, S. Iwasaki, Y. Murakami, S. Kishimoto, S. Maruyama and T. Mizutani, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 235427 CrossRef.
- O. Kiowski, S. Lebedkin, F. Hennrich, S. Malik, H. Rösner, K. Arnold, C. Sürgers and M. M. Kappes, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 075421 CrossRef.
- K. S. Mistry, B. A. Larsen and J. L. Blackburn, ACS Nano, 2013, 7, 2231 CrossRef CAS PubMed.
- Y. Ohno, S. Maruyama and T. Mizutani, in Carbon Nanotubes, ed. J. M. Marulanda, InTech, 2010, p. 109 Search PubMed.
- S. Ghosh, S. M. Bachilo, R. A. Simonette, K. M. Beckingham and R. B. Weisman, Science, 2010, 330, 1656 CrossRef CAS PubMed.
- J. Li, T. Sano, Y. Hirayama, T. Tomita, H. Fujii and K. Wakisaka, Jpn. J. Appl. Phys., 2006, 45, 5232 CrossRef CAS.
- L. Yang and J. Han, Phys. Rev. Lett., 2000, 85, 154 CrossRef CAS.
- K. Arnold, S. Lebedkin, O. Kiowski, F. Hennrich and M. M. Kappes, Nano Lett., 2004, 4, 2349 CrossRef CAS.
- S. B. Cronin, A. K. Swan, M. S. Ünlü, B. B. Goldberg, M. S. Dresselhaus and M. Tinkham, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 035425 CrossRef.
- L. E. Horsburgh, A. P. Monkman and I. D. W. Samuel, Synth. Met., 1999, 101, 113 CrossRef CAS.
- S.-P. Liu, S.-C. Ng and H. S. O. Chan, Synth. Met., 2005, 149, 1 CrossRef CAS PubMed.
- M. Grell, D. D. C. Bradley, X. Long, T. Chamberlain, M. Inbasekaran, E. P. Woo and M. Soliman, Acta Polym., 1998, 49, 439 CrossRef CAS.
- J. Gao, W. Gomulya and M. A. Loi, Chem. Phys., 2013, 413, 35 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Absorption spectra of PFOPy- and PFD-dispersed PLV-SWCNTs in toluene (before ultracentrifugation), simulated configurations of the SWCNT and each oligomer, a graphene sheet map showing the tube species of PFOPy-extracted SWCNTs, absorption spectra of PFOPy-extracted PLV-SWCNTs in toluene and p-xylene (after ultracentrifugation), and data on the PL peak positions of PFD-wrapped PLV-SWCNTs. See DOI: 10.1039/c3nr03812b |
| This journal is © The Royal Society of Chemistry 2014 |