Self-activation and activation of Cu(0) wire for SET-LRP mediated by fluorinated alcohols
Shampa R.
Samanta
a,
Hao-Jan
Sun
a,
Athina
Anastasaki
ab,
David M.
Haddleton
b and
Virgil
Percec
*a
aRoy & Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6323, USA. E-mail: percec@sas.upenn.edu
bDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK
First published on 1st August 2013
Abstract
Herein we report the self-activation and activation of Cu(0) wire used to form a catalyst in single-electron transfer living radical polymerization (SET-LRP) in two fluorinated alcohols employed as solvents, 2,2,2-trifluoroethanol (TFE) and 2,2,3,3-tetrafluoropropanol (TFP). Self-activation and activation of Cu(0) wire by TFP exhibit a stronger increase of the apparent rate constant of propagation, kappp, compared to TFE. SET-LRP of methyl acrylate (MA) in DMSO catalyzed with TFP-activated Cu(0) wire showed a comparable kappp value to that of the hydrazine-activated Cu(0) wire-catalyzed counterpart. SET-LRP catalyzed with Cu(0) wire pretreated with TFE or TFP, and the in situactivation of Cu(0) wire while using TFE or TFP as solvent, proceeded with no initial induction period and exhibits predictable molecular weight evolution with conversion and narrow molecular weight distribution.
Introduction
In the field of Living Radical Polymerization (LRP), Cu(0) mediated single-electron transfer living radical polymerization (SET-LRP)1 is one of the most robust techniques. It is noteworthy that SET-LRP can mediate even the LRP of vinyl chloride1–6 which is claimed to be inert towards Cu(I) catalyzed atom transfer radical polymerization (ATRP).7 In addition, polymerization of a large diversity of monomers, including acrylates,1,3,8–24 methacrylates,3,25–34acrylamides,35–41 methacrylamides,42acrylonitrile,43,44 and other monomers45,46 at ambient temperature with retention of near-perfect chain-end functionality9,10,47–51 can be achieved by SET-LRP. The crucial step in SET-LRP is the disproportionation13,50,52–56 of the in situ generated Cu(I)X into Cu(0) and Cu(II)X2 in the presence of N-containing donor ligands (L),1–3,57,58 which can be achieved in various polar solvents, such as H2O,11,42,45,59–61dimethyl sulfoxide (DMSO),9,10,13,28,31,45,50,62–64dimethyl formamide (DMF),56,60dimethyl acetamide (DMAC),56,60 ethylene carbonate,56,60alcohols,29,34,62,65,66 their binary mixtures,60,67 and even blood serum.68In the development of SET-LRP, we have discovered that Cu(0) in the form of nano-powder or wire12,64,69 can be used to form a catalyst for the SET-LRP and the apparent rate of propagation (kappp) is directly proportional to the surface area of the copper used in the reaction irrespective of its macrostructure.12 This also supports the fact that the “nascent” Cu(0) nanopowder prepared from in situ disproportionation of Cu(I)X/L is the most active form of copper for SET-LRP which leads to kappp higher than those reported for the smallest commercial Cu(0) nanopowders or the highest surface area of the Cu(0) wire investigated.13 However, in a recent report70 our group has demonstrated that commercial Cu(0) wire can be activated for SET-LRP by pretreatment with hydrazine under inert gas. This process removes the oxide layer present on the surface of the wire via the reduction of CuO or CuO2 to Cu(0). A simultaneous increase of the porosity and of the surface of the Cu(0) wire is accomplished.70 This activation results in SET-LRP with a kappp value almost twice of that obtained using non-activated Cu(0) wire and also an improved control of molecular weight evolution and distribution in addition to the removal of an induction time observed for the non-activated Cu(0) wire. These findings suggest that the activation of the Cu(0) catalyst is an excellent tool to obtain fast and more efficient SET-LRP which eventually expands its applicability even for less reactive monomers. Treatment of commercial Cu(0) wire with a concentrated acid71 such as nitric acid, hydrochloric acid and acetic acid also results in the activation of the Cu(0) wire by dissolution and removal of the CuO or CuO2 layer from the surface of the wire. Hydrazine treatment activates the Cu(0) wire by reducing the copper oxide present on its surface to Cu(0) and subsequently increases the surface area of the catalyst and accelerates the rate of polymerization by almost two fold.54,70 By contrast, the acid dissolution method71 shows no significant enhancement of the kappp value compared to its non-activated Cu(0) wire-catalyzed counterpart since in this case the reduced wire dimensions resulting from the consumption of the top layer copper atoms are not accompanied by an enhancement of surface roughness. In addition, prolonged etching of the Cu(0) film by strong acids results in the disappearance of the fine structure of the Cu wire surface.71,72 Nevertheless, the removal of the induction time and a more controlled molecular weight evolution are obtained as a result of the absence of the oxide layer from the Cu(0) wire surface by both hydrazine-activation and acid-dissolution methods.70,71
Recently our laboratory reported that34,65,66 two fluorinated alcohols, 2,2,2-trifluoroethanol (TFE) and 2,2,3,3-tetrafluoropropanol (TFP), show efficient disproportionation of Cu(I)Br in the presence of Me6-TREN resulting in “nascent” Cu(0) nanoparticles and Cu(II)Br2–Me6-TREN complexes and provide excellent SET-LRP media for both hydrophobic and hydrophilic acrylates and methacrylates with excellent control of molecular weight distribution and near-perfect retention of chain-end functionality.34,65,66 This demonstrates the absence of any detectable amount of bimolecular coupling reaction of bromine terminated polymers during SET-LRP of the acrylate and methacrylate monomers in TFE and TFP.73 While exploring these fluorinated alcohols as solvents for SET-LRP it was revealed that they have the ability to activate the Cu(0) wire used as a catalystin situ towards SET-LRP.
Pre-treatment of the Cu(0) wire by TFP and TFE results in an enhanced kappp value for SET-LRP of MA in DMSO compared to that of the non-activated wire under similar conditions and removal of the induction time. As a result, whilst TFE and TFP were used as solvents for SET-LRP with commercial Cu(0) wire as catalyst they act as self-activating solvents. In this report, we have investigated the effect of activation of the Cu(0) wire by TFP and TFE for SET-LRP of MA with methyl 2-bromopropionate (MBP) as initiator, hexamethylated tris(2-aminoethyl)amine (Me6-TREN) as ligand, and DMSO as solvent, as well as the in situactivation of Cu(0) wire while using TFP and TFE as solvents under similar reaction conditions (Scheme 1).
 | ||
| Scheme 1 SET-LRP of methyl acrylate (MA) initiated with methyl 2-bromopropionate (MBP), Me6-TREN as ligand, and catalyzed with the non-activated or activated Cu(0) wire with TFE, TFP or hydrazine. | ||
Results and discussion
Self-activation of Cu(0) wire by TFE and TFP in situ
SET-LRP of MA with MBP as an initiator and catalyzation with 4.5 cm of non-activated, hydrazine activated and HCl-activated Cu(0) wire in the presence of Me6-TREN ligand with a molar ratio of [MA]0/[MBP]0/[Me6-TREN]0 = 222/1/0.1 in TFP (Fig. 1), and 2.0 cm of non-activated, hydrazine activated Cu(0) wire in TFE were conducted at 25 °C. In kinetic experiments, evolution of the number average molecular weight (Mn) with conversion and molecular weight distribution of the resulting polymer demonstrated that in all the cases polymerization was living with an excellent agreement between the theoretical and experimental molecular weights. The experimental values of the apparent rate constant of propagation (kappp), theoretical molecular weight (Mth) vs. Mn, and initiator efficiency (Ieff) are shown in the kinetic plots, Fig. 1(a)–(f) for TFP and Fig. 2(a)–(d) for TFE. It is noteworthy that while TFP was used as the solvent, upon activation of the Cu(0) wire by hydrazine, kappp increased only by 1.25 times (from 0.012 min−1 to 0.015 min−1, Fig. 1(a) and (c)). In the case of TFE this increment is 1.30 times (from 0.0085 min−1 to 0.011 min−1, Fig. 2(a) and (c)).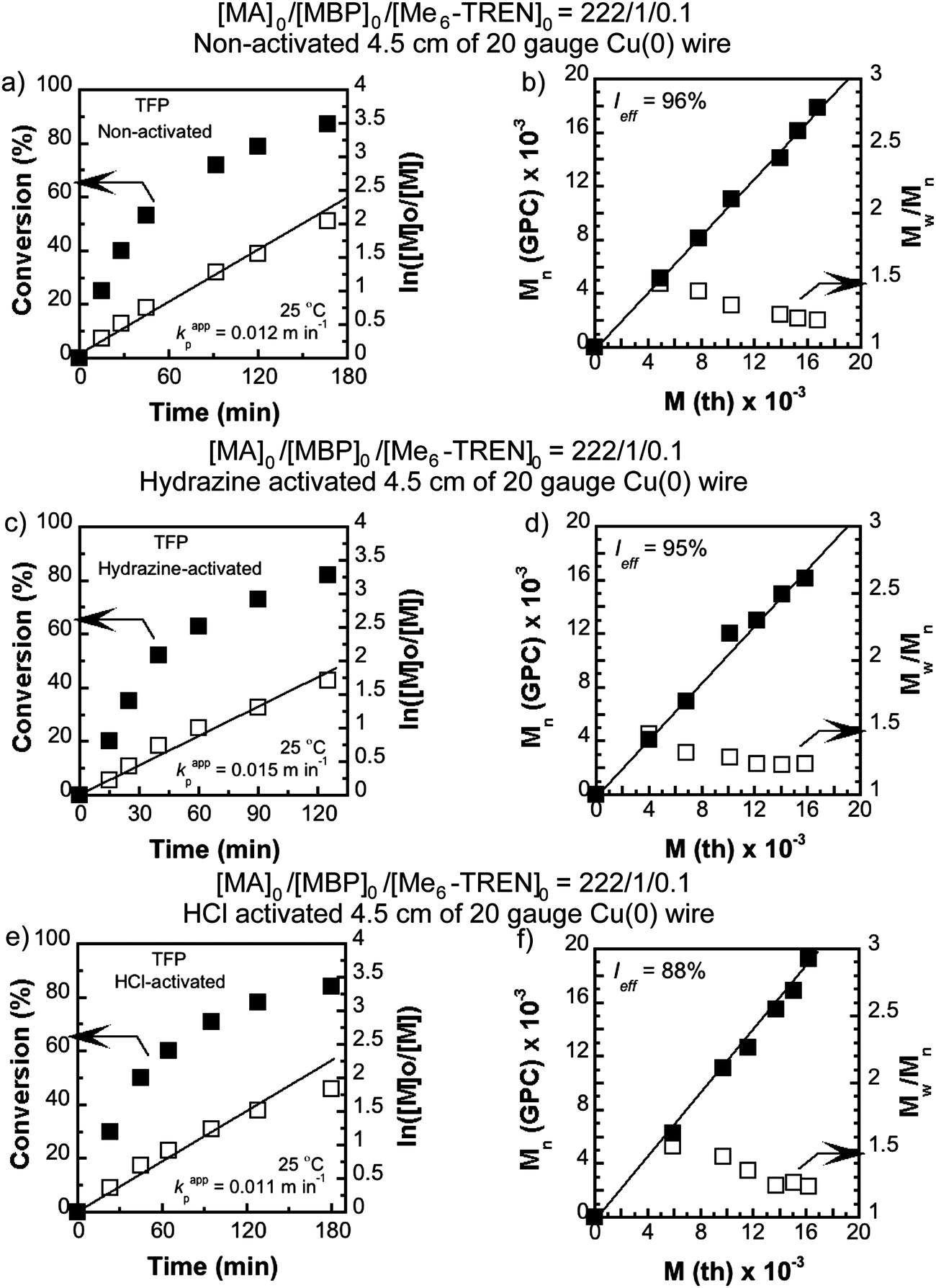 | ||
| Fig. 1 Kinetic plots (left) and Mn and Mw/Mnvs. theoretical Mth (right) for SET-LRP reactions of MA performed in TFP mediated at 25 °C. Conditions: (a and b) MA = 1 mL, TFP = 0.5 mL, and [MA]0/[MBP]0/[Me6-TREN]0 = 222/1/0.1, non-activated 4.5 cm activated Cu(0) wire, (c and d) MA = 1 mL, TFP = 0.5 mL, and [MA]0/[MBP]0/[Me6-TREN]0 = 222/1/0.1, hydrazine-activated 4.5 cm activated Cu(0) wire, and (e and f) MA = 1 mL, TFP = 0.5 mL, and [MA]0/[MBP]0/[Me6-TREN]0 = 222/1/0.1, HCl-activated 4.5 cm activated Cu(0) wire. | ||
 | ||
| Fig. 2 Kinetic plots (left) and Mn and Mw/Mnvs. theoretical Mth (right) for SET-LRP reactions of MA performed in TFP mediated at 25 °C. Conditions: (a and b) MA = 1 mL, TFE = 0.5 mL, and [MA]0/[MBP]0/[Me6-TREN]0 = 222/1/0.1, non-activated 2.0 cm activated Cu(0) wire and (c and d) MA = 1 mL, TFE = 0.5 mL, and [MA]0/[MBP]0/[Me6-TREN]0 = 222/1/0.1, hydrazine-activated 2.0 cm activated Cu(0) wire. | ||
In this regard it should be noted that whilst DMSO was used as the solvent70 the increment of kappp due to activation by hydrazine was almost two fold for 12.5 cm of the Cu(0) wire (from 0.073 min−1 to 0.139 min−1).70 From this observation we conclude that TFE and TFP act as further activating agents for the Cu(0) wire towards SET-LRP probably by modifying the surface of the Cu(0) wire. To further prove this hypothesis SET-LRP of MA was conducted in TFP under similar conditions with 4.5 cm of the HCl activated Cu(0) wire. In this case no further increment of kappp (0.011 min−1, Fig. 1(e)) was observed as compared to the non-activated wire. In comparison, using DMSO as solvent,71 HCl-activation of 4.5 cm Cu(0) wire resulted in an 1.20 fold increment of kappp (from 0.069 min−1 to 0.083 min−1). This result indicates that when TFP is used as the solvent, no further increment in kappp is obtained by surface modification of the Cu(0) wire via acid dissolution.
Activation of Cu(0) wire by TFE and TFP during SET-LRP in DMSO
To further explore the ability of TFE and TFP to activate the Cu(0) wire by such a surface modification, SET-LRP of MA was conducted in DMSO using the TFE and TFP-activated Cu(0) wire (12.5 cm). Activation was carried out by immersing the wire in 2 mL of deoxygenated TFE or TFP for ∼30 min prior to using it in the reaction. The wire was removed from the solvents (TFE or TFP), washed with distilled THF and absolute alcohol followed by drying under reduced pressure for at least 30 minutes.Application of these pretreated Cu(0) wire derived catalysts demonstrated that kappp in the case of the TFP-activated Cu(0) wire was as high as 0.110 min−1 (Fig. 3(a)) compared to 0.139 min−1 for the hydrazine-activated counterpart.70 This is a 1.50 times increment in kappp (from 0.073 min−1 compared to 0.110 min−1)70 compared to the non-activated wire as catalyst. In comparison, the TFE-activated Cu(0) wire gave rise to a kappp value of 0.083 min−1 (Fig. 3(c)) which is only a 1.14 times increment due to activation.
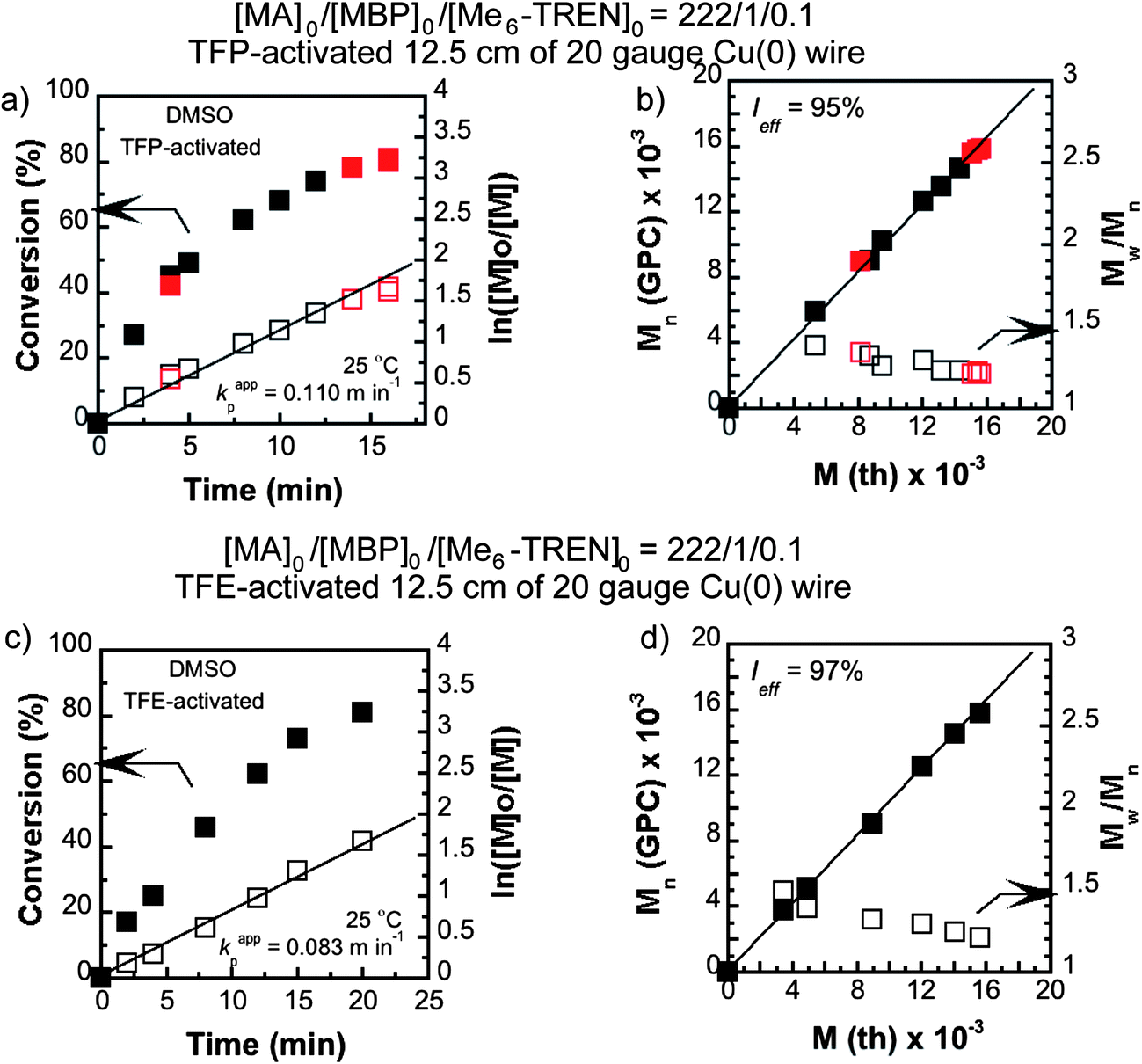 | ||
| Fig. 3 Kinetic plots (left) and Mn and Mw/Mnvs. theoretical Mth (right) for SET-LRP reactions of MA performed in DMSO mediated at 25 °C. Conditions: (a and b) MA = 1 mL, DMSO = 0.5 mL, and [MA]0/[MBP]0/[Me6-TREN]0 = 222/1/0.1, TFP-activated 12.5 cm activated Cu(0) wire and (c and d) MA = 1 mL, DMSO = 0.5 mL, and [MA]0/[MBP]0/[Me6-TREN]0 = 222/1/0.1, TFE-activated 12.5 cm activated Cu(0) wire. | ||
Scanning Electron Microscopy (SEM) experiments were used to analyze the surface morphology of the Cu(0) wire before and after activation by treatment with TFP and TFE (Fig. 4). Similar to our previous study of activation of the Cu(0) wire by treatment with hydrazine hydrate,54 in our current study we observed oriented scratches and cracks on the Cu(0) wire surface, which resulted from processing, under a low magnification of 8000 on both non-activated (Fig. 4(a)) and activated, by TFP and TFE treatment (Fig. 4(c) and (e)). In addition, the SEM image of the non-activated Cu(0) wire at higher magnification also showed a smooth surface (Fig. 4(b)). However, after activation with TFP and TFE, SEM images of the Cu(0) wire surface at higher magnification showed low density dents (Fig. 4(d) and (f)). Interestingly, a similar observation was reported when the Cu(0) wire was activated by hydrazine hydrate.54,75,76 However, it should be noted that compared to hydrazineactivation of the Cu(0) wire which results in a more dramatic increment in the rate of polymerization of SET-LRP,70 TFE and TFP activation resulted in a much less increment in the roughness of surface morphology.54 Nevertheless, this demonstrates that the surface morphology of the commercial Cu(0) wire changes by treatment with TFP and TFE and results in an enhancement in the surface area.54,74–76 We hypothesize that the acceleration of the rate of SET-LRP of MA in DMSO when catalyzed by the TFP or TFE-activated Cu(0) wire compared to its non-activated counterpart is partly because of the increased surface area. This also provides the self-activation property of TFP and TFE by which the commercial Cu(0) wire used as catalyst for SET-LRP in these solvents gets activated in situ.
 | ||
| Fig. 4 SEM images of commercial Cu(0) wire (a and b) before activation, (c and d) after activation by TFP treatment, and (e and f) after activation by TFE treatment at low magnification (left) and high magnification (right). Cu(0) length = 12.5 cm and diameter = 0.812 mm. | ||
Conclusions
The self-activation and activation of the Cu(0) wire used as a catalyst in SET-LRP by TFE and TFP were demonstrated. SET-LRP of MA in TFP catalyzed with the non-activated Cu(0) wire showed comparable kappp values to that of the hydrazine-activated counterpart. In addition, SET-LRP of MA in DMSO catalyzed with 12.5 cm Cu(0) wire exhibits an 1.5 times increment in the kappp value due to activation by TFP. In contrast, activation of the Cu(0) wire by TFE is comparably less effective and results in only a 1.14 times increment in the kappp value for SET-LRP of MA in DMSO under similar conditions. However, while using TFE or TFP as a solvent for SET-LRP, the polymerization proceeded with no observable initial induction periods and predictable molecular weight evolution with conversion throughout the reaction. According to our knowledge there are no literature data reporting the dissolution of CuO and Cu2O by fluorinated solvents. Nevertheless, we speculate that due to their acidic character both TFE and TFP change the surface morphology of the Cu(0) wire by a small extent of dissolution of copper oxide. Hence, the self-activation and activation properties of these solvents for the Cu(0) wire catalyst in SET-LRP along with their ability to polymerize both hydrophobic and hydrophilic monomers with excellent control by this methodology provide a powerful tool for the synthesis of polymers with complex architecture and a simpler method for the activation of the Cu(0) wire than the current activation methodologies involving both hydrazine and acids.Experimental
Materials
Cu wire (20 gauge from Fisher), methyl 2-bromopropionate (MBP) (99%, Acros) and hydrazine hydrate (100%, hydrazine 64%, Acros) used for the activation of Cu wire were used as received. DMSO (99.9%, certified ACS, Fisher) was distilled under reduced pressure before use. Hexamethylated tris(2-aminoethyl)amine (Me6-TREN) was synthesized as described in the literature.77 Activation of copper(0) wire (20 gauge wire, 0.812 mm diameter from Fischer) by hydrazine hydrate70 and by acid dissolution71 was performed according to the procedures elaborated in our laboratory. Activation of copper(0) wire (20 gauge wire, 0.812 mm diameter from Fischer) by TFP was carried out by both distilled and as received TFP (SynQuest Laboratories), and we obtained results with no significant difference. Activation of copper(0) wire (20 gauge wire, 0.812 mm diameter from Fischer) by TFE was carried out by as-received TFE (>99%, AlfaAesar).Techniques
500 MHz 1H-NMRspectra were recorded on a Bruker DRX500 NMR instrument at 20 °C in CDCl3 with tetramethylsilane (TMS) as the internal standard. Gel Permeation Chromatography (GPC) analysis was performed on a Perkin-Elmer Series 10 high-performance liquid chromatograph, equipped with an LC-100 column oven at 30 °C, a Nelson Analytical 900 Series integration data station, a Perkin-Elmer 785 UV-visdetector (254 nm), a Varian star 4090 refractive index (RI) detector, and two AM gel columns (500 Å, 5 μm; and 1000 Å, 5 μm). HPLC grade THF (Fisher) was used as the eluent at a flow rate of 1 mL min−1. The number-average molecular weight (Mn) and molecular weight distribution (Mw/Mn) were determined with poly(methyl methacrylate) (PMMA) standards purchased from American Polymer Standards.The Scanning Electron Microscopy (SEM) experiments were performed with a JEOL 7500F HRSEM equipped with a cold field emission gun operating at a probe current of 1 pA to 2 nA and an accelerating voltage of 0.1–30 kV. Images were collected under SEI mode using an Everhardt–Thornley secondary electron detector, and the sample to detector distance was monitored by an IR-CCD chamber camera. Elemental analysis was conducted with Oxford Inca Thin Window 102 mm EDS. Cu(0) wire samples were cut into sizes of 1–2 cm and placed on a clean Al stub before being mounted on a single stub sample stage. The sample was then placed in the SEM sample chamber without further carbon or metal coating. The sample chamber was equipped with a dry vacuum system and a liquid nitrogen anticontamination device for providing a clean specimen environment and the chamber vacuum was maintained below 1 × 10−4 Pa. The voltage was set to 15 kV with the working distance kept between 4.3 mm and 6.9 mm. The magnification was set to 8000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 for low magnification images and 50000–100000 for high magnification images. Elemental analysis was performed at the same time and area where the images were collected.
000 for low magnification images and 50000–100000 for high magnification images. Elemental analysis was performed at the same time and area where the images were collected.
Typical procedure for polymerization kinetics
Monomer MA was charged to a 25 mL Schlenk tube along with an initiator MBP, ligand Me6-TREN, solvent, and stirring bar. The reaction mixture was then deoxygenated by six freeze–pump–thaw cycles using liquid nitrogen as a freezing mixture. After these cycles the Schlenk tube was opened under positive argon pressure to add the activated Cu(0) wire. It was further deoxygenated and refilled with argon three times and transferred to a thermostatted oil bath operating at the desired temperature. The introduction of the Cu(0) wire defines t = 0. Approximately one or two drops of samples were periodically removed from the reaction mixture by purging the side arm with argon for more than 2 min by using a deoxygenated syringe and needle. Samples were dissolved in CDCl3, and the conversion was measured by 1H-NMR spectroscopy. This solution was then passed through a small basic Al2O3 chromatographic column to remove any residual “nascent” Cu(0) catalyst and Cu(II) deactivator before it was analyzed by GPC for Mn and Mw/Mn determination.Acknowledgements
Financial support by the National Science Foundation (DMR-1120901 and DMR-1066116) and the P. Roy Vagelos Chair at Penn are gratefully acknowledged. A.A. acknowledges the Warwick Transatlantic Fellowship (WTF) scheme for funding. D.M.H. is a Royal Society/Wolfson fellow.References
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS PubMed.
- V. Percec, A. V. Popov, E. Ramirez-Castillo, M. Monteiro, B. Barboiu, O. Weichold, A. D. Asandei and C. M. Mitchell, J. Am. Chem. Soc., 2002, 124, 4940–4941 CrossRef CAS PubMed.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- V. Percec, A. V. Popov, E. Ramirez-Castillo and O. Weichold, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3283–3299 CrossRef CAS.
- T. Hatano, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 164–172 CrossRef CAS.
- V. Percec, A. V. Popov, E. Ramirez-Castillo, J. F. J. Coelho and L. A. Hinojosa-Falcon, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 6267–6282 CrossRef CAS.
- J. Queffelec, S. G. Gaynor and K. Matyjaszewski, Macromolecules, 2000, 33, 8629–8639 CrossRef CAS.
- S. Zhai, B. Wang, C. Feng, Y. Li, D. Yang, J. Hu, G. Lu and X. Huang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 647–655 CrossRef CAS.
- N. H. Nguyen, M. E. Levere and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 860–873 CrossRef CAS.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4684–4695 CrossRef CAS.
- X. Leng, N. Nguyen, B. van Beusekom, D. A. Wilson and V. Percec, Polym. Chem., 2013, 4, 2995–3004 RSC.
- N. H. Nguyen, B. M. Rosen, G. Lligadas and V. Percec, Macromolecules, 2009, 42, 2379–2386 CrossRef CAS.
- X. Jiang, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 403–409 CrossRef CAS.
- S. Zhai, J. Shang, D. Yang, S. Wang, J. Hu, G. Lu and X. Huang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 811–820 CrossRef CAS.
- S. Zhai, X. Song, D. Yang, W. Chen, J. Hu, G. Lu and X. Huang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4055–4064 CrossRef CAS.
- A. Anastasaki, C. Waldron, V. Nikolaou, P. Wilson, R. McHale, T. Smith and D. M. Haddleton, Polym. Chem., 2013, 4, 4113–4119 RSC.
- C. Boyer, A. Atme, C. Waldron, A. Anastasaki, P. Wilson, P. B. Zetterlund, D. Haddleton and M. R. Whittaker, Polym. Chem., 2013, 4, 106–112 RSC.
- M. J. Monteiro, T. Guliashvili and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1835–1847 CrossRef CAS.
- D. Haddleton, J. Burns, C. Houben, C. Waldron, A. Anastasaki and A. Lapkin, Polym. Chem., 2013, 4, 4809–4813 RSC.
- G. Lu, Y. Li, H. Gao, H. Guo, X. Zheng and X. Huang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1099–1106 CrossRef CAS.
- G. Lu, Y. Li, H. Guo, W. Du and X. Huang, Polym. Chem., 2013, 4, 3132–3139 RSC.
- S. Zhai, X. Song, C. Feng, X. Jiang, Y. Li, G. Lu and X. Huang, Polym. Chem., 2013, 4, 4134–4144 RSC.
- G. Lu, Y. Li, B. Dai, C. Xu and X. Huang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1880–1886 CrossRef CAS.
- C. Boyer, A. Derveaux, P. B. Zetterlund and M. R. Whittaker, Polym. Chem., 2012, 3, 117–123 RSC.
- G. Chen, P. M. Wright, J. Geng, G. Mantovani and D. M. Haddleton, Chem. Commun., 2008, 1097–1099 RSC.
- M. W. Jones, M. I. Gibson, G. Mantovani and D. M. Haddleton, Polym. Chem., 2011, 2, 572–574 RSC.
- S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4889–4893 CrossRef CAS.
- S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2243–2250 CrossRef CAS.
- S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2236–2242 CrossRef CAS.
- S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4884–4888 CrossRef CAS.
- N. H. Nguyen, X. Leng and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2013, 4, 2760–2766 RSC.
- X. Song, Y. Zhang, D. Yang, L. Yuan, J. Hu, G. Lu and X. Huang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3328–3337 CrossRef CAS.
- Y. Deng, Y. Li, J. Dai, M. Lang and X. Huang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4747–4755 CrossRef CAS.
- S. R. Samanta, A. Anastasaki, C. Waldron, D. M. Haddleton and V. Percec, Polym. Chem., 2013 10.1039/c3py00902e.
- N. H. Nguyen, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1752–1763 CrossRef CAS.
- X. Tang, X. Liang, Q. Yang, X. Fan, Z. Shen and Q. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4420–4427 CrossRef CAS.
- Y. Deng, J. Z. Zhang, Y. Li, J. Hu, D. Yang and X. Huang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4451–4458 CrossRef CAS.
- C. Feng, Y. Li, D. Yang, Y. Li, J. Hu, S. Zhai, G. Lu and X. Huang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 15–23 CrossRef CAS.
- C. Feng, Z. Shen, Y. Li, L. Gu, Y. Zhang, G. Lu and X. Huang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1811–1824 CrossRef CAS.
- C. Feng, Z. Shen, D. Yang, Y. Li, J. Hu, G. Lu and X. Huang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4346–4357 CrossRef CAS.
- A. Ding, G. Lu, H. Guo, X. Zheng and X. Huang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1091–1098 CrossRef CAS.
- N. Nga Hang, C. Rodriguez-Emmenegger, E. Brynda, Z. Sedlakova and V. Percec, Polym. Chem., 2013, 4, 2424–2427 RSC.
- B. Barboiu and V. Percec, Macromolecules, 2001, 34, 8626–8636 CrossRef CAS.
- X.-H. Liu, G.-B. Zhang, B.-X. Li, Y.-G. Bai and Y.-S. Li, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5439–5445 CrossRef CAS.
- N. H. Nguyen, J. Kulis, H.-J. Sun, Z. Jia, B. van Beusekom, M. E. Levere, D. A. Wilson, M. J. Monteiro and V. Percec, Polym. Chem., 2013, 4, 144–155 RSC.
- Q. Zhang, J. Collins, A. Anastasaki, R. Wallis, D. A. Mitchell, C. R. Becer and D. M. Haddleton, Angew. Chem., Int. Ed., 2013, 52, 4435–4439 CrossRef CAS PubMed.
- G. Lligadas, J. S. Ladislaw, T. Guliashvili and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2007, 46, 278–288 CrossRef.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS PubMed.
- C. Boyer, A. H. Soeriyadi, P. B. Zetterlund and M. R. Whittaker, Macromolecules, 2011, 44, 8028–8033 CrossRef CAS.
- N. H. Nguyen, M. E. Levere, J. Kulis, M. J. Monteiro and V. Percec, Macromolecules, 2012, 45, 4606–4622 CrossRef CAS.
- F. Nyström, A. H. Soeriyadi, C. Boyer, P. B. Zetterlund and M. R. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5313–5321 CrossRef.
- M. E. Levere, N. H. Nguyen, H.-J. Sun and V. Percec, Polym. Chem., 2013, 4, 686–694 RSC.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4227–4240 CAS.
- N. H. Nguyen, H.-J. Sun, M. E. Levere, S. Fleischmann and V. Percec, Polym. Chem., 2013, 4, 1328–1332 RSC.
- B. M. Rosen, X. Jiang, C. J. Wilson, N. H. Nguyen, M. J. Monteiro and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5606–5628 CrossRef CAS.
- M. E. Levere, N. H. Nguyen, X. Leng and V. Percec, Polym. Chem., 2013, 4, 1635–1647 RSC.
- A. Anastasaki, C. Waldron, P. Wilson, R. McHale and D. M. Haddleton, Polym. Chem., 2013, 4, 2672–2675 RSC.
- B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4950–4964 CrossRef CAS.
- Q. Zhang, P. Wilson, Z. Li, R. McHale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, J. Am. Chem. Soc., 2013, 135, 7355–7363 CrossRef CAS PubMed.
- N. H. Nguyen, B. M. Rosen, X. Jiang, S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5577–5590 CrossRef CAS.
- N. H. Nguyen, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1752–1763 CrossRef CAS.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2745–2754 CrossRef CAS.
- G. Lligadas, B. M. Rosen, M. J. Monteiro and V. Percec, Macromolecules, 2008, 41, 8360–8364 CrossRef CAS.
- X. Jiang, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2716–2721 CrossRef CAS.
- S. R. Samanta, M. E. Levere and V. Percec, Polym. Chem., 2013, 4, 3212–3224 RSC.
- S. R. Samanta, A. Anastasaki, C. Waldron, D. M. Haddleton and V. Percec, Polym. Chem., 2013 10.1039/c3py00901g.
- X. Jiang, S. Fleischmann, N. H. Nguyen, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5591–5605 CrossRef CAS.
- Q. Zhang, Z. Li, P. Wilson and D. M. Haddleton, Chem. Commun., 2013, 49, 6608–6610 RSC.
- G. Lligadas, B. M. Rosen, C. A. Bell, M. J. Monteiro and V. Percec, Macromolecules, 2008, 41, 8365–8371 CrossRef CAS.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5109–5119 CrossRef CAS.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4241–4252 CAS.
- S. K. Miller, A. Baiker, M. Meier and A. Wokaun, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 1305–1312 RSC.
- B. Otazaghine, C. Boyer, J.-J. Robin and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2377–2394 CrossRef CAS.
- F. C. Aldred and F. Happey, Nature, 1947, 160, 267–268 CrossRef CAS.
- D. M. Littrell, D. H. Bowers and B. J. Tatarchuk, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 3271–3282 RSC.
- W. D. Bond, J. Phys. Chem., 1962, 66, 1573–1577 CrossRef CAS.
- M. Ciampolini and N. Nardi, Inorg. Chem., 1966, 5, 41–44 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |