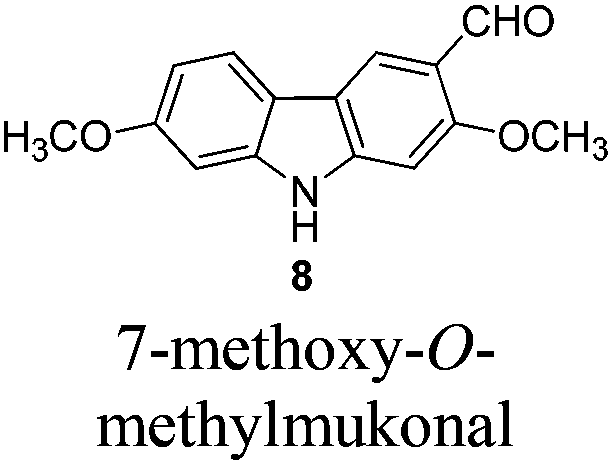
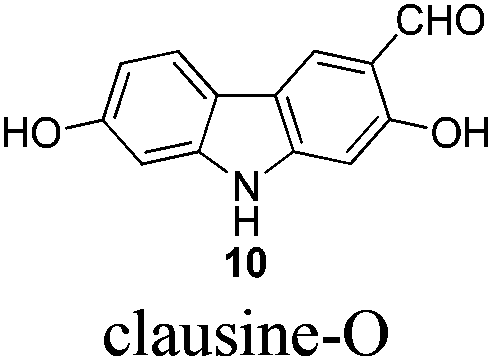
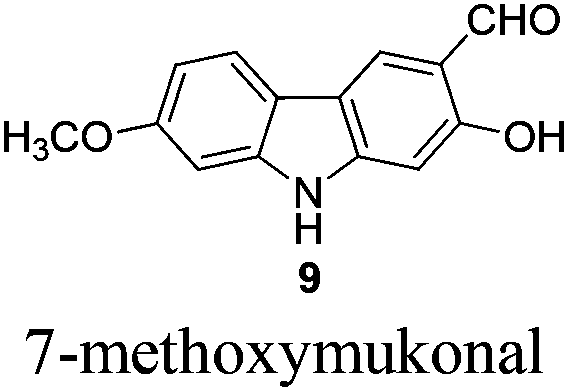
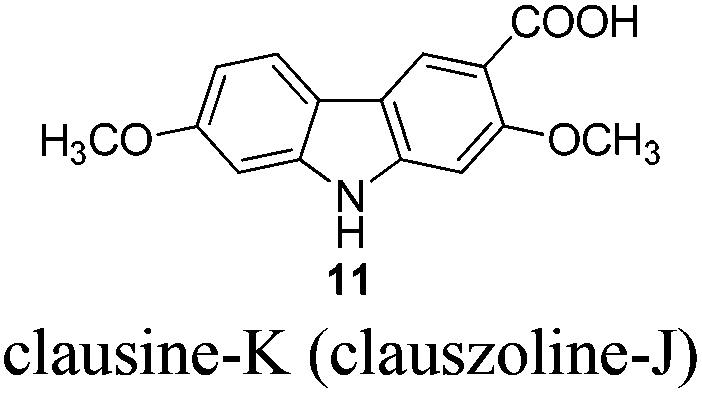
Gram scale synthesis of 7-methoxy-O-methylmukonal, clausine-O, clausine-K, clausine-H, 7-methoxymukonal, and methyl 2-hydroxy-7-methoxy-9H-carbazole-3-carboxylate†
Dengke
Ma
,
Jianxin
Dai
,
Youai
Qiu
,
Chunling
Fu
* and
Shengming
Ma
*
Laboratory of Molecular Recognition and Synthesis, Department of Chemistry, Zhejiang University, Hangzhou 310027, Zhejiang, People's Republic of China. E-mail: masm@sioc.ac.cn; Fax: (+86) 21-62609305
First published on 18th June 2014
Abstract
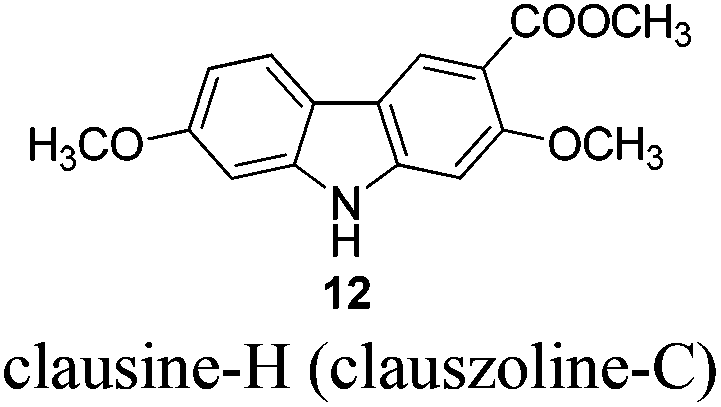
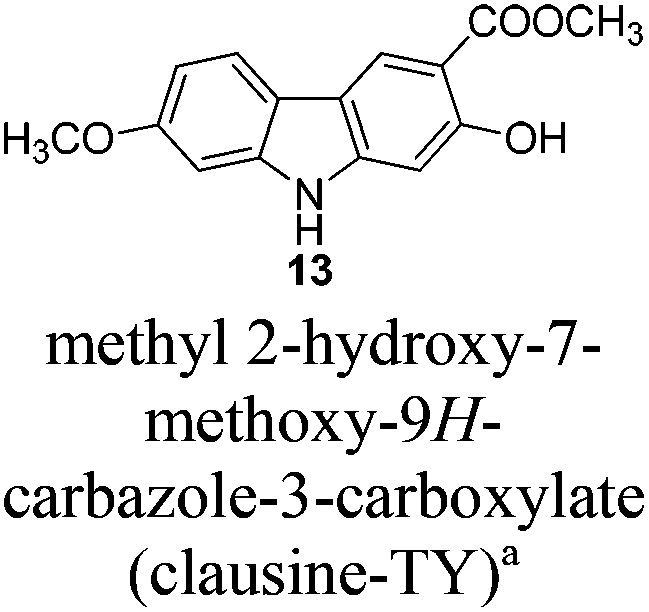
Naturally occurring 2,7-dioxygenated carbazole alkaloids, 7-methoxy-O-methylmukonal, clausine-O, clausine-K (clauszoline-J), clausine-H (clauszoline-C), 7-methoxymukonal and methyl 2-hydroxy-7-methoxy-9H-carbazole-3-carboxylate, have been synthesized via an efficient Au-catalyzed cyclization reaction using readily available 1-benzyl-6-methoxy-1H-indole-2-carbaldehyde and 1-methoxypropa-1,2-diene as the starting materials on the gram scale. Among them, 7-methoxymukonal and methyl 2-hydroxy-7-methoxy-9H-carbazole-3-carboxylate were prepared for the first time. In addition, the structure of methyl 2-hydroxy-7-methoxy-9H-carbazole-3-carboxylate we prepared was further confirmed by X-ray single crystal diffraction study via its ethylation reaction, which indicated that clausine-TY reported by Taufiq-Yap et al. in 2007 should be reassigned.
1. Introduction
Carbazole alkaloids have attracted more and more attention nowadays since many have been isolated from different natural sources and showed valuable and potential biological activities.1 Of particular interest are 2,7-dioxygenated carbazole alkaloids which play an important role in the carbazole alkaloid family. Among all the 2,7-dioxygenated carbazole alkaloid sources, Murraya siamensis, Clausena excavata and Clausena harmandiana represent the richest ones. It is interesting to note that most of these plants have been used as herbal medicine traditionally. For instance, people from the Sukhothai Province of Thailand have used the roots of Murraya siamensis to cure eye sores, snakebites and tuberculosis for a long time.2 Therefore, these isolated compounds from them became important research subjects of biologists and pharmacognosists. Gladly, most of these representative compounds indeed showed exciting biological activity through various kinds of bioassays. Isolation and biological activities of all these carbazole alkaloids are listed with the related references for both isolation and bioassay (Table 1).| Name | Isolation | Biological activity |
|---|---|---|
| a The structure of this isolated compound must be wrong since the data of the compound with the shown structure we prepared in this study are different from those reported by Taufiq-Yap et al. | ||
|
The roots of Murraya siamensis by Lange and co-workers in 19902 | Anti-HIV-1 activity1,3 |
| The rhizomes and roots of Clausena excavata by Kongkathip et al. in 20053 | IC50 = 96 μM against Vero cells4 | |
|
The root bark of Clausena excavata by Wu et al. in 19995 | Activity against the M. tuberculosis H37Rv strain (MIC = 89 μg mL−1)4 |
| Inhibit aggregation of HepG2 liver carcinoma cells (IC50 = 30 μg mL−1)6 | ||
| IC50 = 59.55 μM against KB (oral cancer) cell lines7 | ||
|
The root bark of Clausena harmandiana by Pummangura and co-workers in 19888 | Antimalarial activity with IC50 = 2.94 μg mL−1 and anti TB activity against Mycobacterium tuberculosis H37Ra with MIC = 25 μg mL−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 9 |
| Antifungal activity with IC50 = 2.8 μg mL−110 |
||
|
The stem bark of Clausena excavata by Wu et al. in 199611 | IC50 = 24.4 ± 2.17 μM growth inhibitory against HepG2 cells6 |
| The root bark of Clausena excavata by Ito et al. in 199712a | Antimycobacterial activity against Mycobacterium tuberculosis H37Ra (MIC90 = 100 μg mL−1 = 369 μM)10 | |
| Anti-HIV-1 activity1 | ||
|
The stem bark of Clausena excavata by Wu et al. in 199611 | Antiplasmodial activity |
| The stem bark of Clausena excavata by Ito et al. in 199613 | IC50 = 5.5–10.7 μg mL−114 |
|
|
The stem bark of Clausena excavata by Taufiq-Yap et al. in 200715 | |
Although these carbazole alkaloids present a variety of biological activities, the isolation from the natural sources was not efficient, and the synthetic routes are limited, especially the synthesis on a large scale. For example, only 16 mg of 7-methoxy-O-methylmukonal was isolated from 1 kg clipped roots of Murraya siamensis by Lange and co-workers in 1990,2 and only 0.5 mg of clausine-O was isolated from 0.8 kg root bark of Clausine excavata during the isolation conducted by Wu et al. in 1999.5
In 2005, Knölker et al. pioneered the synthesis of some 2,7-dioxygenated carbazole alkaloids, 7-methoxy-O-methylmukonal, clausine-H, clausine-K and clausine-O, via the iron-mediated arylamine oxidative cyclization.16 Starting from tricarbonyl[η5-1-methoxycyclohexa-1,4-dienylium]iron tetrafluoroborate S1-c with 3-methoxy-4-methylaniline S1-d through an electrophilic substitution reaction followed by oxidative cyclization gave the carbazole framework. However, as one of the key starting materials, S1-c was prepared from a stoichiometric amount of pentacarbonyliron S1-b and methoxycyclohexadiene S1-a.17 Furthermore, a large amount of the oxidant 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (4.3 equiv. DDQ) was used to convert the 3-methyl group to the 3-formyl functionality, affording 7-methoxy-O-methylmukonal S1-e. Using compound S1-f as the critical precursor, clausine-H, clausine-K and clausine-O were synthesized. In the synthesis of clausine-H, a large excess of some environmentally unfriendly and toxic reagents, such as MnO2 (30 equiv.) and KCN (4.5 equiv.), was applied, which makes room for new approaches (Scheme 1). Therefore, efficient synthetic routes for these 2,7-dioxygenated carbazole alkaloids on a large scale are still of high interest. In 2011, our group developed an efficient and general Au-catalyzed cyclization of 1-(3-indolyl)-2,3-allenols via a carbenoid mechanism and applied it to synthesize some naturally occurring carbazole alkaloids.18 We have successfully synthesized siamenol, clausine-N, clausine-C, clauszoline-K, clausine-M, 3-formyl-7-hydroxycarbazole, isomukonidine, clausine-L, mukonidine, glycosinine, mukonal and clausine-V. Herein, we report the synthesis of six 2,7-dioxygenated carbazole alkaloids on the gram scale utilizing this method by addressing the issue of challenging monobromination and monodemethylation based on the readily convertible nature of the C–Br bond.
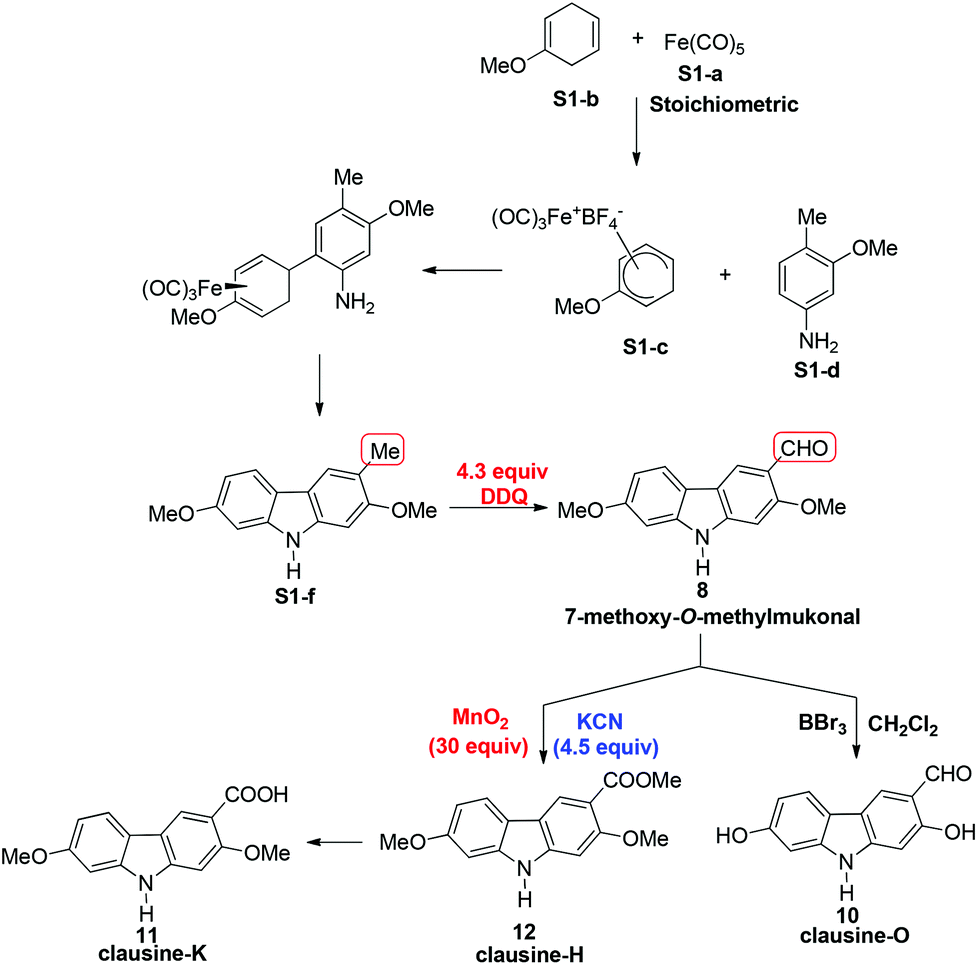 | ||
| Scheme 1 | ||
2. Results and discussion
We conducted the retrosynthetic analysis of these target compounds with the concept of replacing the C–Me bond (S1-f in Scheme 1) with the C–Br bond in Scheme 1 for much easier transformations. Thus, the 3-formyl/carboxyl/carboxylate-substituted 2,7-dioxygenated natural carbazole alkaloids listed in Table 1 could be synthesized by derivatizing 9-benzyl-3-bromo-2,7-dimethoxy-9H-carbazole 5, which may be prepared readily by the cyclization of (6-methoxyindoyl)-2,3-allenol 3 followed by highly selective monobromination at the C-3 position (eq. in Table 2). Then selective demethylation of 7-methoxy-O-methylmukonal 8 and clausine-H 12 at the C-2 position may lead to 7-methoxymukonal 9 and methyl 2-hydroxy-7-methoxy-9H-carbazole-3-carboxylate 13. However, some challenges emerged in this route: (1) selective monobromination (challenge 1, Scheme 2); (2) selective monodemethylation (challenge 2, Scheme 2).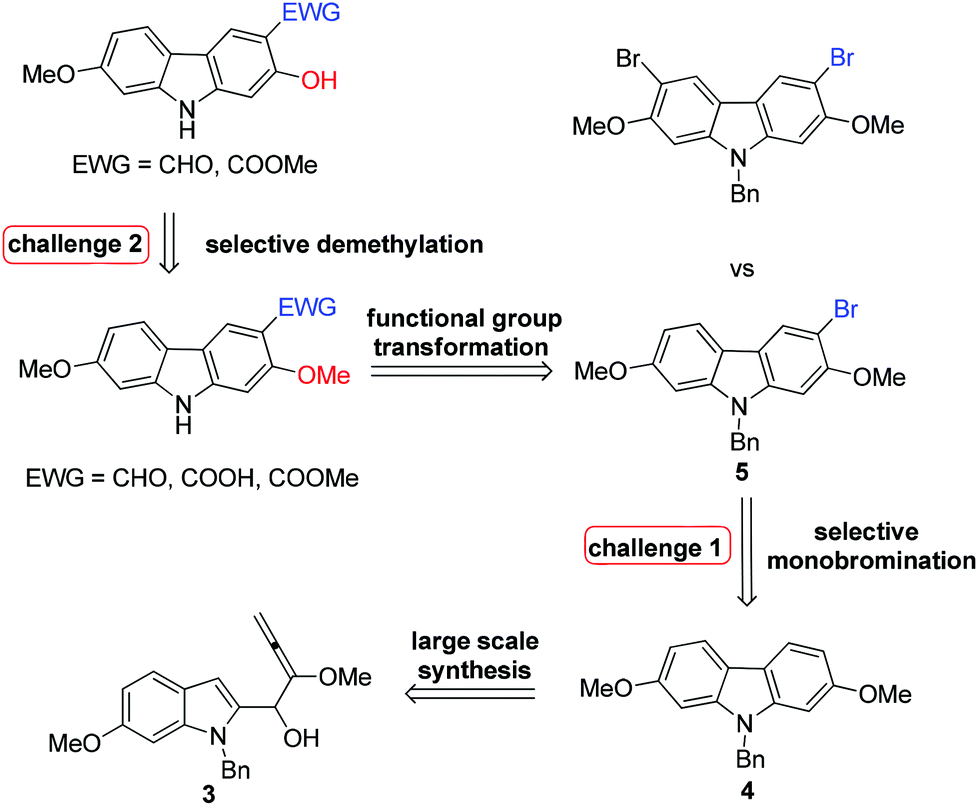 | ||
| Scheme 2 | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | X | Solvent | T (°C) | Time (h) | Yieldb % | Ratio 5:6 |
Recovery % | |
| 5 | 6 | |||||||
| a All reactions are conducted on the 0.6 mmol scale. b Determined by NMR using dibromomethane as the internal standard. c NBS was dissolved in 0.3 mL toluene and added dropwise in 5 min. | ||||||||
| 1 | 1 | CCl4 | 15 | 1 | 74 | 12 | 86:14 |
14 |
| 2 | 1 | CCl4 | 0 | 1 | 67 | 14 | 83:17 |
16 |
| 3c | 1 | CCl4 | 15 | 1 | 73 | 11 | 87:13 |
14 |
| 4 | 0.9 | CH2Cl2 | 0 | 0.5 | 80 | 5 | 94:6 |
15 |
| 5 | 0.9 | CHCl3 | 0 | 0.5 | 80 | 4 | 95:5 |
13 |
| 6 | 0.95 | CHCl3 | 0 | 0.5 | 82 | 6 | 93:7 |
12 |
| 7 | 1 | CHCl3 | 0 | 0.5 | 88 | 7 | 93:7 |
5 |
| 8 | 0.9 | CHCl3 | −20 | 0.5 | 81 | 3 | 96:4 |
10 |
| 9 | 0.92 | CHCl3 | −20 | 0.5 | 83 | 3 | 97:3 |
9 |
| 10 | 0.94 | CHCl3 | −20 | 0.5 | 86 | 4 | 96:4 |
10 |
| 11 | 0.96 | CHCl3 | −20 | 0.5 | 87 | 5 | 95:5 |
6 |
| 12 | 0.98 | CHCl3 | −20 | 0.5 | 90 | 5 | 95:5 |
6 |
| 13 | 1.0 | CHCl3 | −20 | 12.5 | 74 | 5 | 94:6 |
4 |
| 14 | 1.05 | CHCl3 | −20 | 1.5 | 86 | 8 | 91:9 |
1 |
| 15 | 1.06 | CHCl3 | −20 | 1.5 | 84 | 8 | 91:9 |
— |
| 16 | 1.07 | CHCl3 | −20 | 0.25 | 87 | 9 | 91:9 |
— |
| 17 | 1.1 | CHCl3 | −20 | 0.25 | 84 | 12 | 88:12 |
— |
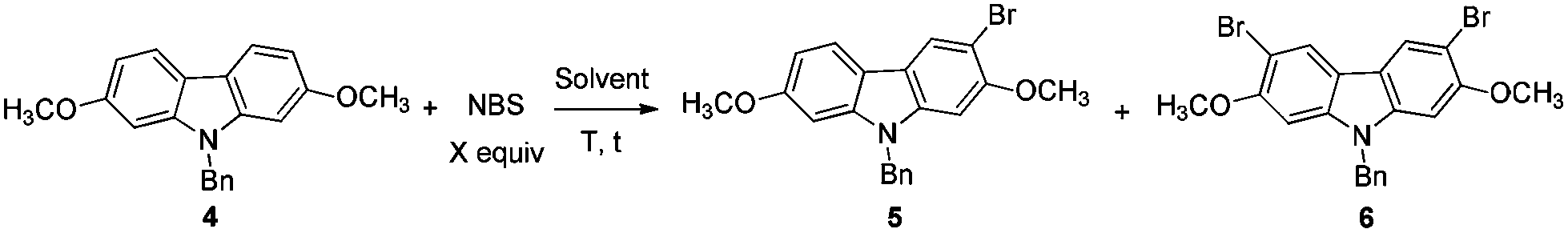
2.1. A large scale synthesis of 9-benzyl-2,7-dimethoxy-9H-carbazole 4
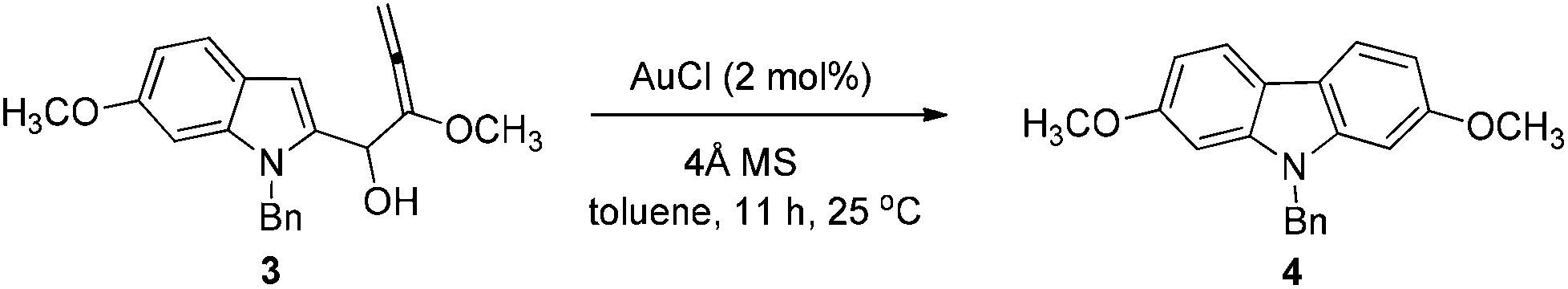
At first, we carried out a reaction on the 4 mmol scale catalyzed by 5% mol AuCl, affording 4 (0.9437 g) in 74% yield. Then we reasoned that a lower loading of catalyst may be realized when the scale was enlarged. However, only 50% of 4 was obtained when the scale of this reaction was promoted to 60 mmol from 4 mmol under 1% mol AuCl catalysis. We reasoned that in situ generated water may reduce the catalytic activity of AuCl. Thus, in the latter studies, the reaction was conducted in the presence of activated 4 Å MS to remove water. To our delight, the yield increased significantly. The same reaction on 60 mmol (15.9 g) was conducted with just 2% mol AuCl in the presence of 8.0 g of 4 Å MS to afford 17.4 g (91% isolated yield) of 4 after simple recrystallization (Scheme 3).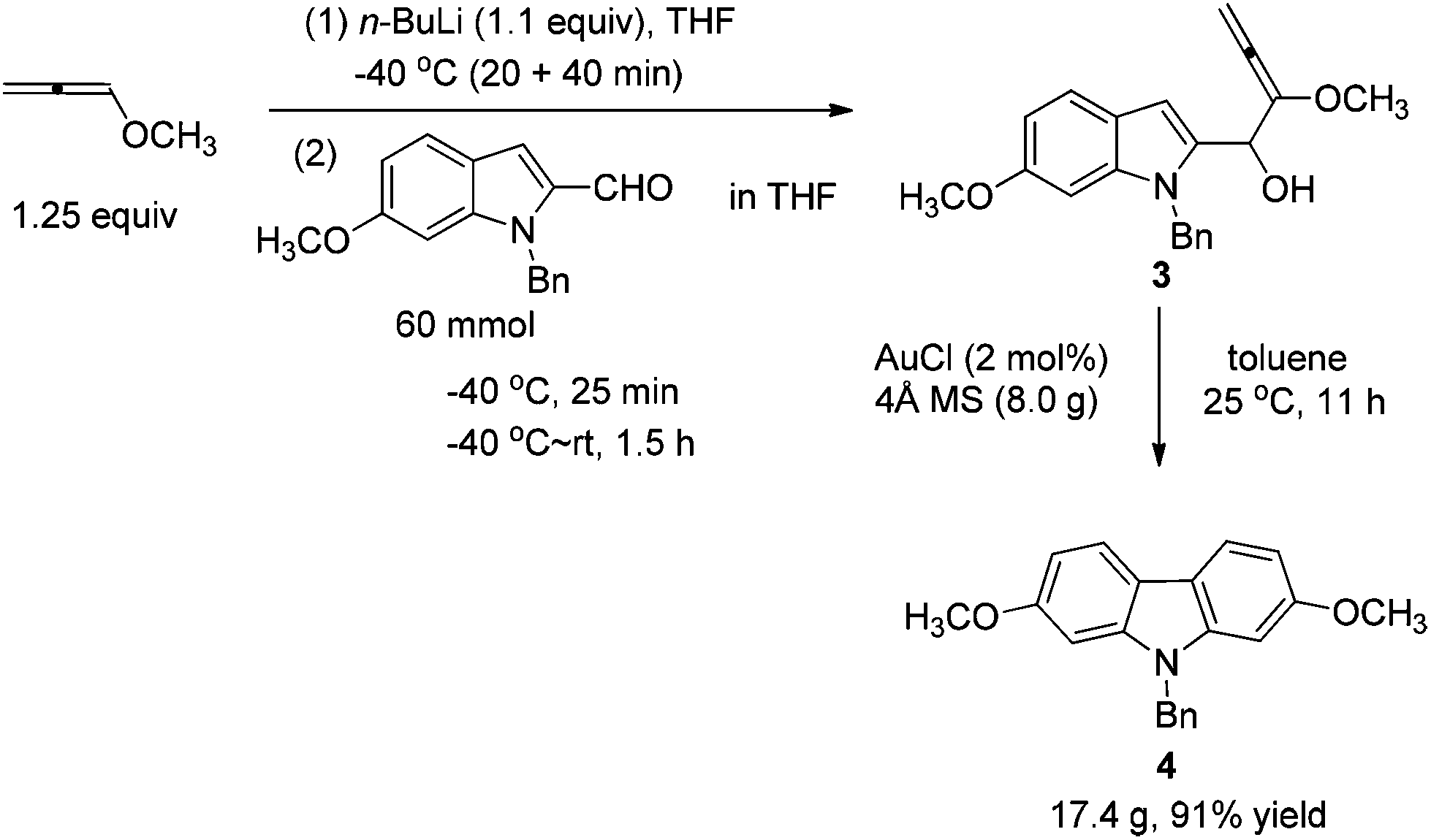 | ||
| Scheme 3 | ||
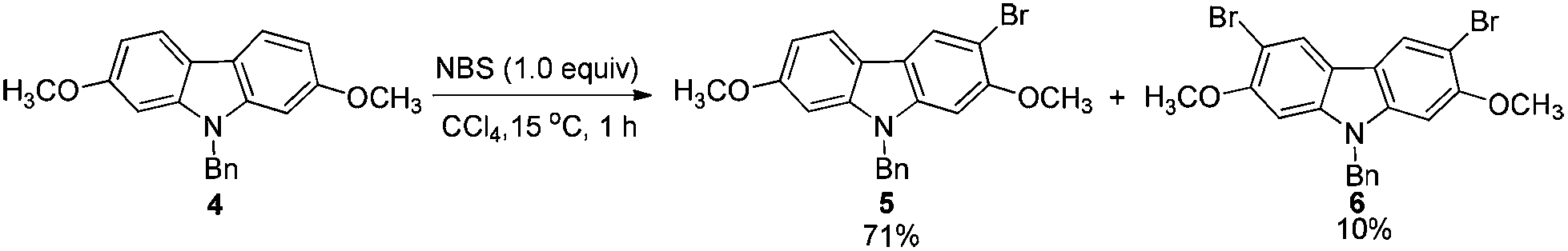
2.2. Highly selective monobromination of 2,7-dimethoxy-9H-carbazole
Because of the symmetric structure of 2,7-dimethoxy-9H-carbazole 4, monobromination at C-3 selectively avoiding the formation of the 3,6-dibromocarbazole byproduct 6 is challenging. Firstly, 4 was treated with 1 equiv. of N-bromosuccinimide (NBS) using carbon tetrachloride as the solvent at room temperature. However, 74% of 5 and 12% of the dibrominated byproduct 6 were obtained, together with 14% recovery of the starting material (entry 1, Table 2). The ratio of 5 and 6 was not obviously improved with dropwise addition of the NBS solution (entry 3, Table 2). The dibrominated product 6 should be afforded upon the reaction of the monobrominated product 5 with NBS. We used the ratio and the solubility in the solvent of carbazole 4 and NBS to solve the problem. Decreasing the reaction temperature led to a worse selectivity (entry 2, Table 2), most probably because of the lower solubility of the starting material 4 at 0 °C. Then some solvents with a better solubility for 4 were tested, and CHCl3 performed better than CH2Cl2 at 0 °C (entries 4 and 5, Table 2). Changing the loading of NBS did not increase the selectivity for the reaction at this temperature (entries 6 and 7, Table 2). A higher yield of 5 and a better selectivity of 5 over 6 were obtained when the reaction was conducted at −20 °C (entry 8, Table 2). The loading of NBS was again screened to obtain a better selectivity. As expected, the selectivity dropped with the increasing loading of NBS (entries 9–17, Table 2). Gratifyingly, the ratio was improved to 97:3 when 0.92 equiv. of NBS was used (entry 9, Table 2). Therefore, we defined the standard conditions of monobromination as follows: bromination with 0.92 equiv. NBS in CHCl3 at −20 °C. With the optimized results in hand, we executed a reaction on 24 mmol (7.6 g) to afford 5 (6.5 g, 16.3 mmol) in 68% yield with a ratio of 5 and 6 as 95:5 as shown by NMR analysis.
2.3. Transformation of the C–Br bond
The target molecules have a formyl (7-methoxy-O-methylmukonal 8), carboxyl acid (clausine-K 11) or carboxylate functionality (clausine-H 12) at the C-3 position. Thus, compound 5 was treated with potassium tert-butoxide in DMSO–THF under an O2 atmosphere to remove the N-benyl group giving 3-bromo-2,7-dimethoxy-9H-carbazole 7 in 77% yield (Scheme 4). Starting from 7, to our delight, 7-methoxy-O-methylmukonal 8 was obtained by its subsequent treatment with KH, n-BuLi and DMF in 62% yield; clausine-K 11 was also synthesized by employing KH to deprotonate the N–H functionality of the carbazole, followed by the addition of n-BuLi to induce the Br–Li exchange. Subsequent carboxylation with gaseous carbon dioxide afforded the target molecule 11 in 68% yield; Using NaHCO3 as the base, with MeI as the methylation reagent, reaction of clausine-K 11 gave the natural product clausine-H in 70% yield as expected. Then clausine-O 10 was synthesized by the treatment of 8 with BBr3 in CH2Cl2 in 71% yield to cleave both of the methyl ethers (Scheme 4).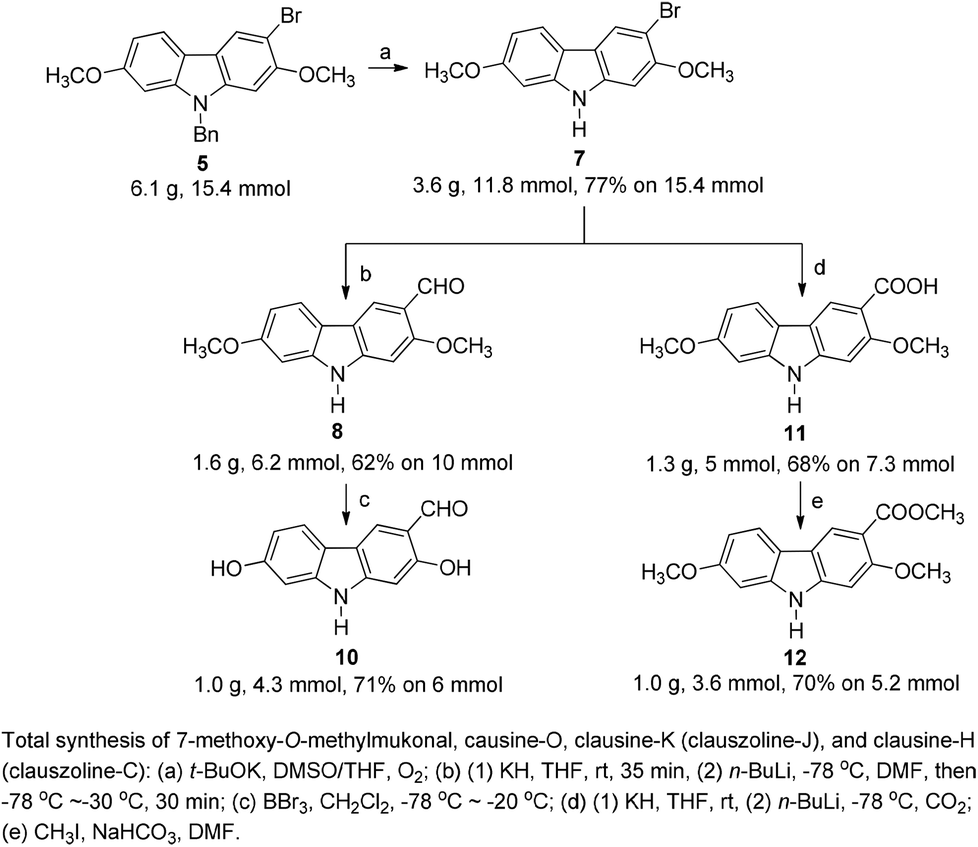 | ||
| Scheme 4 | ||
2.4. Selective demethylation of 2,7-dimethoxy-9H-carbazoles en route to 2-hydroxy-7-methoxy-9H-carbazoles
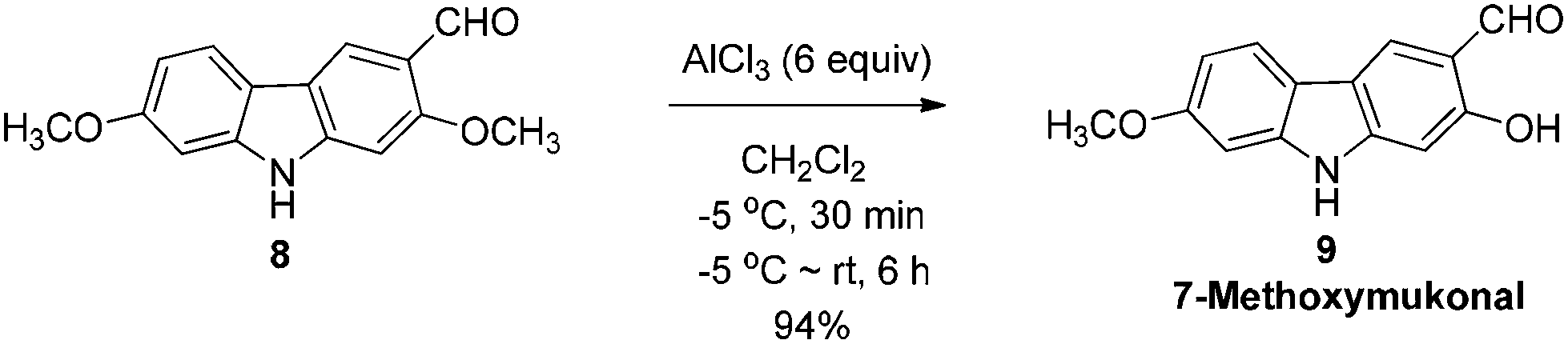
The next challenge is the transformation of 7-methoxy-O-mukonal 8 and clausine-H 12 to 7-methoxymukonal 9 and methyl 2-hydroxy-7-methoxy-9H-carbazole-3-carboxylate 13 selectively by just removing the methyl group at the C-2 position. As an efficient demethylation reagent, BBr3 failed for this procedure since demethylations occurred at both C-2 and C-7 positions to afford clausine-O 10. Fortunately, after systematic screening, we identified that AlCl3 is well-suited for such a purpose. The chelation of AlCl3 with the oxygen atom of the carbonyl group makes it selectively cleave the ether bond adjacent to the carbonyl group. 9 and 13 were synthesized in 94% yield and in 92% yield, respectively, as single products from the selective demethylation (Scheme 5). On the basis of NMR analysis, no demethylation occurred to afford the corresponding 7-hydroxy-2-methoxycarbazole (monodemethylation at the C-7 position) or 2,7-dihydroxycarbazole (demethylation at both C-2 and C-7 positions).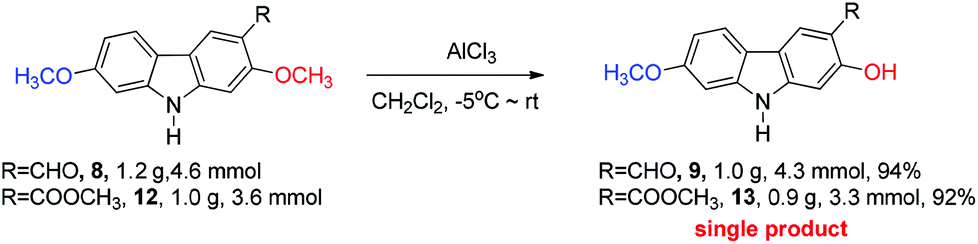 | ||
| Scheme 5 | ||
2.5. Structural confirmation of the product we prepared
The spectroscopic data of 7-methoxy-O-methylmukonal, 7-methoxymukonal, clausine-O, clausine-K and clausine-H are in full agreement with those of reported natural products (see ESI† for the 1H and 13C NMR spectra of all the prepared natural products). However, the NMR data of 13 are not in accordance with clausine-TY reported by Taufiq-Yap et al. (Table 3).15 Although this compound is a solid, we were not able to obtain a single crystal for X-ray diffraction study. Thus, the structure of this synthesized compound was further confirmed by X-ray single crystal diffraction of methyl 2-ethoxy-9-ethyl-7-methoxy-9H-carbazole-3-carboxylate 14 (Fig. 1),19 which was obtained via ethylation of 13 (Scheme 6). Thus, the structure of isolated clausine-TY must be revised.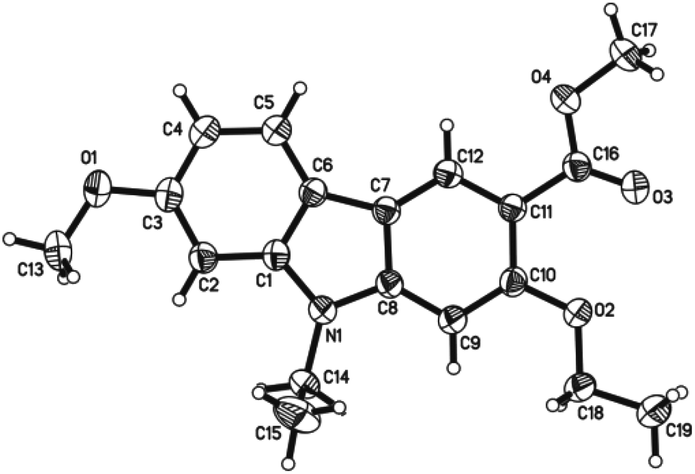 | ||
| Fig. 1 X-ray crystal structure of 14. | ||
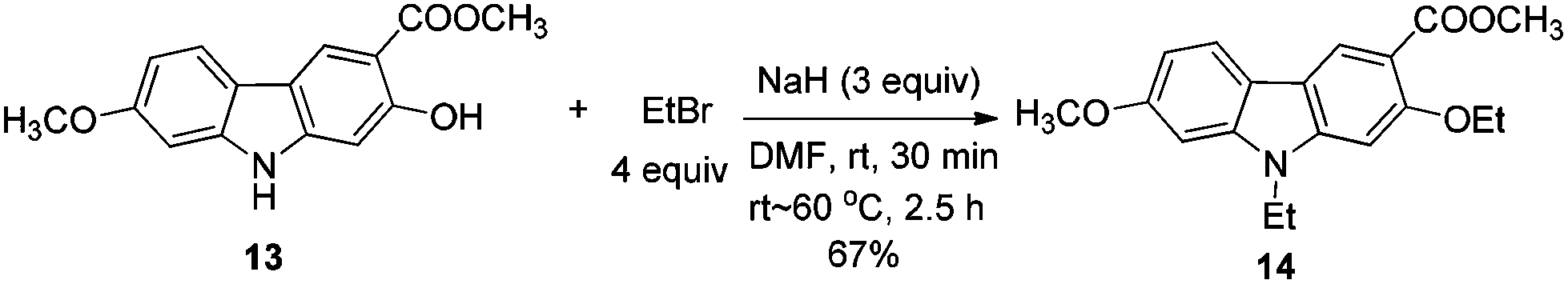 | ||
| Scheme 6 | ||
| δ 1 Ha | δ 13 Cb | ||
|---|---|---|---|
| Reported data | Our data | Reported data | Our data |
| a 400 MHz, acetone-d6. b 100 MHz, acetone-d6. | |||
| 10.87 (s, 1H, OH) | 11.07 (s, 1H, OH) | 166.6 | 173.0 |
| 10.54 (s, 1H, NH) | 10.44 (bs, 1H, NH) | 160.1 | 161.6 |
| 8.65 (s, 1H, ArH) | 8.51 (s, 1H, ArH) | 158.1 | 160.8 |
| 8.02 (d, J = 8.6 Hz, 1H, ArH) | 7.98 (d, J = 8.8 Hz, 1H, ArH) | 145.3 | 147.5 |
| 7.24 (s, 1H, ArH) | 7.04 (d, J = 2.0 Hz, 1H, ArH) | 143.2 | 144.0 |
| 7.05 (s, 1H, ArH) | 6.92 (s, 1H, ArH) | 125.2 | 122.8 |
| 6.88 (dd, J1 = 8.6 Hz, J2 = 2.2 Hz, 1H, ArH) | 6.85 (dd, J1 = 8.6 Hz, J2 = 2.2 Hz, 1H, ArH) | 121.5 | 122.0 |
| 3.87 (s, 3H, OCH3) | 4.02 (s, 3H, OCH3) | 118.6 | 118.8 |
| 3.87 (s, 3H, OCH3) | 3.89 (s, 3H, OCH3) | 117.8 | 118.4 |
| 111.6 | 109.9 | ||
| 109.6 | 106.4 | ||
| 96.1 | 98.4 | ||
| 94.7 | 96.8 | ||
| 57.2 | 56.5 | ||
| 55.8 | 53.2 | ||
Total yields of these naturally occurring carbazole alkaloids on the gram scale are: 7-methoxy-O-methylmukonal (5 steps with 30% overall yield), 7-methoxymukonal (6 steps with 28% overall yield), clausine-O (6 steps with 21% overall yield), clausine-K (clauszoline-J) (5 steps with 32% overall yield), clausine-H (clauszoline-C) (6 steps with 23% overall yield), and methyl 2-hydroxy-7-methoxy-9H-carbazole-3-carboxylate (7 steps with 21% overall yield).
3. Conclusions
In summary, we have realized the total synthesis of natural carbazole alkaloids including 7-methoxy-O-methylmukonal, 7-methoxymukonal, clausine-O, clausine-K (clauszoline-J), clausine-H (clauszoline-C) and methyl 2-hydroxy-7-methoxy-9H-carbazole-3-carboxylate via an efficient Au-catalyzed cyclization route as the key step from readily available starting materials methoxypropadiene and 1-benzyl-6-methoxy-1H-indole-2-carbaldehyde. Among them, 7-methoxymukonal and methyl 2-hydroxy-7-methoxy-9H-carbazole-3-carboxylate were synthesized for the first time.20 It is noted that this method enjoys an outstanding regioselectivity, excellent atom economy and mild reaction conditions, which leads to the gram scale production easily, providing the probability of large scale application in synthesis and medicinal chemistry. However, in this study, some reagents are still being used in a large excess, there is an opportunity for additional improvements. Further studies including biological activity studies of the derivatives and other carbazole alkaloid synthesis are being carried out in our laboratory.4. Experimental section
4.1. General information
1H and 13C NMR spectra were recorded using a Bruker AM 300 MHz spectrometer. IR spectra were recorded using a Perkin–Elmer 983G instrument. Elemental analyses were performed with a Carlo-Erba EA1110 elementary analysis instrument. Mass spectrometry was performed with an HP 5989A system. High-resolution mass spectrometry was performed with a Finnigan MAT 8430 or a Bruker APEXIII instrument. Toluene, THF, and Et2O were distilled from Na/benzophenone before use. CH2Cl2, DMF and DMSO were distilled from CaH2 before use. Unless otherwise indicated, chemicals and solvents were purchased from commercial suppliers.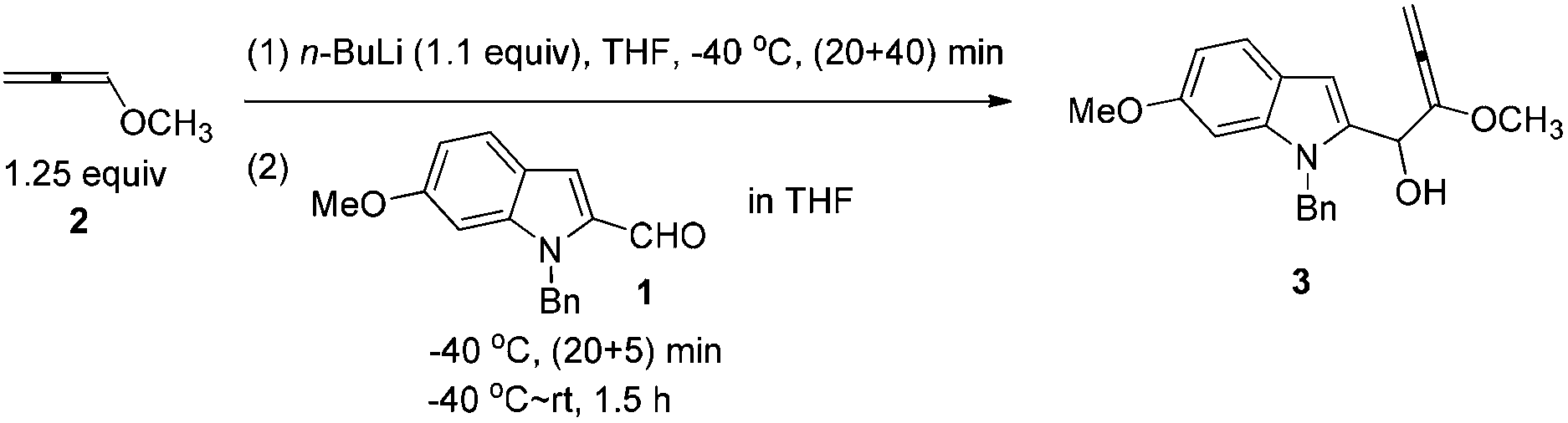
To a solution of 1-methoxypropa-1,2-diene 221 (5.2638 g, 75.2 mmol) in THF (200 mL) was added dropwise n-BuLi (26.4 mL, 2.5 M in hexane, 66 mmol) at −40 °C with stirring under a nitrogen atmosphere for 20 min. After being stirred for 40 min at −40 °C, a solution of 1-benzyl-6-methoxy-1H-indole-2-carbaldehyde 1 (15.9012 g, 60 mmol) in THF (40 mL) was added dropwise at this temperature for 20 min. Then the mixture was allowed to stir for 5 min at −40 °C and then warmed up to room temperature. After 1.5 h, the reaction was complete as monitored by TLC. A saturated aqueous solution of NH4Cl (60 mL) was added dropwise to the mixture at 0 °C using an ice water bath in 5 min to quench the reaction. The aqueous layer was extracted with diethyl ether (40 mL × 3). The combined organic layer was dried over anhydrous Na2SO4. After filtration and evaporation, the product 3 was then subjected to the next step.
To a dry 250 mL three neck round bottom flask were added sequentially AuCl (279.1 mg, 1.2 mmol), toluene (60 mL), and 4 Å MS (8.0012 g) under N2. Then a solution of 3 (prepared above) in toluene (60 mL) was added dropwise with stirring in 35 min at 25 °C under a nitrogen atmosphere. After stirring for 11 h, the reaction was complete as monitored by TLC. Filtration through a short column of silica gel (Ø46 mm × 50 mm) (eluent: Et2O (60 mL × 3); then ethyl acetate (30 mL × 4)), evaporation and recrystallization afforded 418b (n-hexane (30 mL)–CH2Cl2 (30 mL) = 1/1 for the first time, affording 16.6247 g of 4; n-hexane (10 mL)–CH2Cl2 (10 mL) = 1/1 for the second time for the solid from the evaporation of the mother liquid, affording 0.7599 g of 4, combined weight: 16.6247 + 0.7599 = 17.3846 g, 91%, two steps): solid; m.p. 146–147 °C (n-hexane–ethyl acetate); 1H NMR (300 MHz, CDCl3) δ 7.87 (d, J = 8.4 Hz, 2H, ArH), 7.31–7.19 (m, 3H, ArH), 7.18–7.10 (m, 2H, ArH), 6.88–6.74 (m, 4H, ArH), 5.37 (s, 2H, PhCH2), 3.83 (s, 6H, 2 × OCH3); 13C NMR (75 MHz, CDCl3) δ 158.3, 142.2, 136.9, 128.8, 127.4, 126.4, 120.1, 117.2, 107.2, 93.8, 55.6, 46.6; IR (KBr) ν (cm−1) 2953, 1602, 1574, 1485, 1470, 1453, 1434, 1358, 1332, 1279, 1258, 1242, 1195, 1167, 1120, 1077, 1052, 1040, 1027; MS (70 eV, EI) m/z (%) 318 (M+ + 1, 22.67), 317 (M+, 100).
A suspension of 4 (0.1903 g, 0.6 mmol) and NBS (0.1068 g, 0.6 mmol) in CCl4 (6 mL) was stirred at 15 °C for 1 h. The reaction was complete as monitored by TLC. Filtration through a short column of silica gel (eluent: ethyl acetate (15 mL × 3)), evaporation and column chromatography on silica gel (petroleum ether–ethyl acetate = 30/l) afforded 5 (0.1676 g, 71%) and 6 (27.4 mg, 10%). 5: solid; m.p. 140–142 °C (n-hexane–dichloromethane); 1H NMR (300 MHz, CDCl3) δ 8.12 (s, 1H, ArH), 7.83 (d, J = 8.4 Hz, 1H, ArH), 7.31–7.22 (m, 3H, ArH), 7.16–7.07 (m, 2H, ArH), 6.84 (dd, J1 = 8.4 Hz, J2 = 2.1 Hz, 1H, ArH), 6.75 (d, J = 2.4 Hz, 1H, ArH), 6.73 (s, 1H, ArH), 5.37 (s, 2H, CH2), 3.87 (s, 3H, CH3), 3.83 (s, 3H, CH3); 13C NMR (75 MHz, CDCl3) δ 158.6, 153.6, 142.0, 140.8, 136.4, 128.8, 127.6, 126.2, 123.6, 120.3, 117.7, 116.0, 107.7, 103.1, 93.7, 92.7, 56.4, 55.6, 46.5; IR (KBr) ν (cm−1) 1603, 1465, 1452, 1436, 1416, 1359, 1310, 1193, 1163, 1133, 1122, 1078, 1040, 1012; MS (70 eV, EI) m/z (%) 397 (M+(81Br), 44.49), 395 (M+(79Br), 45.01), 91 (100). Elemental analysis calcd for C21H18BrNO2: C, 63.65; H, 4.58; N, 3.53. Found: C, 63.99; H, 4.75; N, 3.48.
6: Solid; m.p. 244–246 °C (decomp) (n-hexane–dichloromethane); 1H NMR (300 MHz, CDCl3) δ 8.08 (s, 2H, ArH), 7.31–7.25 (m, 3H, ArH), 7.11–7.05 (m, 2H, ArH), 6.71 (s, 2H, ArH), 5.39 (s, 2H, CH2), 3.88 (s, 6H, 2 × OCH3); 13C NMR (75 MHz, CDCl3) δ 154.2, 140.9, 136.1, 129.0, 127.8, 126.2, 123.9, 116.8, 103.8, 92.8, 56.5, 46.7; IR (KBr) ν (cm−1) 3012, 2996, 2964, 2940, 2849, 2830, 1600, 1495, 1452, 1420, 1371, 1356, 1315, 1293, 1253, 1196, 1167, 1082, 1041; MS (70 eV, EI) m/z (%) 477 (M+(81,81Br), 51.44), 475 (M+(81,79Br), 100), 473 (M+(79,79Br), 52.11). Elemental analysis calcd for C21H17Br2NO2: C, 53.08; H, 3.61; N, 2.95. Found: C, 53.05; H, 3.61; N, 2.79.
To a 500 mL round bottom flask were added 4 (7.6082 g, 24 mmol) and CHCl3 (240 mL) subsequently. Then NBS (3.9304 g, 22.08 mmol) was added with stirring at −20 °C. After addition, the mixture was stirred for 30 min at this temperature. The reaction was complete as monitored by TLC. Filtration through a short column of silica gel (eluent: ethyl acetate (30 mL × 3)), evaporation and column chromatography on silica gel afforded 5 (petroleum ether–ethyl acetate = 30/1 for the first round, affording 4.1096 g of 5; petroleum ether–ethyl acetate = 30/1 for the second round (impure part), affording 2.3539 g of 5), combined weight: 4.1096 + 2.3539 = 6.4635 g, 68%): 1H NMR (300 MHz, CDCl3) δ 8.10 (s, 1H, ArH), 7.80 (d, J = 8.4 Hz, 1H, ArH), 7.29–7.20 (m, 3H, ArH), 7.11–7.04 (m, 2H, ArH), 6.82 (dd, J1 = 8.4 Hz, J2 = 2.1 Hz, 1H, ArH), 6.71 (d, J = 2.1 Hz, 1H, ArH), 6.68 (s, 1H, ArH), 5.30 (s, 2H, CH2), 3.83 (s, 3H, CH3), 3.81 (s, 3H, CH3); 13C NMR (75 MHz, CDCl3) δ 158.6, 153.6, 141.9, 140.8, 136.4, 128.8, 127.5, 126.2, 123.6, 120.3, 117.7, 116.0, 107.7, 103.1, 93.6, 92.7, 56.4, 55.6, 46.4.
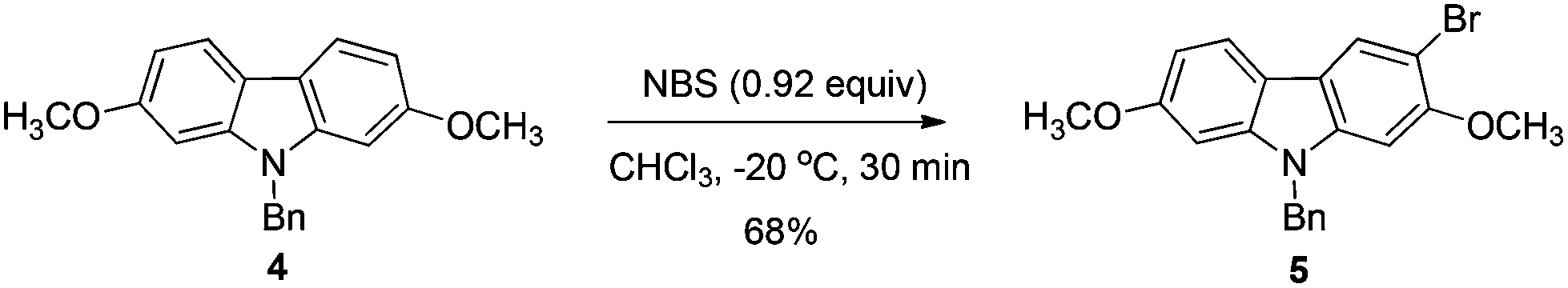
A solution of 5 (6.0895 g, 15.4 mmol) and t-BuOK (17.2483 g, 154 mmol) in THF (80 mL) and DMSO (80 mL) with an O2 balloon was stirred at room temperature. After 22.5 h, the reaction was complete as monitored by TLC. The resulting mixture was diluted with 50 mL of ethyl acetate, and quenched with water (50 mL) at 0 °C using an ice water bath. The aqueous layer was extracted with ethyl acetate (60 mL × 3), the combined organic layer was washed with water (30 mL × 3) and saturated brine (20 mL), and then dried over anhydrous Na2SO4. After filtration and evaporation, the obtained solid was washed with CHCl3 (10 mL) via filtration to afford 7 (3.6059 g, 77%): solid; m.p. 213–215 °C (n-hexane–ethyl acetate); 1H NMR (300 MHz, DMSO-d6) δ 11.18 (bs, 1H, NH), 8.24 (s, 1H, ArH), 7.94 (d, J = 8.4 Hz, 1H, ArH), 7.14 (s, 1H, ArH), 7.00 (d, J = 1.8 Hz, 1H, ArH), 6.79 (dd, J1 = 8.4 Hz, J2 = 2.1 Hz, 1H, ArH), 3.95 (s, 3H, CH3), 3.86 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ 158.9, 153.7, 142.1, 140.9, 124.1, 121.4, 118.4, 116.4, 108.9, 102.6, 95.8, 95.6, 57.2, 56.2; IR (KBr) ν (cm−1) 3377, 1611, 1567, 1499, 1455, 1435, 1328, 1301, 1270, 1254, 1230, 1197, 1162, 1112, 1039, 1031; MS (70 eV, EI) m/z (%) 307 (M+(81Br), 96.59), 305 (M+(79Br), 100). Elemental analysis calcd for C14H12BrNO2: C, 54.92; H, 3.95; N, 4.58. Found: C, 54.89; H, 4.16; N, 4.54.
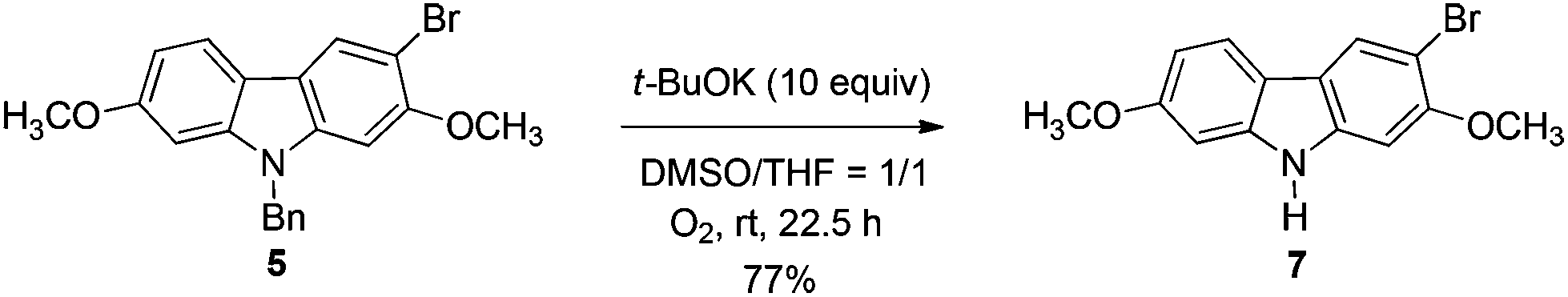
To a suspension of freshly washed (with anhydrous diethyl ether) and dried potassium hydride (161.6 mg, 4 mmol) in THF (25 mL) was added 7 (618.9 mg, 2 mmol). After being stirred for 35 min at room temperature, the mixture was cooled to −78 °C, and n-BuLi (0.92 mL, 2.4 M in hexane, 2.2 mmol) was added dropwise to the mixture within 5 min. After being stirred at −78 °C for another 30 min, anhydrous DMF (1.00 mL, d = 0.9445 g mL−1, 949 mg, 13 mmol) was added dropwise within 5 min. The mixture was allowed to warm up to −30 °C in 10 min, and after 30 min at −30 °C, it was allowed to warm to room temperature (5 min). The resulting mixture was quenched with water (10 mL) at 0 °C and extracted with ethyl acetate (20 mL × 3). The organic layer was washed with dilute hydrochloric acid (5%, 10 mL), and dried over anhydrous Na2SO4. After filtration and evaporation, the residue was purified by column chromatography on silica gel (petroleum ether–ethyl acetate = 2/1–1/2–1/5 for the first round, petroleum ether–ethyl acetate = 3/1–1/1–1/2 for the second round (impure part)) to give 7-methoxy-O-methylmukonal 816 (combined weight: 335.8 mg, 65%): solid; m.p. 222–223 °C (n-hexane–ethyl acetate) (lit.16 225 °C); 1H NMR (300 MHz, acetone-d6) δ 10.56 (bs, 1H, NH), 10.49 (s, 1H, CHO), 8.42 (s, 1H, ArH), 8.04 (d, J = 8.7 Hz, 1H, ArH), 7.16 (s, 1H, ArH), 7.08 (d, J = 0.9 Hz, 1H, ArH), 6.89 (dd, J1 = 8.4 Hz, J2 = 1.8 Hz, 1H, ArH), 4.04 (s, 3H, CH3), 3.90 (s, 3H, CH3); 13C NMR (75 MHz, acetone-d6) δ 189.3, 162.5, 160.8, 147.3, 143.8, 122.3, 120.9, 120.3, 119.0, 118.7, 110.3, 97.0, 94.7, 57.1, 56.6; 1H NMR (300 MHz, DMSO-d6) δ 11.53 (bs, 1H, NH), 10.40 (s, 1H, CHO), 8.40 (s, 1H, ArH), 8.03 (d, J = 8.4 Hz, 1H, ArH), 7.08 (s, 1H, ArH), 7.03 (s, 1H, ArH), 6.83 (d, J = 8.1 Hz, 1H, ArH), 4.01 (s, 3H, CH3), 3.87 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ 189.0, 161.2, 159.4, 146.5, 142.8, 121.8, 120.6, 118.6, 117.8, 117.4, 109.4, 96.2, 94.0, 56.9, 56.3; IR (KBr) ν (cm−1) 3253, 1665, 1640, 1605, 1510, 1471, 1430, 1372, 1356, 1325, 1274, 1251, 1206, 1157, 1036; MS (70 eV, EI) m/z (%) 256 (M+ + 1, 14.46), 255 (M+, 100).

Gram scale synthesis: following this procedure, the reaction of 7 (3.0564 g, 10 mmol), potassium hydride (0.8109 g, 20 mmol), n-BuLi (4.6 mL, 2.4 M in hexane, 11 mmol) and anhydrous DMF (5.00 mL, d = 0.9445 g mL−1, 4.745 g, 65 mmol) (DMF was added within 10 min) in THF (100 mL) afforded 7-methoxy-O-methylmukonal 8 (petroleum ether–CH2Cl2 = 1/1 to CH2Cl2) (1.5693 g, 62%): 1H NMR (300 MHz, DMSO-d6) δ 11.50 (bs, 1H, NH), 10.40 (s, 1H, CHO), 8.40 (s, 1H, ArH), 8.02 (d, J = 8.4 Hz, 1H, ArH), 7.09 (s, 1H, ArH), 7.03 (d, J = 1.8 Hz, 1H, ArH), 6.83 (dd, J1 = 8.6 Hz, J2 = 1.7 Hz, 1H, ArH), 4.01 (s, 3H, CH3), 3.87 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ 189.0, 161.2, 159.4, 146.5, 142.8, 121.7, 120.6, 118.6, 117.8, 117.3, 109.4, 96.3, 94.0, 56.9, 56.2.
To a dry Schlenk tube were added AlCl3 (804.0 mg, 6 mmol), 8 (255.0 mg, 1 mmol), and CH2Cl2 (30 mL) at −5 °C under a nitrogen atmosphere. The mixture was stirred at −5 °C for 30 min and then allowed to warm up to room temperature. After 6 h, the reaction was complete as monitored by TLC. The resulting mixture was quenched with 10 mL of a saturated aqueous solution of NaHCO3 at 0 °C and then diluted with 20 mL of ethyl acetate. The aqueous layer was extracted with ethyl acetate (20 mL × 3). The organic layer was washed with water (10 mL) and dried over anhydrous Na2SO4. After filtration and evaporation, the residue was pure without further purification to give 7-methoxymukonal 9
 2,9 (227.2 mg, 94%): solid; m.p. 221–222 °C (n-hexane–ethyl acetate) (lit.8 226–227 °C, lit.9 208–209 °C); 1H NMR (600 MHz, acetone-d6) δ 11.48 (s, 1H, OH), 10.60 (bs, 1H, NH), 10.00 (s, 1H, CHO), 8.34 (s, 1H, ArH), 7.98 (d, J = 8.4 Hz, 1H, ArH), 7.08 (d, J = 2.4 Hz, 1H, ArH), 6.90 (s, 1H, ArH), 6.89 (dd, J1 = 9.0 Hz, J2 = 1.8 Hz, 1H, ArH), 3.91 (s, 3H, CH3); 13C NMR (75 MHz, acetone-d6) δ 197.4, 162.0, 161.0, 148.0, 144.1, 127.9, 122.1, 119.5, 118.4, 116.9, 110.4, 98.1, 97.2, 56.6; 1H NMR (600 MHz, CDCl3) δ 11.43 (s, 1H, OH), 9.92 (s, 1H, CHO), 8.12 (bs, 1H, NH), 8.05 (s, 1H, ArH), 7.85 (d, J = 8.4 Hz, 1H, ArH), 6.89 (s, 1H, ArH), 6.88 (d, J = 8.4 Hz, 1H, ArH), 6.84 (s, 1H, ArH), 3.90 (s, 3H, OCH3); 13C NMR (75 MHz, CDCl3) δ 195.2, 160.6, 159.2, 145.7, 141.5, 126.0, 120.5, 117.9, 116.9, 115.4, 108.9, 96.9, 95.6, 55.7; 1H NMR (300 MHz, DMSO-d6) δ 11.45 (bs, 1H), 11.00 (s, 1H), 10.15 (s, 1H, CHO), 8.34 (s, 1H, ArH), 7.95 (d, J = 8.7 Hz, 1H, ArH), 7.00 (d, J = 1.8 Hz, 1H, ArH), 6.92 (s, 1H, ArH), 6.83 (dd, J1 = 8.6 Hz, J2 = 2.3 Hz, 1H, ArH), 3.87 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ 193.7, 160.3, 159.4, 146.9, 143.0, 124.3, 121.5, 118.0, 117.3, 116.5, 109.2, 97.3, 96.4, 56.3; IR (KBr) ν (cm−1) 3265, 2852, 2834, 1616, 1510, 1470, 1392, 1353, 1317, 1275, 1247, 1219, 1183, 1157, 1105, 1028; MS (70 eV, EI) m/z (%) 242 (M+ + 1, 16.31), 241 (M+, 100).
2,9 (227.2 mg, 94%): solid; m.p. 221–222 °C (n-hexane–ethyl acetate) (lit.8 226–227 °C, lit.9 208–209 °C); 1H NMR (600 MHz, acetone-d6) δ 11.48 (s, 1H, OH), 10.60 (bs, 1H, NH), 10.00 (s, 1H, CHO), 8.34 (s, 1H, ArH), 7.98 (d, J = 8.4 Hz, 1H, ArH), 7.08 (d, J = 2.4 Hz, 1H, ArH), 6.90 (s, 1H, ArH), 6.89 (dd, J1 = 9.0 Hz, J2 = 1.8 Hz, 1H, ArH), 3.91 (s, 3H, CH3); 13C NMR (75 MHz, acetone-d6) δ 197.4, 162.0, 161.0, 148.0, 144.1, 127.9, 122.1, 119.5, 118.4, 116.9, 110.4, 98.1, 97.2, 56.6; 1H NMR (600 MHz, CDCl3) δ 11.43 (s, 1H, OH), 9.92 (s, 1H, CHO), 8.12 (bs, 1H, NH), 8.05 (s, 1H, ArH), 7.85 (d, J = 8.4 Hz, 1H, ArH), 6.89 (s, 1H, ArH), 6.88 (d, J = 8.4 Hz, 1H, ArH), 6.84 (s, 1H, ArH), 3.90 (s, 3H, OCH3); 13C NMR (75 MHz, CDCl3) δ 195.2, 160.6, 159.2, 145.7, 141.5, 126.0, 120.5, 117.9, 116.9, 115.4, 108.9, 96.9, 95.6, 55.7; 1H NMR (300 MHz, DMSO-d6) δ 11.45 (bs, 1H), 11.00 (s, 1H), 10.15 (s, 1H, CHO), 8.34 (s, 1H, ArH), 7.95 (d, J = 8.7 Hz, 1H, ArH), 7.00 (d, J = 1.8 Hz, 1H, ArH), 6.92 (s, 1H, ArH), 6.83 (dd, J1 = 8.6 Hz, J2 = 2.3 Hz, 1H, ArH), 3.87 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ 193.7, 160.3, 159.4, 146.9, 143.0, 124.3, 121.5, 118.0, 117.3, 116.5, 109.2, 97.3, 96.4, 56.3; IR (KBr) ν (cm−1) 3265, 2852, 2834, 1616, 1510, 1470, 1392, 1353, 1317, 1275, 1247, 1219, 1183, 1157, 1105, 1028; MS (70 eV, EI) m/z (%) 242 (M+ + 1, 16.31), 241 (M+, 100).
Gram scale synthesis: following this procedure, the reaction of 8 (1.1685 g, 4.58 mmol) and AlCl3 (3.6689 g, 27.48 mmol) in CH2Cl2 (140 mL) in a 250 mL three neck round bottom flask afforded 7-methoxymukonal 9 (1.0392 g, 94%): 1H NMR (300 MHz, acetone-d6) δ 11.48 (s, 1H, OH), 10.62 (bs, 1H, NH), 10.00 (s, 1H, CHO), 8.33 (s, 1H, ArH), 7.98 (d, J = 8.7 Hz, 1H, ArH), 7.07 (d, J = 2.1 Hz, 1H, ArH), 6.90 (s, 1H, ArH), 6.89 (dd, J1 = 8.7 Hz, J2 = 2.4 Hz, 1H, ArH), 3.90 (s, 3H, OCH3); 13C NMR (75 MHz, acetone-d6) δ 197.3, 162.0, 161.0, 148.0, 144.1, 127.8, 122.1, 119.5, 118.4, 116.9, 110.4, 98.1, 97.2, 56.6.
To a solution of 8 (252.6 mg, 1 mmol) in CH2Cl2 (25 mL) was added a solution of BBr3 (9.0 mL, 1.0 M in CH2Cl2, 9 mmol) at −78 °C within 10 min under a nitrogen atmosphere. The mixture was stirred at −78 °C for 1 h and then allowed to warm up to −20 °C. After 18 h, the reaction was complete as monitored by TLC. The resulting mixture was quenched with 30 mL of a saturated aqueous solution of NaHCO3 at −20 °C, and the aqueous layer was extracted with ethyl acetate (20 mL × 3). The organic layer was washed with water (10 mL) and dried over anhydrous Na2SO4. After filtration and evaporation, the residue was purified by column chromatography on silica gel (petroleum ether–ethyl acetate = 2/1–1/1) to give clausine-O 105,16 (171.5 mg, 76%): solid; m.p. >280 °C (decomp) (n-hexane–ethyl acetate) (lit.5 >280 °C, lit.16 >300 °C (decomp)); 1H NMR (300 MHz, acetone-d6) δ 11.49 (s, 1H, OH), 10.56 (bs, 1H, NH), 10.00 (s, 1H, CHO), 8.55 (s, 1H, OH), 8.32 (s, 1H, ArH), 7.92 (d, J = 8.4 Hz, 1H, ArH), 6.98 (d, J = 2.1 Hz, 1H, ArH), 6.87 (s, 1H, ArH), 6.83 (dd, J1 = 8.3 Hz, J2 = 2.0 Hz, 1H, ArH); 13C NMR (75 MHz, acetone-d6) δ 197.3, 162.0, 158.5, 148.0, 144.3, 127.5, 122.2, 119.8, 117.7, 116.7, 111.1, 99.0, 98.0; 1H NMR (300 MHz, DMSO-d6) δ 11.34 (bs, 1H, NH), 10.96 (s, 1H, OH), 10.14 (s, 1H, CHO), 9.54 (s, 1H, OH), 8.28 (s, 1H, ArH), 7.85 (d, J = 8.1 Hz, 1H, ArH), 6.94–6.76 (m, 2H, ArH), 6.68 (d, J = 8.4 Hz, 1H, ArH); 13C NMR (75 MHz, DMSO-d6) δ 193.6, 160.1, 157.4, 146.8, 143.2, 123.7, 121.5, 118.4, 116.3, 116.1, 110.1, 98.1, 97.1; IR (KBr) ν (cm−1) 3370, 3267, 1617, 1578, 1467, 1443, 1375, 1327, 1282, 1258, 1217, 1200, 1176, 1150, 1116; MS (70 eV, EI) m/z (%) 228 (M+ + 1, 13.40), 227 (M+, 100).
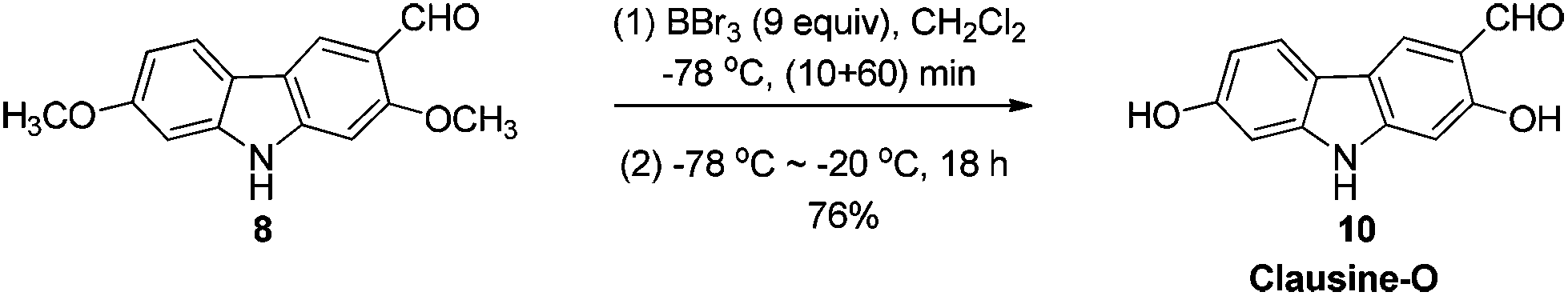
Gram scale synthesis: following this procedure, the reaction of 8 (1.5306 g, 6 mmol) and BBr3 (54 mL, 1.0 M in CH2Cl2, 54 mmol) (within 20 min) in CH2Cl2 (150 mL) in a 250 mL three neck round bottom flask at −78 °C for 90 min, and then at −20 °C for 16.5 h afforded clausine-O 10 (petroleum ether–ethyl acetate = 2/1) (0.9659 g, 71%): 1H NMR (300 MHz, DMSO-d6) δ 11.34 (bs, 1H, NH), 10.95 (s, 1H, OH), 10.14 (s, 1H, CHO), 9.54 (s, 1H, OH), 8.28 (s, 1H, ArH), 7.85 (d, J = 8.4 Hz, 1H, ArH), 6.85–6.81 (m, 2H, ArH), 6.67 (dd, J1 = 8.4 Hz, J2 = 2.1 Hz, 1H, ArH); 13C NMR (75 MHz, DMSO-d6) δ 193.7, 160.1, 157.5, 146.8, 143.3, 123.8, 121.5, 118.4, 116.3, 116.2, 110.1, 98.1, 97.1.
To a suspension of freshly washed (with anhydrous diethyl ether) and dried potassium hydride (324.4 mg, 8 mmol) in THF (20 mL) was added 7 (308.7 mg, 1 mmol). After being stirred for 35 min at room temperature, the resulting mixture was cooled to −78 °C, and n-BuLi (0.46 mL, 2.4 M in hexane, 1.1 mmol) was added within 5 min. After being stirred at −78 °C for another 30 min, CO2 (dried by passing through concentrated H2SO4) was bubbled through the reaction mixture for 40 min. The resulting mixture was then allowed to warm up to room temperature. After 3 h, the reaction was complete as monitored by TLC. The mixture was diluted with 10 mL of ethyl acetate, acidified with dilute hydrochloric acid (aq. 10%) to pH < 4, stirred for 10 min, extracted with ethyl acetate (20 mL × 3), washed with water (5 mL), and dried over anhydrous Na2SO4. After filtration and evaporation, the residue was purified by column chromatography on silica gel (petroleum ether–ethyl acetate = 1/1 to ethyl acetate–CH2Cl2 = 5/1–3/1–2/1) to give clausine-K (clauszoline-J) 1111,12,16 (198.4 mg, 73%): solid; m.p. 239–240 °C (n-hexane–ethyl acetate) (lit.11 250–256 °C, lit.12b,16 239–240 °C); 1H NMR (300 MHz, DMSO-d6) δ 12.19 (bs, 1H, NH), 11.35 (s, 1H, COOH), 8.44 (s, 1H, ArH), 7.99 (d, J = 8.4 Hz, 1H, ArH), 7.07 (s, 1H, ArH), 7.01 (s, 1H, ArH), 6.81 (dd, J1 = 8.4 Hz, J2 = 1.2 Hz, 1H, ArH), 3.93 (s, 3H, CH3), 3.86 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ 168.5, 159.0, 158.3, 144.4, 142.6, 124.1, 121.5, 117.2, 116.7, 113.2, 109.1, 96.0, 94.8, 56.9, 56.2; IR (KBr) ν (cm−1) 3315, 1664, 1618, 1577, 1464, 1445, 1412, 1384, 1345, 1322, 1289, 1273, 1245, 1235, 1200, 1163, 1114, 1083, 1038, 1024; MS (70 eV, EI) m/z (%) 272 (M+ + 1, 17.72), 271 (M+, 100).
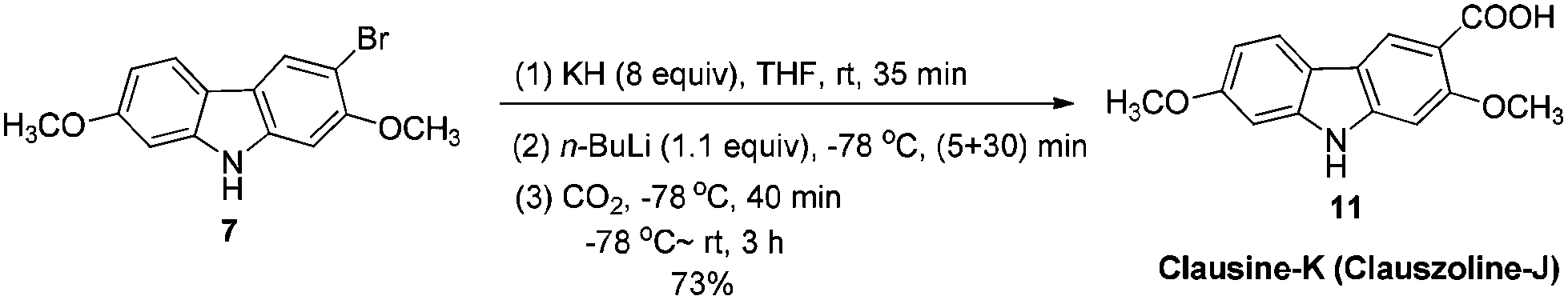
Gram scale synthesis: following this procedure, the reaction of 7 (2.2370 g, 7.3 mmol), potassium hydride (2.3363 g, 58.4 mmol), n-BuLi (3.2 mL, 2.5 M in hexane, 8 mmol) and CO2 (bubbled for 50 min) in THF (140 mL) afforded clausine-K (clauszoline-J) 11 (petroleum ether–ethyl acetate = 1/1 to ethyl acetate–CH2Cl2 = 5/1–3/1–2/1) (1.3469 g, 68%): 1H NMR (300 MHz, DMSO-d6) δ 12.21 (bs, 1H, NH), 11.34 (s, 1H, COOH), 8.46 (s, 1H, ArH), 7.99 (d, J = 8.7 Hz, 1H, ArH), 7.08 (s, 1H, ArH), 7.02 (d, J = 2.1 Hz, 1H, ArH), 6.81 (dd, J1 = 8.4 Hz, J2 = 1.2 Hz, 1H, ArH), 3.93 (s, 3H, CH3), 3.86 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ 168.5, 159.0, 158.3, 144.4, 142.6, 124.1, 121.4, 117.3, 116.7, 113.2, 109.1, 96.0, 94.9, 56.9, 56.2.
To a dry three neck round bottom flask equipped with a reflux condensing tube were added 11 (270.6 mg, 1 mmol), NaHCO3 (334.7 mg, 4 mmol), and DMF (20 mL) sequentially at 25 °C under N2. Then MeI (0.29 mL, d = 2.28 g mL−1, 639 mg, 4.5 mmol) was added within 5 min. After 13 h, the reaction was complete as monitored by TLC. The resulting mixture was quenched with water (20 mL). The aqueous layer was extracted with ethyl acetate (20 mL × 3), the combined organic layer was washed with water (10 mL × 2) and dried over anhydrous Na2SO4. After filtration and evaporation, the residue was purified by column chromatography on silica gel (petroleum ether–ethyl acetate = 5/1–4/1–1/3) to give clausine-H (clauszoline-C) 1211,12,16 (200.6 mg, 70%): solid; m.p. 192–193 °C (n-hexane–ethyl acetate) (lit.11 192–194 °C, lit.12b 184–185 °C, lit.16 191–192 °C); 1H NMR (300 MHz, acetone-d6) δ 10.40 (bs, 1H, NH), 8.45 (s, 1H, ArH), 7.99 (d, J = 8.4 Hz, 1H, ArH), 7.15 (s, 1H, ArH), 7.07 (d, J = 0.9 Hz, 1H, ArH), 6.87 (dd, J1 = 8.1 Hz, J2 = 0.6 Hz, 1H, ArH), 3.94 (s, 3H, CH3), 3.90 (s, 3H, CH3), 3.88 (s, 3H, CH3); 13C NMR (75 MHz, acetone-d6) δ 168.3, 160.6, 159.8, 145.5, 143.7, 124.6, 121.9, 118.7, 118.2, 114.6, 110.0, 96.9, 96.0, 57.4, 56.6, 52.4; IR (KBr) ν (cm−1) 3293, 1700, 1617, 1578, 1460, 1437, 1383, 1346, 1322, 1276, 1229, 1193, 1156, 1114, 1089, 1041, 1027; MS (70 eV, EI) m/z (%) 286 (M+ + 1, 10.22), 285 (M+, 53.59), 43 (100).
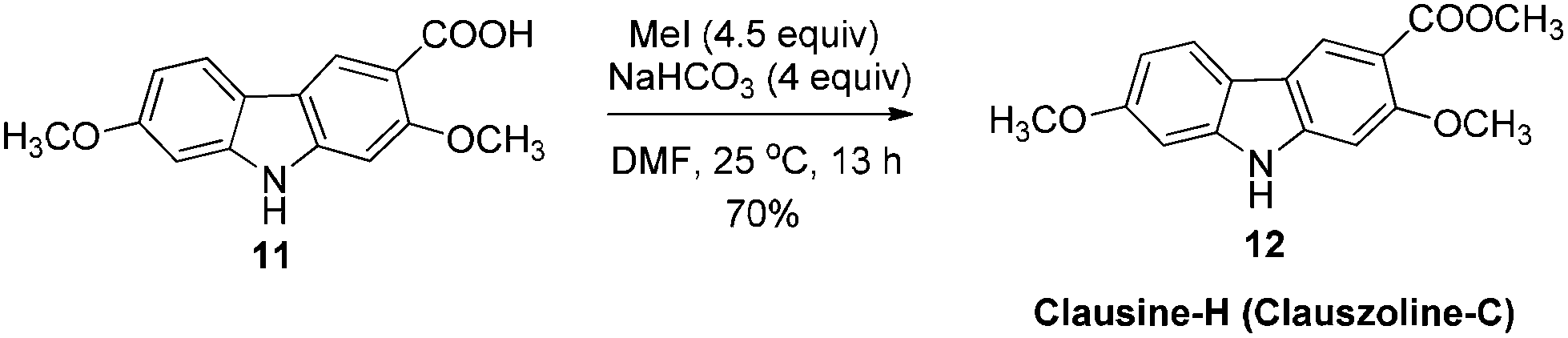
Gram scale synthesis: following this procedure, the reaction of 11 (1.4087 g, 5.2 mmol), NaHCO3 (1.7489 g, 20.8 mmol), and MeI (1.5 mL, d = 2.28 g mL−1, 3.408 g, 24 mmol) in DMF (100 mL) in a 250 mL three neck round bottom flask for 22 h afforded clausine-H (clauszoline-C) 12 (petroleum ether–ethyl acetate = 5/1–3/1–1/3) (1.0379 g, 70%): 1H NMR (300 MHz, acetone-d6) δ 10.43 (bs, 1H, NH), 8.45 (s, 1H, ArH), 7.99 (d, J = 8.7 Hz, 1H, ArH), 7.15 (s, 1H, ArH), 7.07 (d, J = 2.1 Hz, 1H, ArH), 6.87 (dd, J1 = 8.7 Hz, J2 = 2.1 Hz, 1H, ArH), 3.94 (s, 3H, CH3), 3.90 (s, 3H, CH3), 3.87 (s, 3H, CH3); 13C NMR (75 MHz, acetone-d6) δ 168.2, 160.5, 159.8, 145.5, 143.6, 124.6, 121.9, 118.6, 118.0, 114.4, 110.0, 96.8, 95.8, 57.2, 56.5, 52.4.
To a dry Schlenk tube were added AlCl3 (535.0 mg, 4 mmol), 12 (285.2 mg, 1 mmol), and CH2Cl2 (30 mL) at −5 °C under a nitrogen atmosphere. The mixture was stirred at −5 °C for 30 min and then allowed to warm up to room temperature. After 4 h, the reaction was complete as monitored by TLC. The resulting mixture was diluted with 20 mL of ethyl acetate and quenched with 15 mL of a saturated aqueous solution of NaHCO3 at 0 °C. The aqueous layer was extracted with ethyl acetate (30 mL × 3). The organic layer was washed with water (10 mL) and dried over anhydrous Na2SO4. After filtration and evaporation, the residue was pure without further purification to give 13 (246.0 mg, 91%): solid; m.p. 175–176 °C (n-hexane–ethyl acetate); 1H NMR (400 MHz, acetone-d6) δ 11.07 (s, 1H, OH), 10.44 (bs, 1H, NH), 8.51 (s, 1H, ArH), 7.98 (d, J = 8.8 Hz, 1H, ArH), 7.04 (d, J = 2.0 Hz, 1H, ArH), 6.92 (s, 1H, ArH), 6.85 (dd, J1 = 8.6 Hz, J2 = 2.2 Hz, 1H, ArH), 4.02 (s, 3H, OCH3), 3.89 (s, 3H, OCH3); 13C NMR (100 MHz, acetone-d6) δ 173.0, 161.6, 160.8, 147.5, 144.0, 122.8, 122.0, 118.8, 118.4, 109.9, 106.4, 98.4, 96.8, 56.5, 53.2; IR (KBr) ν (cm−1) 3318, 1672, 1624, 1598, 1509, 1466, 1442, 1377, 1329, 1275, 1248, 1216, 1195, 1163, 1126, 1088, 1027; MS (70 eV, EI) m/z (%) 272 (M+ + 1, 9.21), 271 (M+, 55.47), 239 (100). Elemental analysis calcd for C15H13NO4: C, 66.41; H, 4.83; N, 5.16. Found: C, 66.58, H, 4.83; N, 4.88.
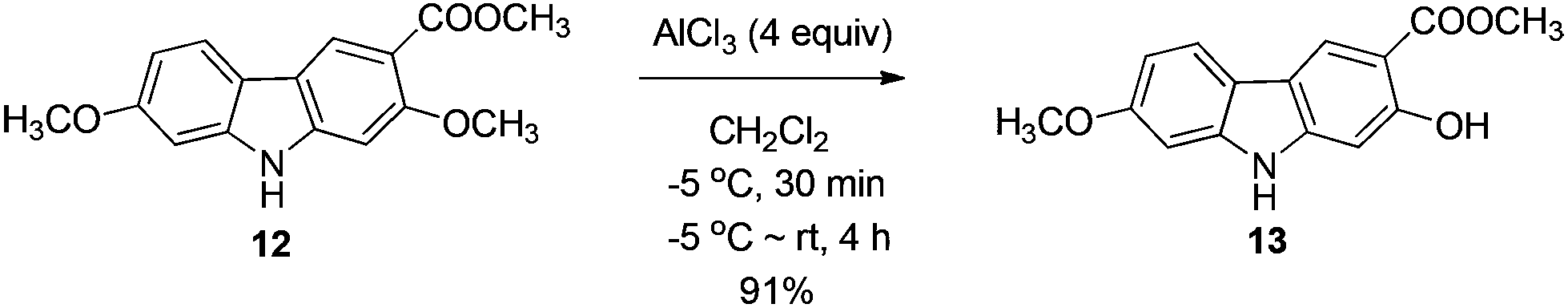
Gram scale synthesis: following this procedure, the reaction of 12 (1.0262 g, 3.6 mmol) and AlCl3 (1.9229 g, 14.4 mmol) in CH2Cl2 (100 mL) in a 250 mL three neck round bottom flask afforded 13 (0.8947 g, 92%): 1H NMR (300 MHz, acetone-d6) δ 11.08 (s, 1H, OH), 10.41 (bs, 1H, NH), 8.48 (s, 1H, ArH), 7.95 (d, J = 8.7 Hz, 1H, ArH), 7.03 (d, J = 1.8 Hz, 1H, ArH), 6.92 (s, 1H, ArH), 6.85 (dd, J1 = 8.6 Hz, J2 = 2.3 Hz, 1H, ArH), 4.01 (s, 3H, OCH3), 3.88 (s, 3H, OCH3); 13C NMR (75 MHz, acetone-d6) δ 172.9, 161.6, 160.7, 147.4, 144.0, 122.7, 121.9, 118.7, 118.4, 109.9, 106.3, 98.4, 96.8, 56.5, 53.2.
To a dry Schlenk tube equipped with a reflux condensing tube were added 13 (56.2 mg, 0.2 mmol), NaH (24.1 mg, 0.6 mmol, 60%), and DMF (3 mL) at room temperature under a nitrogen atmosphere. The mixture was stirred at room temperature for 30 min. Then EtBr (60 μL, d = 1.46 g mL−1, 87.2 mg, 0.8 mmol) and DMF (1 mL) were added and the mixture was warmed up to 60 °C and reacted for 2.5 h. The reaction was complete as monitored by TLC. The resulting mixture was cooled to room temperature and quenched with 5 mL of water at 0 °C. The aqueous layer was extracted with ethyl acetate (10 mL × 3), the combined organic layer was washed with saturated NH4Cl (6 mL) and dried over anhydrous Na2SO4. After filtration and evaporation, the residue was purified by column chromatography on silica gel (petroleum ether–ethyl acetate = 5/1–2/1) to give 14 (45.7 mg, 67%): solid; m.p. 156–158 °C (n-hexane–ethyl acetate); 1H NMR (300 MHz, CDCl3) δ 8.50 (s, 1H, ArH), 7.87 (d, J = 8.1 Hz, 1H, ArH), 6.89–6.77 (m, 3H, ArH), 4.27–4.15 (m, 4H, 2 × CH2), 3.93 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 1.54 (t, J = 7.1 Hz, 3H, CH3), 1.40 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (75 MHz, CDCl3) δ 167.2, 158.7, 157.9, 143.7, 141.9, 123.8, 120.5, 117.0, 116.4, 112.3, 107.5, 93.7, 93.4, 65.5, 55.7, 51.6, 37.6, 14.8, 13.4; IR (KBr) ν (cm−1) 2983, 2944, 2933, 2882, 2835, 1712, 1600, 1582, 1568, 1471, 1439, 1399, 1377, 1365, 1354, 1339, 1320, 1278, 1261, 1204, 1177, 1127, 1078, 1053, 1026, 1001; MS (70 eV, EI) m/z (%) 328 (M+ + 1, 21.78), 327 (M+, 100). Elemental analysis calcd for C19H21NO4: C, 69.71; H, 6.47; N, 4.28. Found: C, 69.69, H, 6.43; N, 4.04.
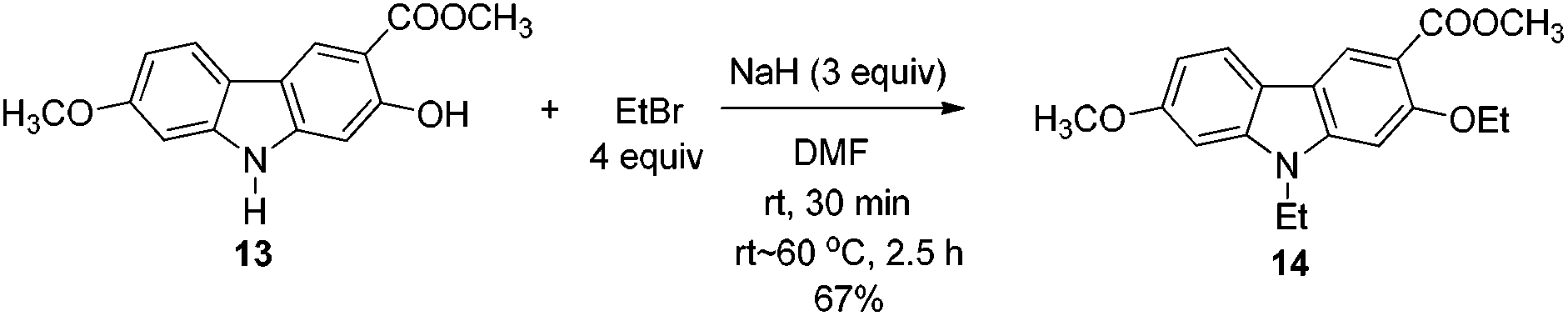
Acknowledgements
Financial support from the National Natural Science Foundation for China (21232006) and the National Basic Research Program of China (2011CB808700) is greatly appreciated. S. Ma is a Qiu shi Adjunct professor at Zhejiang University. We thank Mr Ruizhi Lü in our group for reproducing the results of entry 9 in Table 2, 8 (7-methoxy-O-methylmukonal) and 9 (7-methoxymukonal).Notes and references
- (a) H.-J. Knölker and K. R. Reddy, Chem. Rev., 2002, 102, 4303 CrossRef; (b) A. W. Schmidt, K. R. Reddy and H.-J. Knölker, Chem. Rev., 2012, 112, 3193 CrossRef CAS PubMed; (c) H.-J. Knölker, Top. Curr. Chem., 2005, 244, 115 Search PubMed; (d) D. P. Chakraborty and S. Roy, in Progress in the Chemistry of Organic Natural Products, ed. W. Herz, H. Grisebach, G. W. Kirby, W. Steglich and C. Tamm, Springer-Verlag, Wien, 1991, vol. 57, p. 71 Search PubMed; (e) D. P. Chakraborty, in The Alkaloids, ed. G. A. Cordell, Academic Press, New York, 1993, vol. 44, p. 257 Search PubMed.
- N. Ruangrungsi, J. Ariyaprayoon, G. L. Lange and M. G. Organ, J. Nat. Prod., 1990, 53, 946 CrossRef CAS.
- B. Kongkathip, N. Kongkathip, A. Sunthitikawinsakul, C. Napaswat and C. Yoosook, Phytother. Res., 2005, 19, 728 CrossRef CAS PubMed.
- T. A. Choi, R. Czerwonka, R. Forke, A. Jäger, J. Knöll, M. P. Krahl, T. Krause, K. R. Reddy, S. G. Franzblau and H.-J. Knölker, Med. Chem. Res., 2008, 17, 374 CrossRef CAS.
- T.-S. Wu, S.-C. Huang, P.-L. Wu and C.-S. Kuoh, Phytochemistry, 1999, 52, 523 CrossRef CAS.
- W. Lin, Y. Wang, S. Lin, C. Li, C. Zhou, S. Wang, H. Huang, P. Liu, G. Ye and X. Shen, Eur. J. Med. Chem., 2012, 47, 214 CrossRef CAS PubMed.
- U. Songsiang, T. Thongthoom, C. Boonyarat and C. Yenjai, J. Nat. Prod., 2011, 74, 208 CrossRef CAS PubMed.
- C. Chaichantipyuth, S. Pummangura, K. Naowsaran, D. Thanyavuthi, J. E. Anderson and J. L. McLaughlin, J. Nat. Prod., 1988, 51, 1285 CrossRef CAS.
- T. Thongthoom, U. Songsiang, C. Phaosiri and C. Yenjai, Arch. Pharm. Res., 2010, 33, 675 CrossRef CAS PubMed.
- A. Sunthitikawinsakul, N. Kongkathip, B. Kongkathip, S. Phonnakhu, J. W. Daly, T. F. Spande, Y. Nimit and S. Rochanaruangtai, Planta Med., 2003, 69, 155 CrossRef CAS PubMed.
- T.-S. Wu, S.-C. Huang, P.-L. Wu and C.-M. Teng, Phytochemistry, 1996, 43, 133 CrossRef CAS PubMed.
- (a) C. Ito, S. Katsuno, H. Ohta, M. Omura, I. Kajiura and H. Furukawa, Chem. Pharm. Bull., 1997, 45, 48 CrossRef CAS; (b) U. Sriphana, Y. Thongsri, C. Prariyachatigul, C. Pakawatchai and C. Yenjai, Arch. Pharm. Res., 2013, 36, 1078 CrossRef CAS PubMed.
- C. Ito, H. Ohta, H. T.-W. Tan and H. Furukawa, Chem. Pharm. Bull., 1996, 44, 2231 CrossRef CAS.
- C. Yenjai, S. Sripontan, P. Sriprajun, P. Kittakoop, A. Jintasirikul, M. Tanticharoen and Y. Thebtaranonth, Planta Med., 2000, 66, 277 CrossRef CAS PubMed.
- Y. H. Taufiq-Yap, T. H. Peh, G. C. L. Ee, M. Rahmani, M. A. Sukari, A. M. Ali and R. Muse, Nat. Prod. Res., 2007, 21, 810 CrossRef CAS PubMed.
- (a) O. Kataeva, M. P. Krahl and H.-J. Knölker, Org. Biomol. Chem., 2005, 3, 3099 RSC; (b) M. P. Krahl, O. Kataeva, A. W. Schmidt and H.-J. Knölker, Eur. J. Org. Chem., 2013, 59 CrossRef CAS; (c) I. Bauer and H.-J. Knölker, Top. Curr. Chem., 2012, 309, 203 CrossRef CAS PubMed.
- (a) A. J. Birch, P. E. Cross, J. Lewis, D. A. White and S. B. Wild, J. Chem. Soc. A, 1968, 332 RSC; (b) H.-J. Knölker, B. P. Ahrens, G. M. Heininger and P. G. Jones, Tetrahedron, 2000, 56, 2259 CrossRef.
- (a) W. Kong, C. Fu and S. Ma, Chem. – Eur. J., 2011, 17, 13134 CrossRef CAS PubMed; (b) Y. Qiu, D. Ma, C. Fu and S. Ma, Org. Biomol. Chem., 2013, 11, 1666 RSC.
- Crystal data for compound 14: C19H21NO4, MW = 327.37, orthorhombic, space group P212121, final R indices [I > 2σ(I)], R1 = 0.0425, wR2 = 0.1074; R indices (all data), R1 = 0.0514, wR2 = 0.1170; a = 8.8435 (4) Å, b = 13.8907 (7) Å, c = 14.1790 (6) Å, α = 90.00°, β = 90.00 (4)°, γ = 90.00°, V = 1741.80 (14) Å3, T = 293 (2) K, Z = 4, reflections collected/unique 11501/1548 (Rint = 0.0422), number of observations [>2σ(I)] 1796, parameters: 222. Supplementary crystallographic data have been deposited at the Cambridge Crystallographic Data Centre, CCDC 988874.
- After we submitted our manuscript for the first time to Org. Chem. Front. on April 18, H.-J. Knölker reported the synthesis of 7-methoxymukonal. Eur. J. Org. Chem., 2014, DOI:10.1002/ejoc.201402201, which was published online on May 16, 2014.
- For an account on the chemistry of alkoxyallenes; see: M. Brasholz, H.-U. Reissig and R. Zimmer, Acc. Chem. Res., 2009, 42, 45 CrossRef CAS PubMed.
- (a) M. Rawat and W. D. Wulff, Org. Lett., 2004, 6, 329 CrossRef CAS PubMed; (b) A. A. Haddach, A. Kelleman and M. V. Deaton-Rewolinski, Tetrahedron Lett., 2002, 43, 399 CrossRef CAS.
- (a) M. P. Moyer, J. F. Shiurba and H. Rapoport, J. Org. Chem., 1986, 51, 5106 CrossRef CAS; (b) C.-A. Harrison, P. M. Jackson, C. J. Moody and J. M. J. Williams, J. Chem. Soc., Perkin Trans. 1, 1995, 1131 RSC; (c) L. Wang, K. W. Woods, Q. Li, K. J. Barr, R. W. McCroskey, S. M. Hannick, L. Gherke, R. B. Credo, Y.-H. Hui, K. Marsh, R. Warner, J. Y. Lee, N. Zielinski-Mozng, D. Frost, S. H. Rosenberg and H. L. Sham, J. Med. Chem., 2002, 45, 1697 CrossRef CAS PubMed.
- Z. Du, J. Lu, H. Yu, Y. Xu and A. Li, J. Chem. Res., 2010, 222 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 988874. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00163j |
| This journal is © the Partner Organisations 2014 |