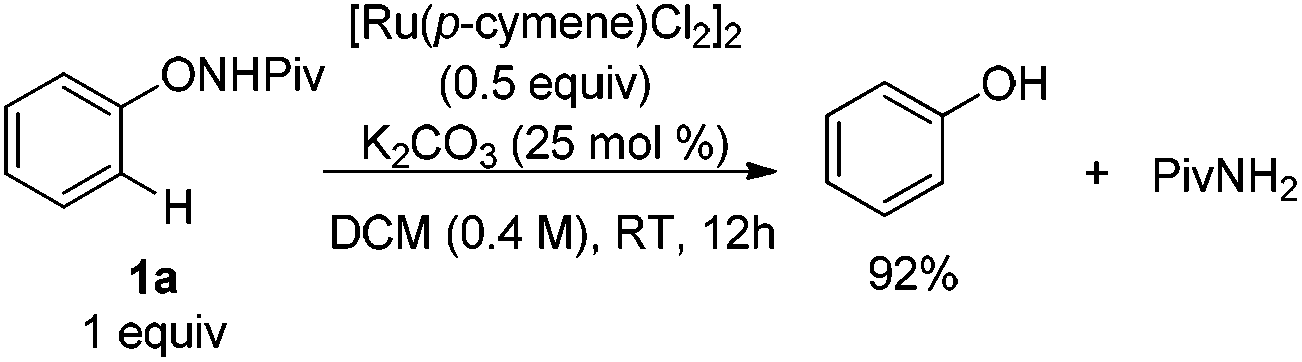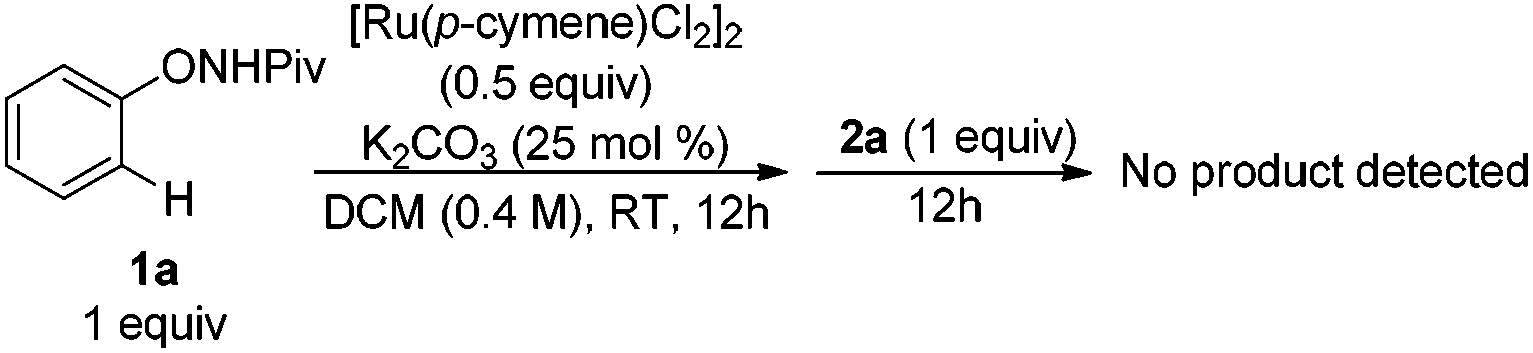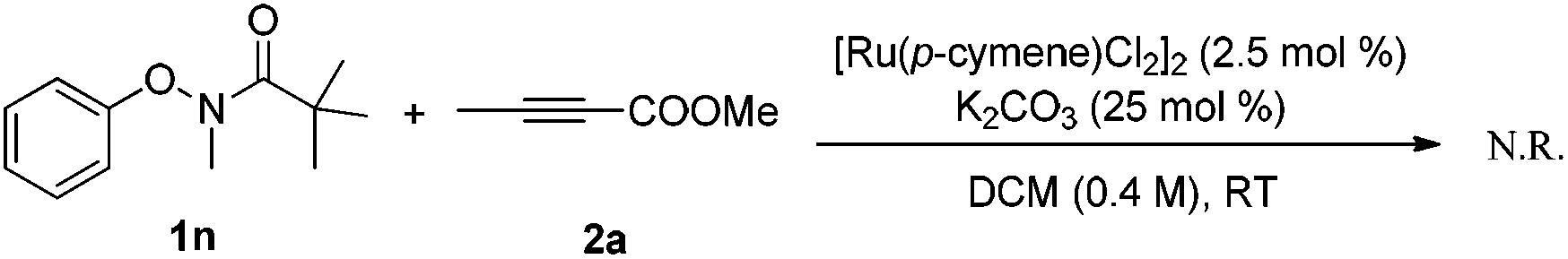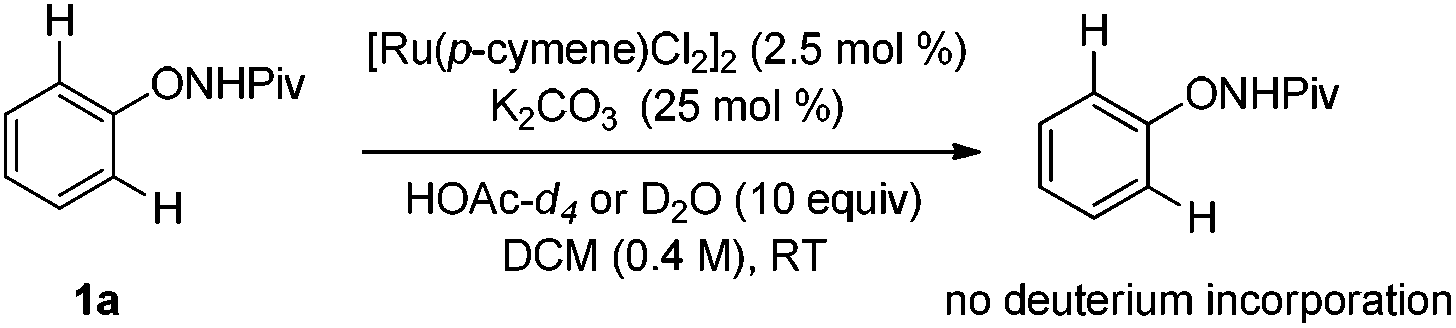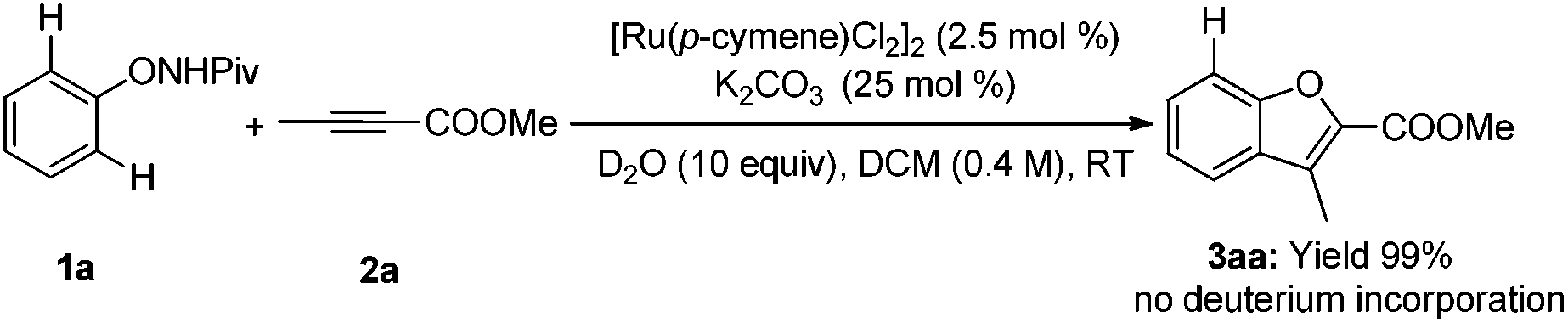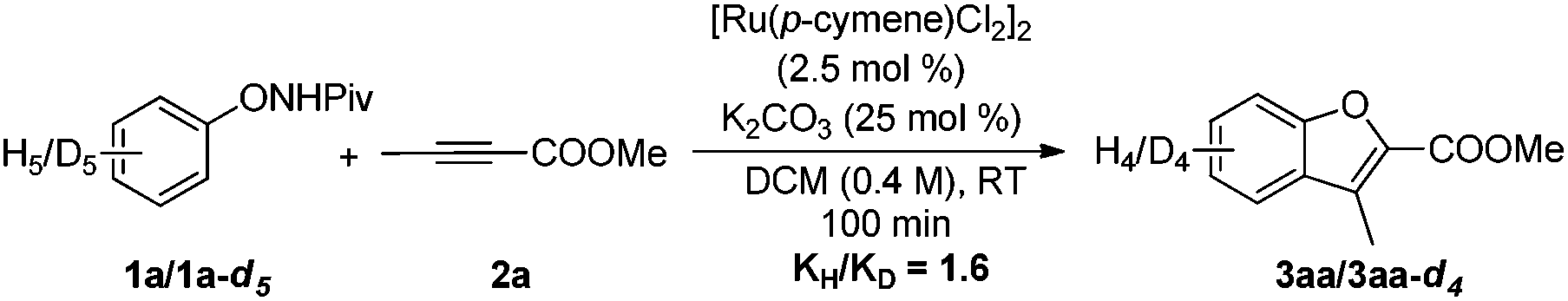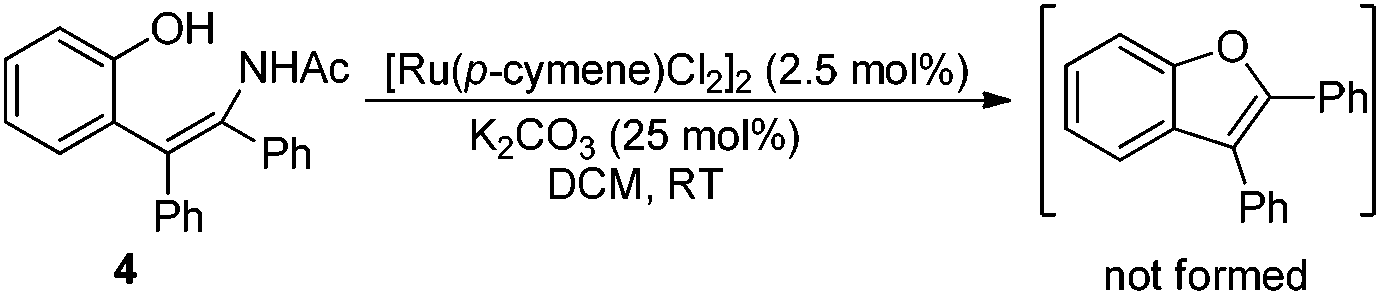DOI:
10.1039/C4QO00196F
(Research Article)
Org. Chem. Front., 2014,
1, 1161-1165
Synthesis of benzofurans via ruthenium-catalyzed redox-neutral C–H functionalization and reaction with alkynes under mild conditions†
Received
14th July 2014
, Accepted 15th September 2014
First published on 16th September 2014
Abstract
Using an oxidizing directing group, a mild RuII-catalyzed direct coupling of N-phenoxypivalamide with internal alkynes was developed, generating the benzofurans in moderate to high yields.
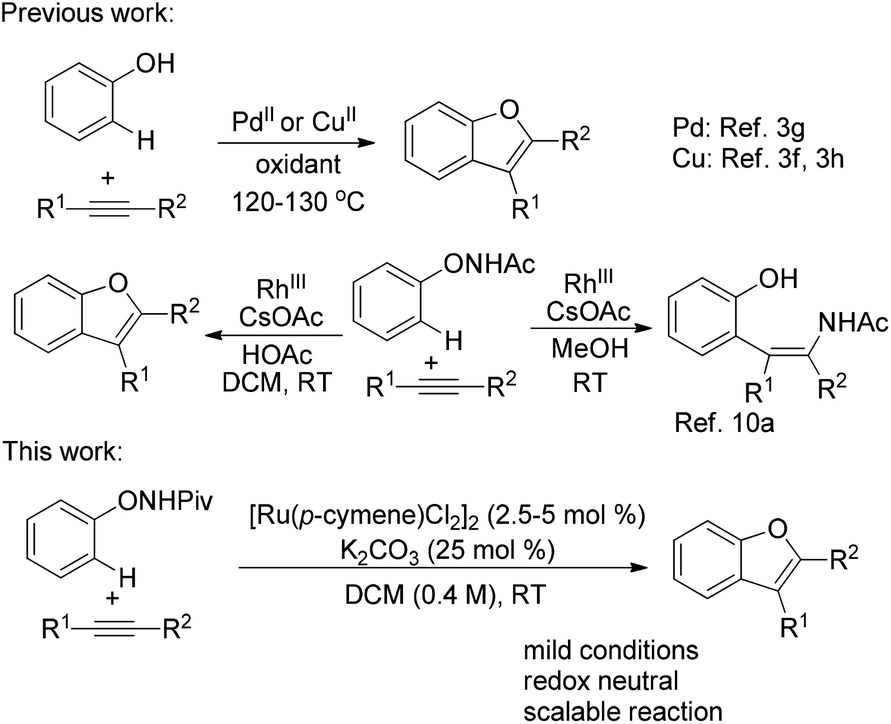
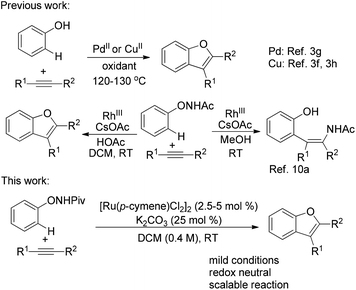
Benzofuran is a privileged structural framework widely existing in natural products, biologically active molecules and pharmaceuticals.1 Accordingly, continued efforts have been devoted to the development of synthetic approaches for this motif.2 Among these approaches, the transition-metal-catalyzed transformation has received considerable attention. In particular, the transition-metal-catalyzed C–H bond activation provides a powerful and straightforward synthetic method by obviating the need for prefunctionalized substrates.3 A representative example is the direct access to benzofurans by the oxidative annulation of phenols and unactivated internal alkynes (Scheme 1).3f–h
 |
| | Scheme 1 Synthesis of benzofurans by annulation of arenes with alkynes. | |
The transition-metal-catalyzed oxidative annulation of various aromatic substrates with alkynes represents a step-economical access to heterocycles.4 Despite the notable advances, the use of an external, mostly metallic oxidant for catalyst regeneration is inevitable and harsh conditions are generally required. To address these drawbacks, an attractive redox-neutral strategy employing an oxidizing directing group which acts as both a directing group and an internal oxidant has been applied in this field.5 Diverse oxidizing directing groups were designed to construct different heterocycles by Pd-, Rh- or Ru-catalyzed transformations.6–8 In these reactions, the regeneration of the active catalyst was accompanied by the cleavage of the N–O or N–N bond.9 In our previous work,10a,b we designed a novel oxidizing directing group for mild Rh-catalyzed C–H functionalizations of N-phenoxyacetamide10 with alkynes to furnish ortho-alkenyl phenols or benzofuran products selectively (Scheme 1). We envisioned that an analogous but more economic process may be achieved by the use of a less expensive catalyst, such as ruthenium.
Ruthenium(II) catalysts have recently attracted increasing interest in C–H functionalization due to their good stability, low cost and high efficiency.11 Herein, we wish to describe the development of a mild RuII catalyzed C–H functionalization of N-phenoxypivalamide with internal alkynes for the synthesis of benzofurans without any external oxidant (Scheme 1).
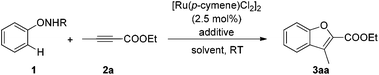
In preliminary experiments, N-phenoxyacetamide (1.2 equiv.) was treated with [Ru(p-cymene)Cl2]2 (2.5 mol%), CsOAc (25 mol%) and ethyl 2-butynoate (2a, 1 equiv.) in DCM at room temperature. The benzofuran product 3aa was obtained in 46% yield (Table 1, entry 1). Further examination indicated that N-phenoxyacetamide was decomposed to phenol as a side product, which decreased the yield of 3aa. Other substrates with different substituents on the nitrogen atom were screened. Fortunately, N-phenoxypivalamide (1a) gave a nearly quantitative yield while other substrates were found to be less effective or ineffective (Table 1, entries 2 and 3).12 K2CO3 appeared to be the ideal additive owing to the shorter reaction time (Table 1, entry 5). The addition of acetic acid (2 equiv.) inhibited the reaction (Table 1, entry 7). To our surprise, the transformation also proceeded well when DBU was used instead of metal carboxylate (Table 1, entry 6). Other solvents were not as good as DCM and no ortho-hydroxyphenyl substituted enamide was detected compared with Rh-catalyzed reactions in our previous work (Table 1, entries 8–11).10a
Table 1 Optimization of the reaction conditionsa
|
|
| Entry |
R |
Additive |
Solvent |
Yieldb (%) |
|
Reaction conditions: 1 (0.12 mmol), 2a (0.1 mmol), [Ru(p-cymene)Cl2]2 (2.5 mol%), and additives (25 mol%) in solvent (0.4 M) at room temperature for 12 h under air. Piv: pivaloyl.
1H NMR yield.
Reaction was run at room temperature for 24–48 h as monitored by TLC.
2 equiv. of HOAc was added.
|
| 1c |
Ac |
CsOAc |
DCM |
46 |
| 2c |
Piv |
CsOAc |
DCM |
99 |
| 3c |
Ts |
CsOAc |
DCM |
9 |
| 4 |
Piv |
CsOAc |
DCM |
61 |
| 5 |
Piv |
K2CO3 |
DCM |
99 |
| 6 |
Piv |
DBU |
DCM |
90 |
| 7d |
Piv |
K2CO3 |
DCM |
12 |
| 8 |
Piv |
K2CO3 |
Dioxane |
83 |
| 9 |
Piv |
K2CO3 |
Toluene |
90 |
| 10 |
Piv |
K2CO3 |
CH3CN |
76 |
| 11 |
Piv |
K2CO3 |
MeOH |
10 |
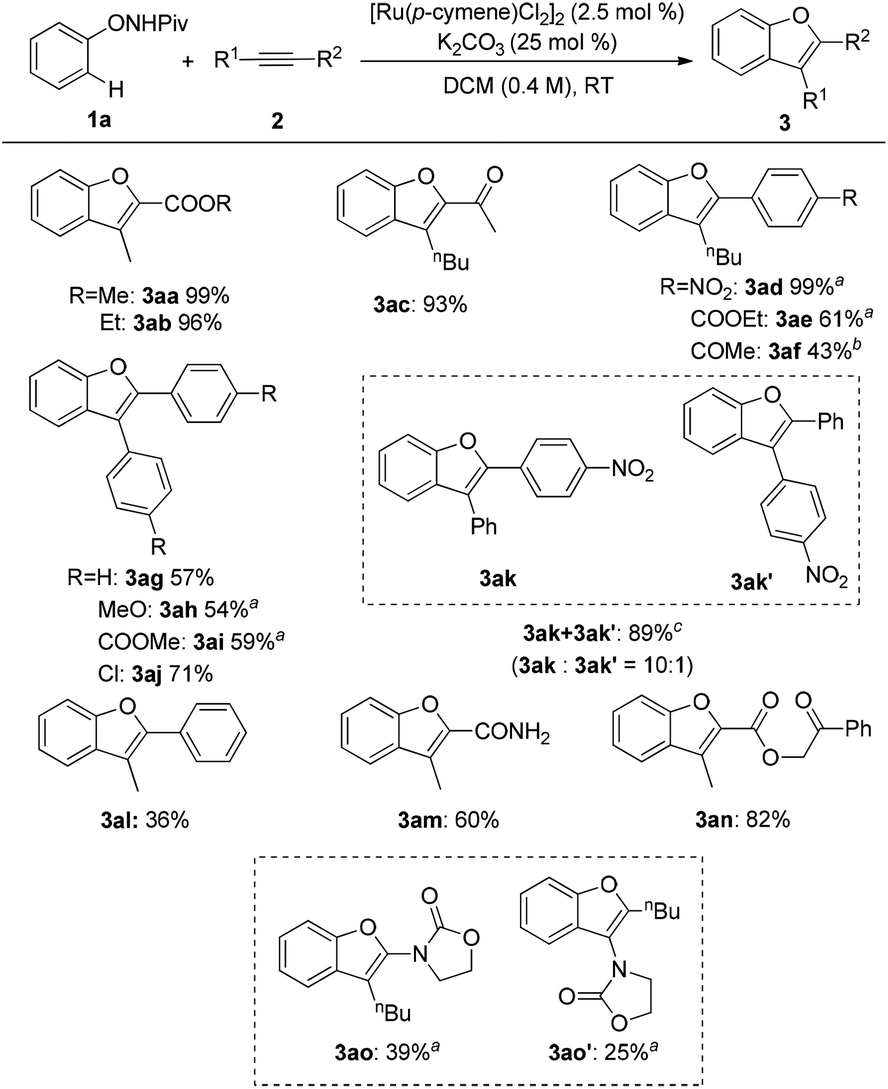
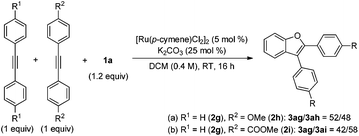
With the optimized conditions in hand, various alkynes were reacted with 1a and the results are shown in Scheme 2. Several functional groups, such as ester (3aa, 3ab, 3an), ketone (3ac) and amide (3am), were well tolerated in this reaction to give good yields of the expected products with a high regioselectivity. Aryl–alkyl disubstituted alkynes seemed to be less effective unless an electron-withdrawing group was equipped (3ad–3af, 3al). Moreover, symmetrical diaryl acetylenes bearing various substituents were compatible with the reaction conditions furnishing the corresponding benzofurans in moderate to good yields (3ag–3aj). Unsymmetrical diaryl acetylene resulted in relatively poor regioselectivity (3ak and 3ak′). It is noteworthy that the ynamide 2o was also a good reactant for this transformation affording moderate yields of two isomers (3ao and 3ao′). Nevertheless, dialkyl alkynes and terminal alkynes gave poor results.
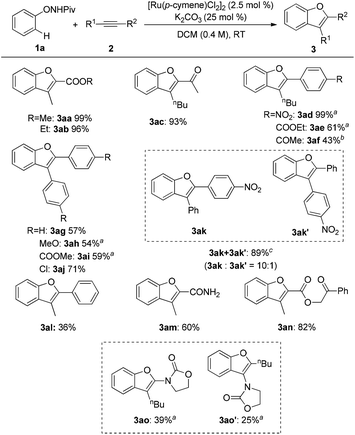 |
| | Scheme 2 Scope of alkynes for the synthesis of the benzofurans. Reaction conditions: 1a (0.24 mmol), 2 (0.2 mmol), [Ru(p-cymene)Cl2]2 (2.5 mol%), and K2CO3 (25 mol%) in DCM (0.4 M) at room temperature under air; isolated yield was reported. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 mol% of the catalyst was used. b10 mol% of catalyst was used. cNMR spectra indicated an inseparable mixture of isomers, the ratio was determined by 1H NMR. 5 mol% of the catalyst was used. b10 mol% of catalyst was used. cNMR spectra indicated an inseparable mixture of isomers, the ratio was determined by 1H NMR. | |
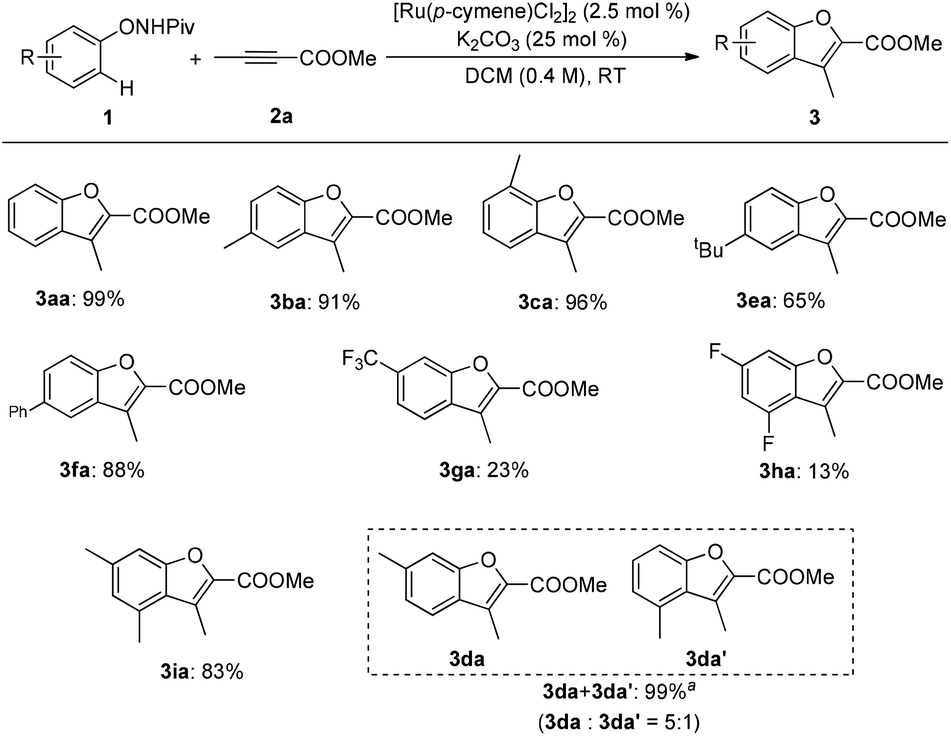
Various substituents on N-phenoxypivalamide were then examined and a significant electronic effect was observed (Scheme 3). With alkyl- or phenyl-substituted N-phenoxypivalamides, the reaction proceeded smoothly and provided the desired products in good to excellent yields (3aa–3fa). In addition, sterically hindered 3,5-dimethyl-substituted N-phenoxypivalamide was also tolerated (3ia). Unfortunately, low yields were obtained with 3-trifluoromethyl- and 3,5-difluoro-substituted N-phenoxypivalamides as the substrates (3ga, 3ha). Of note, when a meta-substituted N-phenoxypivalamide was employed, the areneruthenation mainly occurred at the less hindered site (3da).
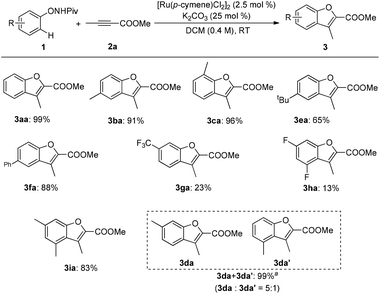 |
| | Scheme 3 Reaction scope for substituted N-phenoxypivalamides. Reaction conditions: 1 (0.24 mmol), 2a (0.2 mmol), [Ru(p-cymene)Cl2]2(2.5 mol%), and K2CO3 (25 mol%) in DCM (0.4 M) at room temperature under air; isolated yield was reported. aNMR spectra indicated an inseparable mixture of isomers, the ratio was determined by 1H NMR. | |
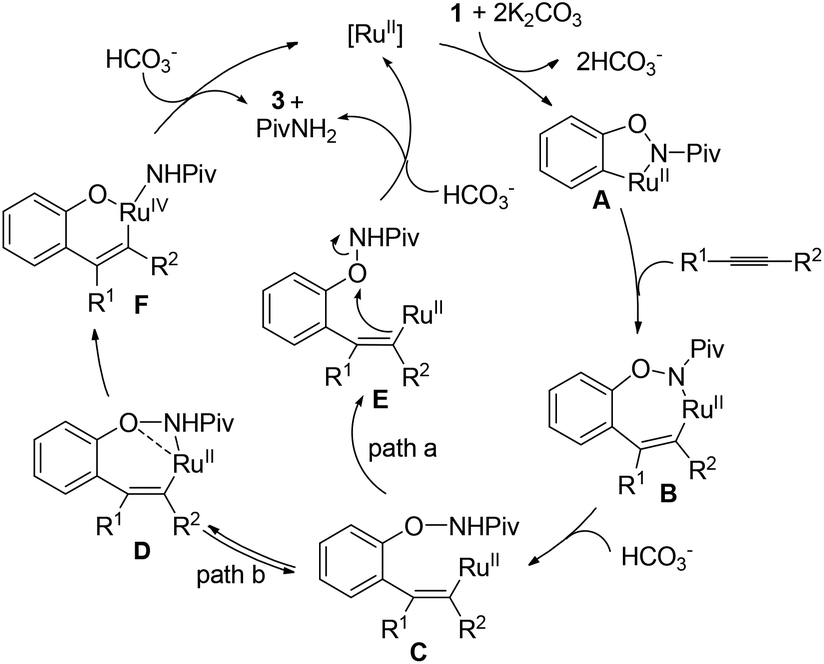
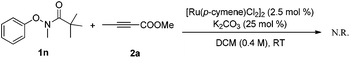
Experiments were then carried out to gain more insights into the reaction mechanism. As we mentioned before, phenol was detected as a side product in this reaction. When the reaction was run with stoichiometric amounts of [Ru(p-cymene)Cl2]2 under the standard conditions, 1a was decomposed to phenol and pivalamide quantitatively after 12 hours (eqn (1)). In another experiment, 1a was treated with [Ru(p-cymene)Cl2]2 under the standard conditions for 12 hours and subsequently 2a was added to the reaction mixture. No benzofuran was observed after several hours (eqn (2)), indicating that the catalytic cycle was not initiated by the cleavage of the O–N bond. In addition, N-methyl-substituted phenoxypivalamide cannot be converted to the desired product, implying that the N–H bond is crucial for this transformation (eqn (3)). Moreover, the regioselectivity of the electron-deficient alkynes (3aa–3af, 3am, 3an) probably excluded the possibility of direct nucleophilic addition of the amido group in 1a to alkynes. From these results, we believed that the catalytic cycle was initiated by the N-ruthenation in the presence of a base and subsequent C–H cleavage.13
| | 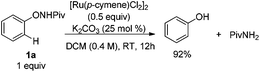 | (1) |
| | 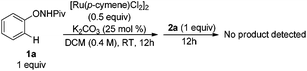 | (2) |
| |  | (3) |
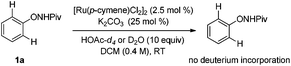
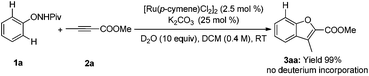
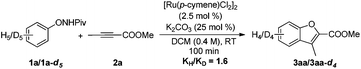
Deuterium-labeling experiments were conducted in the presence of HOAc-d4 or D2O under the standard conditions. No deuterium incorporation was found at the ortho-positions of 1a in the absence of alkyne (eqn (4)). Similarly, when the reaction between 1a and 2a was performed in the presence of D2O, 3aa was obtained smoothly and no deuterium incorporation was observed (eqn (5)). These results implied that the C–H activation might be irreversible under the reaction conditions.8a,14 Furthermore, the KIE was determined to be
| |  | (4) |
| |  | (5) |
| |  | (6) |
| |  | (7) |
| |  | (8) |
kH/
kD ≈ 1.6 (
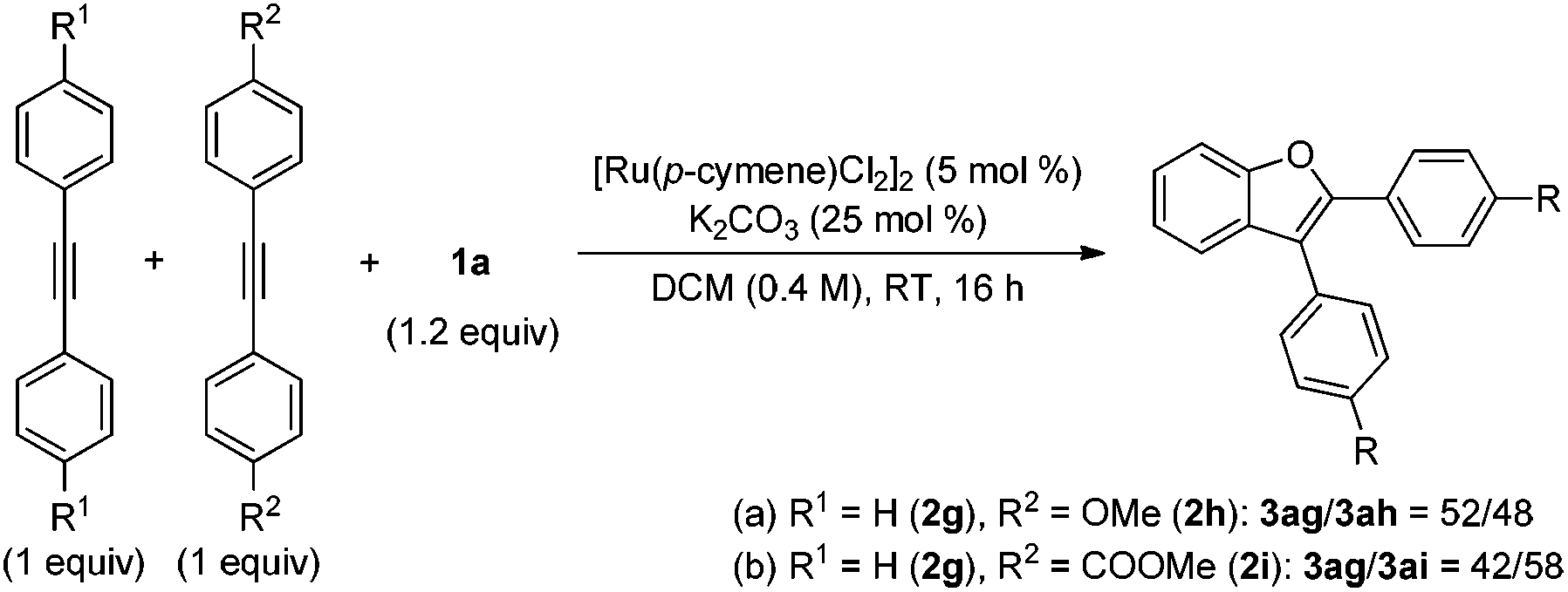
eqn (6)), indicating that the C–H bond cleavage process is not involved in the rate determining step. Competition experiments between substituted diphenylacetylenes revealed that the more electron-deficient alkynes afford higher yields, which is consistent with the reported Ru-catalyzed annulation of arenes with alkynes (
eqn (7)).
8a,15
Based on the results obtained above and the literature,8a,b the mechanism hypotheses are proposed (Scheme 4). The radical mechanism might be excluded because the presence of TEMPO (1 equiv.) or catalytic amount of BHT did not affect the reaction under standard conditions. In addition, enamide 4 failed to afford the benzofuran product under the standard reaction conditions (eqn (8)) indicating that the benzofuran product was not derived from enamide.16 The reaction might be initiated with N-ruthenation in the presence of a base followed by an irreversible C–H cleavage by RuII to yield a five-membered ruthenacycle intermediate A. Subsequent alkyne insertion generates a seven-membered intermediate B, which upon protolysis would yield the intermediate C. Then, intramolecular substitution might occur to form the C–O bond and break the O–N bond simultaneously (path a). According to our previous work10a and the literature,17 the non-oxidative cleavage of the O–N bond in phenoxyamide usually proceeded with the aid of acid. However, the addition of acetic acid inhibited the reaction (Table 1, entry 7), which makes the concerted non-oxidative process less reasonable. Alternatively, the intermediate C would lead reversibly to the intermediate D, in which oxygen and nitrogen atoms might both coordinate to ruthenium. The following intramolecular oxidative addition and reductive elimination involving RuIV intermediate F would provide the benzofuran product 3 and regenerate Ru(II).11b,18 Further studies to understand the reaction pathway are ongoing in our laboratory.
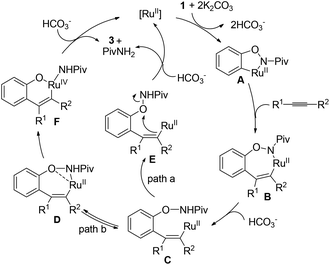 |
| | Scheme 4 Mechanism hypotheses. | |
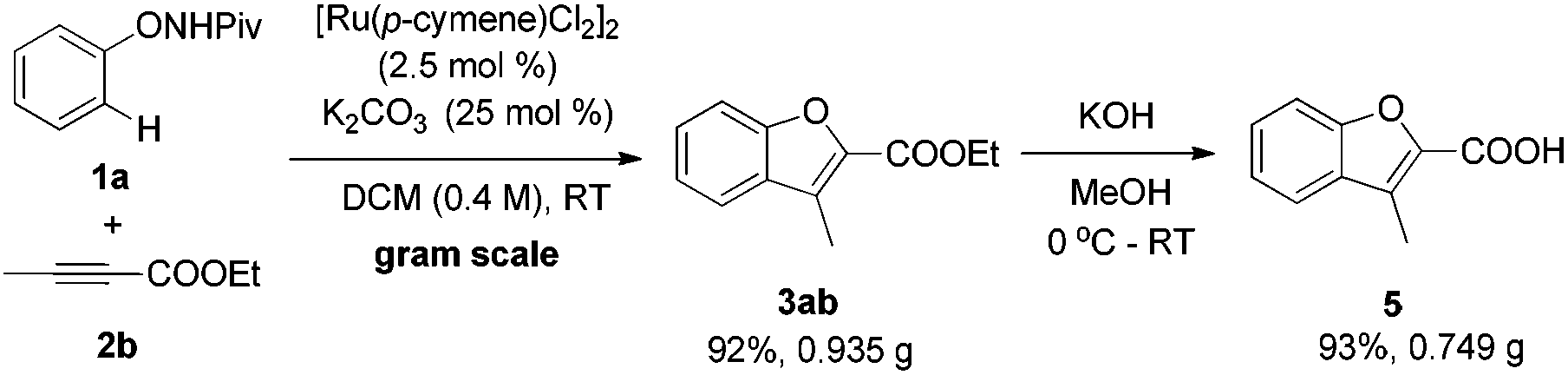
Finally, a relatively large-scale reaction was conducted. Under standard conditions, the desired benzofuran product could be produced conveniently on a gram scale without obvious decrease in yield (eqn (9)). Further hydrolysis of 3ab will result in 3-methylbenzofuran-2-carboxylic acid 5, which is a useful building block for complex heterocyclic compounds.19
| | 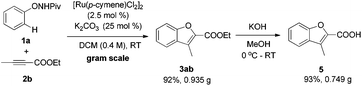 | (9) |
In summary, we have developed a mild Ru-catalyzed C–H functionalization for the synthesis of benzofuran derivatives. Using –ONHPiv as a novel oxidizing directing group, no external oxidant is needed, and a clean and economic process was developed. More detailed investigations to understand the reaction mechanism and further studies to explore new transformation properties of the novel directing group are in progress.
Acknowledgements
We thank the National Basic Research Program of China (2015CB856600), the National Natural Science Foundation of China (21202184, 21232006), and the Chinese Academy of Sciences for financial support.
Notes and references
-
(a) B. Carlsson, B. N. Singh, M. Temciue, S. Nilsson, Y.-L. Li, C. Mellin and J. Malm, J. Med. Chem., 2002, 45, 623 CrossRef CAS PubMed;
(b) A. D. Patil, A. J. Freyer, L. Killmer, P. Offen, B. Carte, A. J. Jurewicz and R. K. Johnson, Tetrahedron, 1997, 53, 5047 CrossRef CAS;
(c) O. Shirota, V. Pathak, S. Sekita, M. Stake, Y. Nagashima, Y. Hirayama, Y. Hakamata and T. Haysahi, J. Nat. Prod., 2003, 66, 1128 CrossRef CAS PubMed.
- For selected reviews, see:
(a) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644 CrossRef CAS PubMed;
(b) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395 CrossRef CAS PubMed;
(c) X.-F. Wu, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1 CrossRef CAS PubMed.
-
(a) X. Guo, R. Yu, H. Li and Z. Li, J. Am. Chem. Soc., 2009, 131, 17387 CrossRef CAS PubMed;
(b) K. Tsuchikama, Y. Hashimoto, K. Endo and T. Shibata, Adv. Synth. Catal., 2009, 351, 2850 CrossRef CAS;
(c) U. Sharma, T. Naveen, A. Maji, S. Manna and D. Maiti, Angew. Chem., Int. Ed., 2013, 52, 12669 CrossRef CAS PubMed;
(d) Y. Moon, Y. Kim, H. Hong and S. Hong, Chem. Commun., 2013, 49, 8323 RSC;
(e) D.-H. Lee, K.-H. Kwon and C. S. Yi, J. Am. Chem. Soc., 2012, 134, 7325 CrossRef CAS PubMed;
(f) R. Zhu, J. Wei and Z. Shi, Chem. Sci., 2013, 4, 3706 RSC;
(g) M. Kuram, M. Bhanuchandra and A. K. Sahoo, Angew. Chem., Int. Ed., 2013, 52, 4607 CrossRef CAS PubMed;
(h) W. Zeng, W. Wu, H. Jiang, L. Huang, Y. Sun, Z. Chen and X. Li, Chem. Commun., 2013, 49, 6611 RSC;
(i) S. Wang, P. Li, L. Yu and L. Wang, Org. Lett., 2011, 13, 5968 CrossRef CAS PubMed.
- For recent reviews on heterocycle synthesis initiated by C–H activation, see:
(a) T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212 CrossRef CAS PubMed;
(b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed;
(c) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC.
- For a review, see: F. W. Patureau and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1977 CrossRef CAS PubMed.
- Pd-catalyzed C–H functionalization using an oxidative directing group, see:
(a) J. Wu, X. Cui, L. Chen, G. Jiang and Y. Wu, J. Am. Chem. Soc., 2009, 131, 13888 CrossRef CAS PubMed;
(b) Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 3676 CrossRef CAS PubMed.
- Rh-catalyzed C–H functionalization using an oxidative directing group, see:
(a) N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908 CrossRef CAS PubMed;
(b) N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449 CrossRef CAS PubMed;
(c) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350 CrossRef CAS PubMed;
(d) H. Wang and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 7318 CrossRef CAS PubMed;
(e) P. C. Too, Y.-F. Wang and S. Chiba, Org. Lett., 2010, 12, 5688 CrossRef CAS PubMed;
(f) X. Zhang, D. Chen, M. Zhao, J. Zhao, A. Jia and X. Li, Adv. Synth. Catal., 2011, 353, 719 CrossRef CAS;
(g) T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 11846 RSC;
(h) X. Xu, Y. Liu and C.-M. Park, Angew. Chem., Int. Ed., 2012, 51, 3972 Search PubMed;
(i) T. K. Hyster, L. Knörr, T. R. Ward and T. Rovis, Science, 2012, 338, 500 CrossRef CAS PubMed;
(j) B. Ye and N. Cramer, Science, 2012, 338, 504 CrossRef CAS PubMed;
(k) H. Wang, C. Grohmann, C. Nimphius and F. Glorius, J. Am. Chem. Soc., 2012, 134, 19592 CrossRef CAS PubMed;
(l) J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2013, 135, 66 CrossRef CAS PubMed;
(m) T. A. Davis, T. K. Hyster and T. Rovis, Angew. Chem., Int. Ed., 2013, 52, 14181 CrossRef CAS PubMed.
- Ru-catalyzed C–H functionalization using an oxidative directing group, see:
(a) B. Li, H. Feng, S. Xu and B. Wang, Chem. – Eur. J., 2011, 17, 12573 CrossRef CAS PubMed;
(b) L. Ackermann and S. Fenner, Org. Lett., 2011, 13, 6548 CrossRef CAS PubMed;
(c) B. Li, J. Ma, N. Wang, H. Feng, S. Xu and B. Wang, Org. Lett., 2012, 14, 736 CrossRef CAS PubMed;
(d) C. Kornhaaß, J. Li and L. Ackermann, J. Org. Chem., 2012, 77, 9190 CrossRef PubMed;
(e) R. K. Chinnagolla, S. Pimparkar and M. Jeganmohan, Org. Lett., 2012, 14, 3032 CrossRef CAS PubMed.
-
(a) D. Zhao, Z. Shi and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12426 CrossRef CAS PubMed;
(b) B. Liu, C. Song, C. Sun, S. Zhou and J. Zhu, J. Am. Chem. Soc., 2013, 135, 16625 CrossRef CAS PubMed;
(c) L. Zheng and R. Hua, Chem. – Eur. J., 2014, 20, 2352 CrossRef CAS PubMed;
(d) C. Wang and Y. Huang, Org. Lett., 2013, 15, 5294 CrossRef CAS PubMed.
-
(a) G. Liu, Y. Shen, Z. Zhou and X. Lu, Angew. Chem., Int. Ed., 2013, 52, 6033 CrossRef CAS PubMed;
(b) Y. Shen, G. Liu, Z. Zhou and X. Lu, Org. Lett., 2013, 15, 3366 CrossRef CAS PubMed;
(c) F. Hu, Y. Xia, F. Ye, Z. Liu, C. Ma, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 1364 CrossRef CAS PubMed;
(d) P. Duan, Y. Yang, R. Ben, Y. Yan, L. Dai, M. Hong, Y. Wu, D. Wang, X. Zhang and J. Zhao, Chem. Sci., 2014, 5, 1574 RSC.
- For reviews on Ru(II)-catalyzed C–H functionalizations, see:
(a) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed;
(b) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed;
(c) L. Ackermann and R. Vicente, Top. Curr. Chem., 2010, 292, 211 CrossRef CAS.
- See Table S1 in the ESI† for details.
- B. Li, H. Feng, N. Wang, J. Ma, H. Song, S. Xu and B. Wang, Chem. – Eur. J., 2012, 18, 12873 CrossRef CAS PubMed.
- T. K. Hyster and T. Rovis, J. Am. Chem. Soc., 2010, 132, 10565 CrossRef CAS PubMed.
- L. Ackermann, A. V. Lygin and N. Hofmann, Angew. Chem., Int. Ed., 2011, 50, 6379 CrossRef CAS PubMed.
- Y. Endo, K. Shudo and T. Okamoto, J. Am. Chem. Soc., 1982, 104, 6393 CrossRef CAS.
- In rhodium catalyzed reaction of N-phenoxyacetamide with alkynes, enamide can be produced selectively. For the detailed mechanism, see: ref. 10a.
- RuIV has been proposed as the key intermediate in RuII catalyzed reactions, for example:
(a) E. F. Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, J. Am. Chem. Soc., 2011, 133, 10161 CrossRef CAS PubMed;
(b) I. Fabre, N. V. Wolff, G. L. Duc, E. F. Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, Chem. – Eur. J., 2013, 19, 7595 CrossRef CAS PubMed For oxidative addition of the N–O bond to Ru(II), see:
(c) J. F. Hull, S. T. Hilton and R. H. Crabtree, Inorg. Chim. Acta, 2010, 363, 1243 CrossRef CAS PubMed For some fully characterized RuIV complexes, see:
(d) Z. Sahli, N. Derrien, S. Pascal, B. Demerseman, T. Roisnel, F. Barrière, M. Achard and C. Bruneau, Dalton Trans., 2011, 40, 5625 RSC;
(e) J. Díez, J. Gimeno, A. Lledós, F. J. Suárez and C. Vicent, ACS Catal., 2012, 2, 2087 CrossRef;
(f) E. K. Huang, W.-M. Cheung, S. L.-F. Chan, H. H. Y. Sung, I. D. Williams and W.-H. Leung, Organometallics, 2013, 32, 4483 CrossRef.
-
(a) S. B. Bedford, M. J. Begley, P. Cornwall, J. P. Foreman and D. W. Knight, J. Chem. Soc., Perkin Trans. 1, 1996, 2827 RSC;
(b) P. C. Ting, J. F. Lee, N. Zorn, H. M. Kim, R. G. Aslanian, M. Lin, M. Smith, S. S. Walker, J. Cook, M. V. Heek and J. Lachowicz, Bioorg. Med. Chem. Lett., 2013, 23, 985 CrossRef CAS PubMed;
(c) J. A. Kaizerman, M. I. Gross, Y. Ge, S. White, W. Hu, J.-X. Duan, E. E. Baird, K. W. Johnson, R. D. Tanaka, H. E. Moser and R. W. Bürli, J. Med. Chem., 2003, 46, 3914 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and compound characterisation. See DOI: 10.1039/c4qo00196f |
|
| This journal is © the Partner Organisations 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.