 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multicomponent azide–alkyne cycloaddition catalyzed by impregnated bimetallic nickel and copper on magnetite†
Juana M.
Pérez
,
Rafael
Cano
and
Diego J.
Ramón
*
Instituto de Síntesis Orgánica (ISO), and Departamento de Química Orgánica, Facultad de Ciencias, Universidad de Alicante, Apdo. 99, E-03080-Alicante, Spain. E-mail: djramon@ua.es; Fax: +35-965903549; Tel: +35-965903548
First published on 21st May 2014
Abstract
A new bimetallic catalyst derived from nickel and copper has been used successfully for the first time in the multicomponent reaction of terminal alkynes, sodium azide, and benzyl bromide derivatives. The presence of both metallic species on the surface of magnetite seems to have a positive and synergetic effect. The catalyst loading is the lowest ever published for a catalyst of copper anchored on any type of iron support. The catalyst could be easily removed from the reaction media just by magnetic decantation and it could be reused up to ten times without any negative effect on the initial results.
Introduction
Although the 1,3-dipolar cycloaddition of azide derivatives and alkynes dates back to the nineteenth century,1 the pioneer and seminal works of the Medal2 and Sharpless3 groups on the copper-catalyzed process were the definitive push for the blossoming of this process. This process allowed access to different 1,2,3-triazoles of great interest to different areas of chemistry and pharmacy, in short reaction times, under mild conditions, and as only one regioisomer.4The tremendous success of the homogenous copper(I) complexes as catalysts has eclipsed the activity of others, such as those derived from ruthenium, platinum, palladium,5 silver6 or nickel,7 as well as the use of other heterogeneous catalysts. However, very recently some heterogeneous catalysts have emerged as an alternative. Thus, the particles of copper,8 or its oxide derivatives,9 different copper salts supported on charcoal,10 on organic materials,11 as well as on inorganic supports12 have been tested for this transformation, with copper loading of these catalysts ranging from 0.5 to 12 mol%. Interestingly, some of the inorganic supports were based on iron, which permitted the development of magnetic catalyst and separation, as it was for the case of copper supported on iron (5 mol%),13 copperferrite (5 mol%),14 or ligand-grafted copper on magnetite (2 mol%).15
The intrinsic instability of organic azides, mainly those of low molecular weight, has been an important drawback in the generalization of this approach for the synthesis of interesting polyvalent structures. However, the use of a multicomponent approach, generating the azide derivative in situ by reaction of sodium azide and the corresponding organic reagent,9c,10b,d–f,11d,f,h,12f,h,14,15b,16 has permitted us to overcome this problem. On the other hand, we have recently developed a new, simple and robust method to immobilize different metal oxides17 onto magnetite,18 and we initially decided to apply the copper impregnated on magnetite catalyst19 to the multicomponent azide–alkyne cycloaddition reaction.
Results and discussion
Simple azide–alkyne cycloaddition
Although our ultimate goal was to get a heterogeneous and recyclable catalyst for the multicomponent version of azide–alkyne cycloaddition, the study was started with the standard two-component reaction between ethynylbenzene (1a) and (azidomethyl)benzene (2a) catalyzed by impregnated copper on magnetite (Table 1).|
|
||||
|---|---|---|---|---|
| Entry | Solvent | T (°C) | t (d) | Yielda3a (4a) (%) |
| a Isolated yield after column chromatography. b Reaction carried out in absence of catalyst. c Reaction performed using only nanomagnetite (21 mol%). | ||||
| 1b | H2O | 110 | 7 | 46 (49) |
| 2 | PhMe | 70 | 2 | 58 (0) |
| 3 | PhMe | 25 | 2 | 35 (7) |
| 4 | PhMe | 110 | 2 | 73 (25) |
| 5 | THF | 70 | 2 | 43 (7) |
| 6 | CHCl3 | 70 | 2 | 52 (6) |
| 7 | MeCN | 70 | 2 | 15 (6) |
| 8 | DMSO | 70 | 2 | 33 (8) |
| 9 | MeOH | 70 | 2 | 50 (0) |
| 10 | H2O | 70 | 1 | 94 (0) |
| 11 | 70 | 2 | 73 (12) | |
| 12c | H2O | 70 | 1 | 82 (1) |
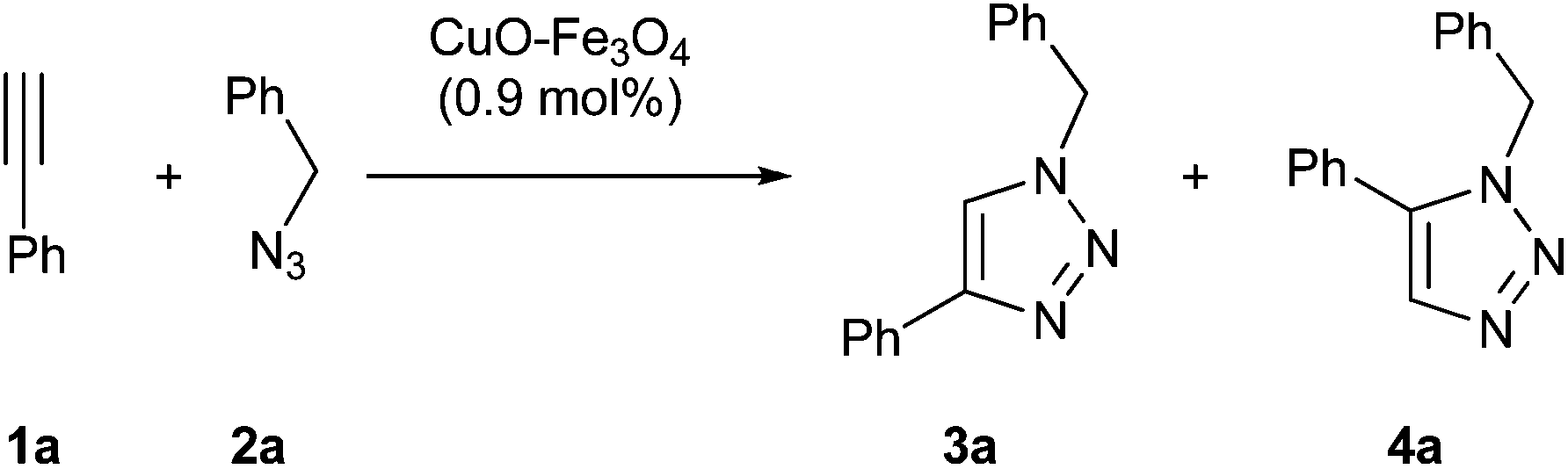
The initial reaction was conducted in absence of catalyst at 110 °C in water, obtaining after 7 days a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of both possible isomers. Then, the reaction was repeated in the presence of copper catalyst in toluene at 70 °C giving exclusively 1-benzyl-4-phenyl-1H-1,2,3-triazole (3a) in a modest yield (entry 2). Both the decrease and the increase of temperature led to the formation of a mixture of regioisomers (entries 3 and 4). Then, the influence of solvent was examined, finding that the highest yield was reached in water (entry 10). Under these conditions, the role of magnetite support was studied and high activity of the supposed inert material was found (entry 12).
:1 mixture of both possible isomers. Then, the reaction was repeated in the presence of copper catalyst in toluene at 70 °C giving exclusively 1-benzyl-4-phenyl-1H-1,2,3-triazole (3a) in a modest yield (entry 2). Both the decrease and the increase of temperature led to the formation of a mixture of regioisomers (entries 3 and 4). Then, the influence of solvent was examined, finding that the highest yield was reached in water (entry 10). Under these conditions, the role of magnetite support was studied and high activity of the supposed inert material was found (entry 12).
Once the activity of copper catalyst was examined, its recycling was studied. After the first trial, the magnetite was collected with a magnet, washed with toluene and ethanol, and dried. The recycled catalyst could be re-used three fold with similar results (82–78%). However, the yield dropped to 35% in the forth use, keeping this level of results during the following 5 cycles (Fig. 1). The phenomenon of leaching was studied by ICP-MS analysis of the resulting reaction solution mixture, and 1.1% of the initial amount of copper was detected (0.007% of iron), which could explained the lost of activity. Moreover, the TEM images of the recycled catalyst showed a small change in copper particle size from 7.1 ± 6.5 nm of the fresh prepared catalyst to 6.4 ± 5.2 nm for the recycled one, which would not affect the reactivity of the recycled catalyst. Finally, it should be pointed out that the BET surface area did not suffer a great change, from 6.2 m2 g−1 for the initial catalyst to 8.4 m2 g−1 for the used one, which is practically the same specific area.
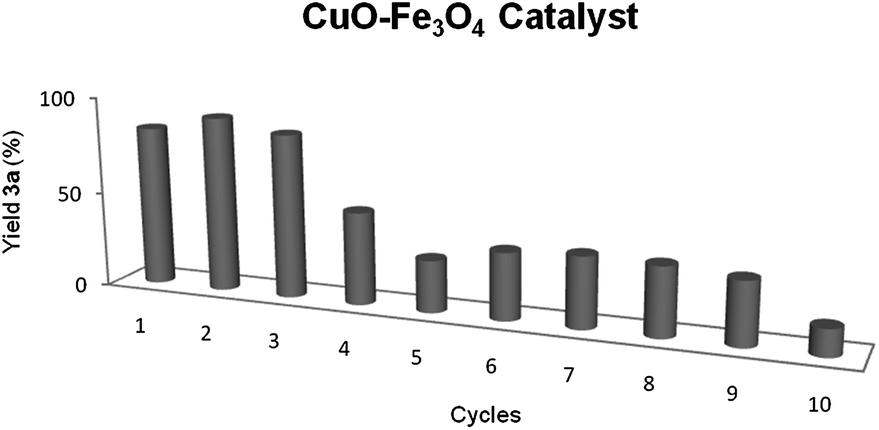 | ||
| Fig. 1 Recycling of CuO–Fe3O4 catalyst for the cycloaddition. | ||
Multicomponent cycloaddition processes
After finding that copper catalyst was effective in the cycloaddition between azides and terminal alkynes, we faced the problem of the multicomponent version,9c,10d,e,11d,h,12h,14,15b,16b,c using benzyl bromide (5a), sodium azide (6) and ethynylbenzene (1a) as reaction model (Table 2). The reaction in water gave a mixture of expected heterocycle 3a together with its regioisomer 4a (compare entry 1 in Table 2 and entry 9 in Table 1). This initial trial showed that the change from simple cycloaddition to the multicomponent reaction one was not so simple. Thus, a new optimization process on this multicomponent reaction was carried out, starting by studying the effect of solvent (entries 1–8 in Table 2). The best result was obtained in absence of solvent, but a small amount of the product arising from homocoupling of terminal alkyne was found.19e The optimal temperature seemed to be 50 °C (entries 8–11), since at higher temperatures different by-products were formed, and at lower temperatures a modest yield was achieved. Finally, the increase of amount of reagents, 5a and 6, increased the yield (compare entry 12 in Table 2 with entry 9 in Table 1).|
|
||||
|---|---|---|---|---|
| Entry | Solvent | T (°C) | t (d) | Yielda3a (4a) (%) |
| a Isolated yield after column chromatography. b 1,4-Diphenylbuta-1,3-diyne was isolated in a 10%. c Reaction performed using 2 equiv. of compounds 5a and 6. | ||||
| 1 | H2O | 70 | 3 | 57 (13) |
| 2 | PhMe | 70 | 3 | 33 (0) |
| 3 | THF | 70 | 3 | 21 (4) |
| 4 | CHCl3 | 70 | 3 | 19 (1) |
| 5 | MeCN | 70 | 3 | 16 (0) |
| 6 | DMSO | 70 | 3 | 25 (3) |
| 7 | MeOH | 70 | 3 | 32 (25) |
| 8 | — | 70 | 3 | 69b (0) |
| 9 | — | 50 | 2 | 71 (0) |
| 10 | — | 25 | 3 | 38 (0) |
| 11 | — | 110 | 2 | 53b (6) |
| 12c | — | 25 | 2 | 83 (0) |
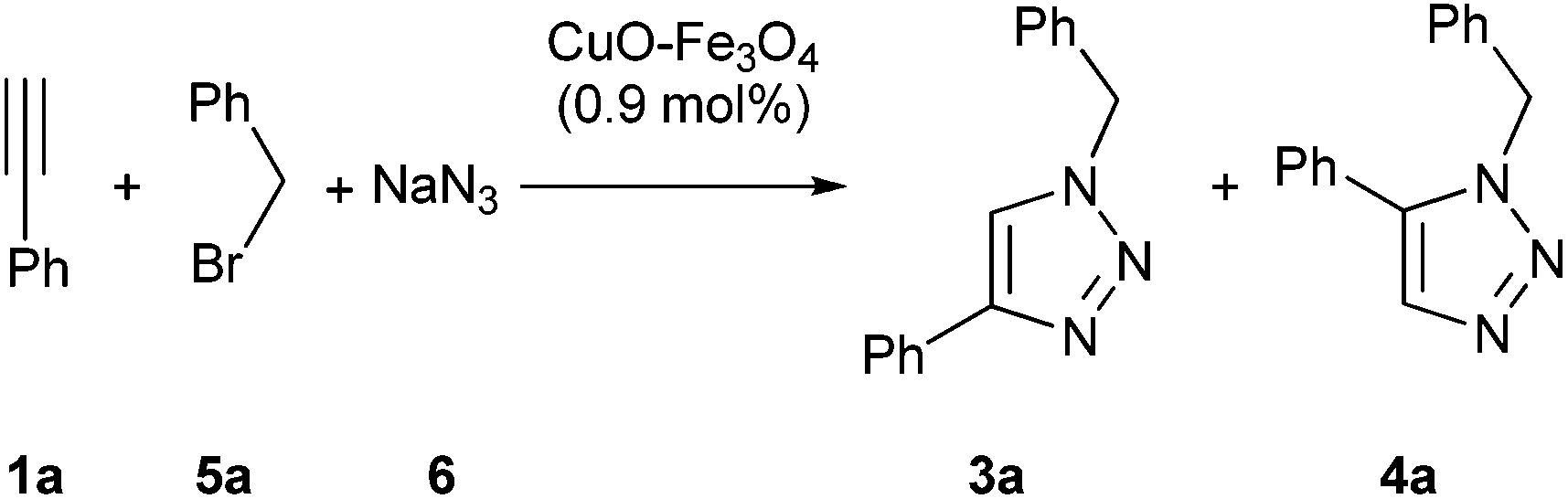
Although copper catalysts have been the most used, other metal catalysts have also shown some activity for this reaction. For this reason we tested a series of impregnated metal catalyst in this multicomponent version (Table 3), starting from the uncatalyzed reaction (entry 1). From all ductile metal oxide, only nickel and copper catalysts showed activity (entries 2–14).
|
|
||
|---|---|---|
| Entry | Catalyst (mol%) | Yielda3a (%) |
| a Isolated yield after column chromatography. b Powder < 5 μm. c Powder < 50 nm. d Reaction performed during 24 h. | ||
| 1 | — | 0 |
| 2 | Fe3O4b (21) | 0 |
| 3 | Fe3O4c (21) | 0 |
| 4 | CoO–Fe3O4 (1.4) | 0 |
| 5 | NiO–Fe3O4 (1.0) | 5 |
| 6 | CuO–Fe3O4 (0.9) | 83 |
| 7 | Ru2O3–Fe3O4 (1.3) | 0 |
| 8 | Rh2O3–Fe3O4 (0.4) | 0 |
| 9 | PdO–Fe3O4 (1.2) | 0 |
| 10 | AgO2–Fe3O4 (1.3) | 0 |
| 11 | OsO2–Fe3O4 (0.5) | 0 |
| 12 | IrO2–Fe3O4 (0.1) | 0 |
| 13 | PtO–Fe3O4 (0.5) | 0 |
| 14 | Au2O3–Fe3O4 (0.1) | 0 |
| 15 | PdO/Cu–Fe3O4 (1.5/0.9) | 42 |
| 16 | NiO/Cu–Fe3O4 (0.9/0.9) | 98 |
| 17 | NiO/Cu–Fe3O4 (0.2/0.2) | 15 |
| 18 | NiO/Cu–Fe3O4 (1.8/1.8) | >99d |
| 19 | NiO–Fe3O4 (1.0) + CuO–Fe3O4 (0.9) | 87 |
| 20 | CuO (0.9) | 78 |
| 21 | NiO (0.9) | 12 |
| 22 | Cu(OH)2 (0.9) | 58 |
| 23 | Ni(OH)2 (0.9) | 11 |
| 24 | NiO (0.9) + CuO (0.9) | 76 |
| 25 | Ni(OH)2 (0.9) + Cu(OH)2 (0.9) | 62 |
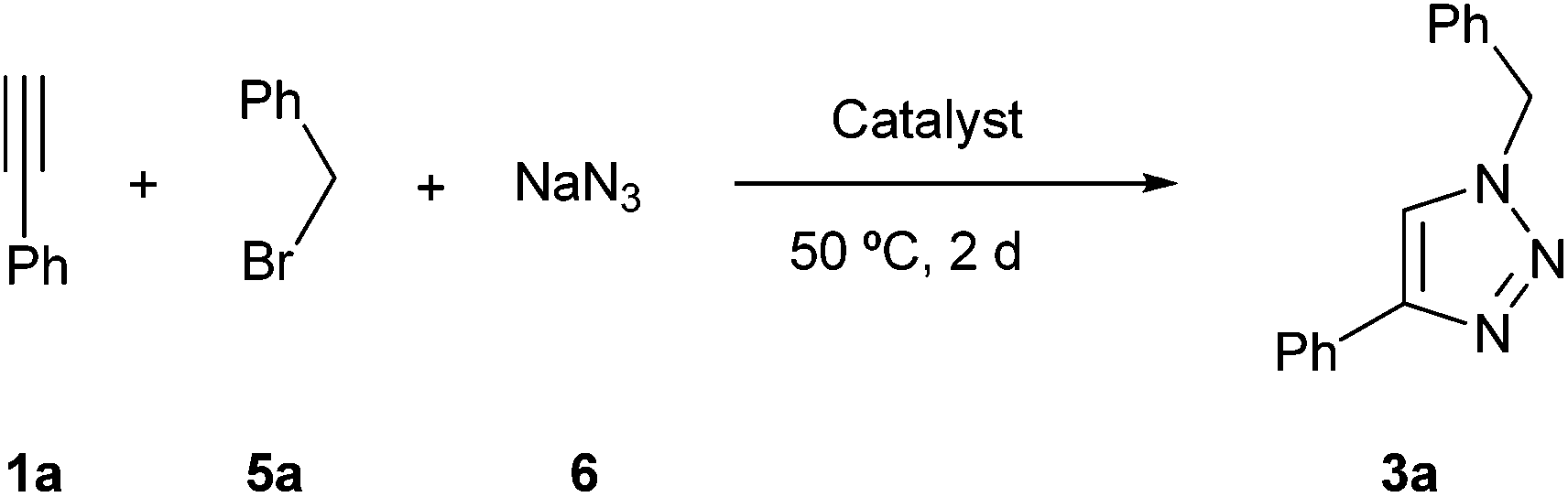
Then, a series of bimetallic derivatives were studied, finding that Pd/Cu system19c could render the expected product 3a (entry 15). Very recently, different bimetallic Ni–Cu/C composite catalysts20 have been tested in the simple cycloaddition of azides and terminal alkynes and these results prompted us to prepare the corresponding bimetallic one impregnated on magnetite. Its reaction gave the expected product with an excellent result (entry 16). The decrease of the amount of Ni–Cu catalyst had an important detrimental effect, meanwhile its increase had a marginal benefit (compare entries 16–18).
Faced with the excellent result obtained with the bimetallic nickel–copper catalyst we wondered if the yield was a result of a simple addition of two independent catalytic sites or was it the result of some type of synergic effect. To answer that question, the reaction was repeated using both catalysts (the copper and the nickel one) with almost the same loading and the result seemed to be the addition of the activity of both catalysts (compare entries 5 and 6 with entry 19). Therefore, we believe that the bimetallic catalyst develops a synergetic effect that makes it superior to the addition of both parts, although the nature of this positive interaction is unknown.
Finally, the unsupported metal catalysts were tested. Thus, the reaction using CuO alone gave the expected product 3a with a good result (Table 3, entry 20), meanwhile the related nickel oxide gave a worse result (entry 21). When the reaction was repeated with the corresponding metal hydroxide derivatives the yields were slightly lower (entries 22 and 23). The equimolecular mixture of both metallic catalysts did not show any improvement of the result obtained by the copper derivative (compare entries 20, 22 and 24, 25, respectively).
The bimetallic Ni–Cu catalyst could be recycled and reused tenfold, just by collection of the catalyst with a magnet, washing with toluene and ethanol, and drying, without any depreciation in its activity (Fig. 2).
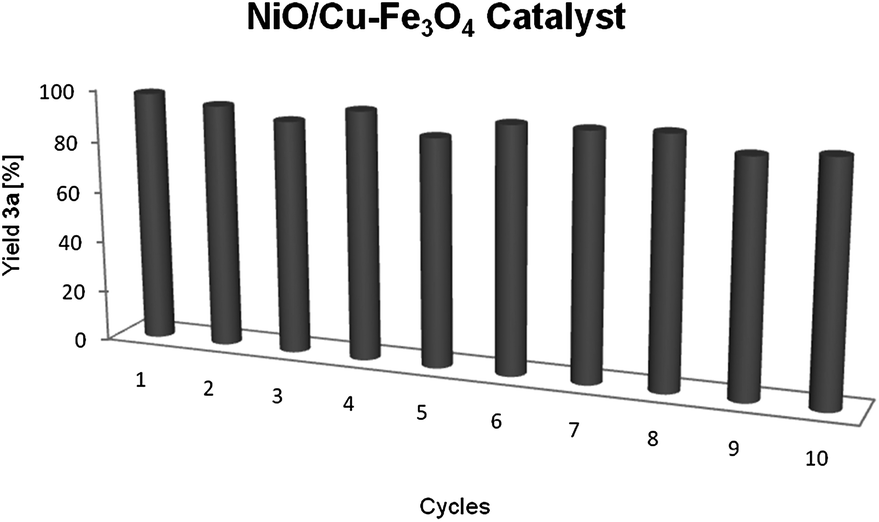 | ||
| Fig. 2 Recycling of NiO/Cu–Fe3O4 catalyst for the multicomponent reaction. | ||
The phenomenon of leaching was studied by ICP-MS analysis of the resulting reaction solution mixture, and 1.1, and 0.2% of the initial amount of copper, and nickel, respectively, was detected (0.006% of iron). The TEM images of the recycled catalyst showed a small change in the particle size from 3.1 ± 1.7 nm of the freshly prepared catalyst to 4.7 ± 2.4 nm for the recycled one. Moreover, XPS data analysis of bimetallic catalyst showed only NiO, CuO and Cu2O species (Fig. 3), which was confirmed by Auger spectroscopy (see ESI†). However, the recycled one showed the presence of Ni(OH)2 as well as Cu(OH)2. These small changes, in particle size and the nickel species seemed not to affect the activity of the bimetallic catalyst, since it could be reused several times with similar activity.
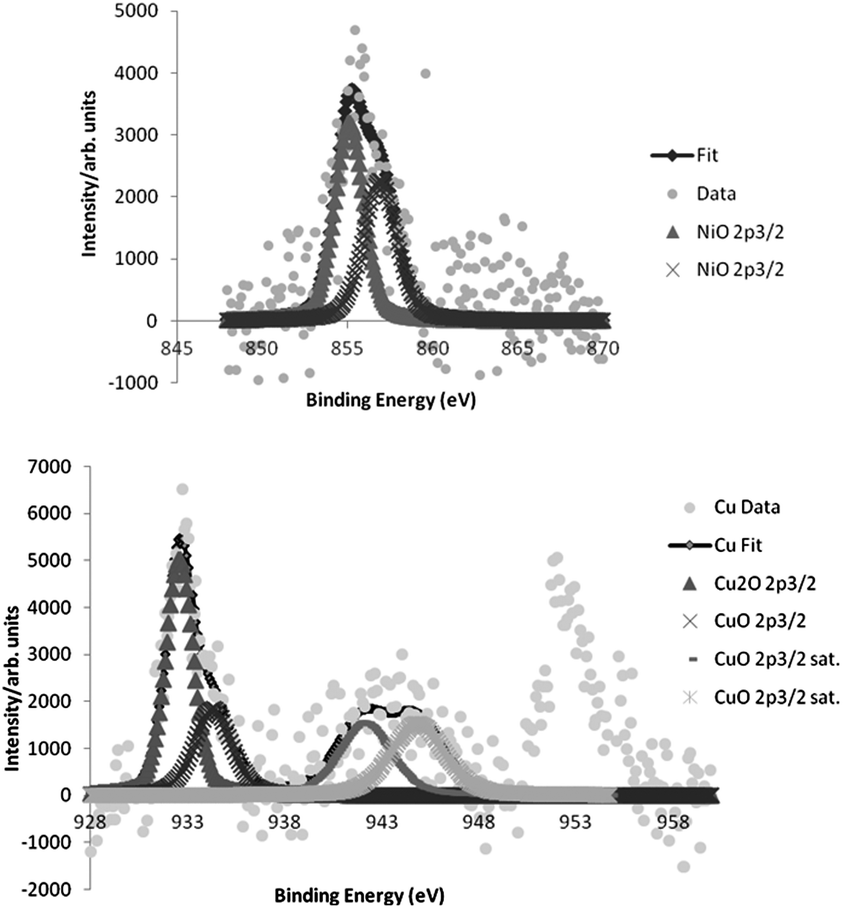 | ||
| Fig. 3 X-Ray photoelectron spectroscopy of the fresh NiO/Cu–Fe3O4 catalyst. | ||
To know if the reaction took place by the leached copper or nickel species to the organic medium, we performed the standard multicomponent reaction (Table 3, entry 16). After that, the catalyst was removed carefully by a magnet at high temperature, and washed with toluene. The solvents of the above solution, without catalyst, were removed under low pressure and alkyne 1a, sodium azide (6) and 4-bromobenzyl alcohol were added to the above residue. The resulting mixture was heated again at 50 °C for 24 h. The analysis of crude mixture, after hydrolysis, revealed the formation of compound 3a in 95% (catalyzed process) and product 3b in less than 1% yield by GC-analysis (compare with entry 2 in Table 4). Therefore, we could exclude that the final leached copper–nickel species were responsible for the reaction results under the standard conditions.
|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | No | Yielda (%) |
| a Isolated yield after column chromatography. b Reaction performed during 4 days. | ||||
| 1 | Ph | Ph | 3a | 98 |
| 2 | Ph | 4-BrC6H4 | 3b | >99 |
| 3 | Ph | 3-BrC6H4 | 3c | >99 |
| 4 | Ph | 2-BrC6H4 | 3d | >99b |
| 5 | Ph | 2-MeC6H4 | 3e | 59b |
| 6 | Ph | 3-MeC6H4 | 3f | 50 |
| 7 | Ph | 3,5-(MeO)2C6H3 | 3g | 89 |
| 8 | 4-ClC6H4 | Ph | 3h | 80b |
| 9 | 4-ClC6H4 | 4-BrC6H4 | 3i | >99 |
| 10 | 2-ClC6H4 | Ph | 3j | 45b |
| 11 | 4-BrC6H4 | Ph | 3k | 42b |
| 12 | 4-BrC6H4 | 3-MeC6H4 | 3l | 90 |
| 13 | 4-MeOC6H4 | Ph | 3m | 42b |
| 14 | 3-MeC6H4 | Ph | 3n | 55b |
| 15 | 3-MeC6H4 | 3-BrC6H4 | 3o | 86 |
| 16 | 3-MeC6H4 | 3-MeC6H4 | 3p | 49 |
| 17 | C6H4(CO)2N | Ph | 3q | 37b |
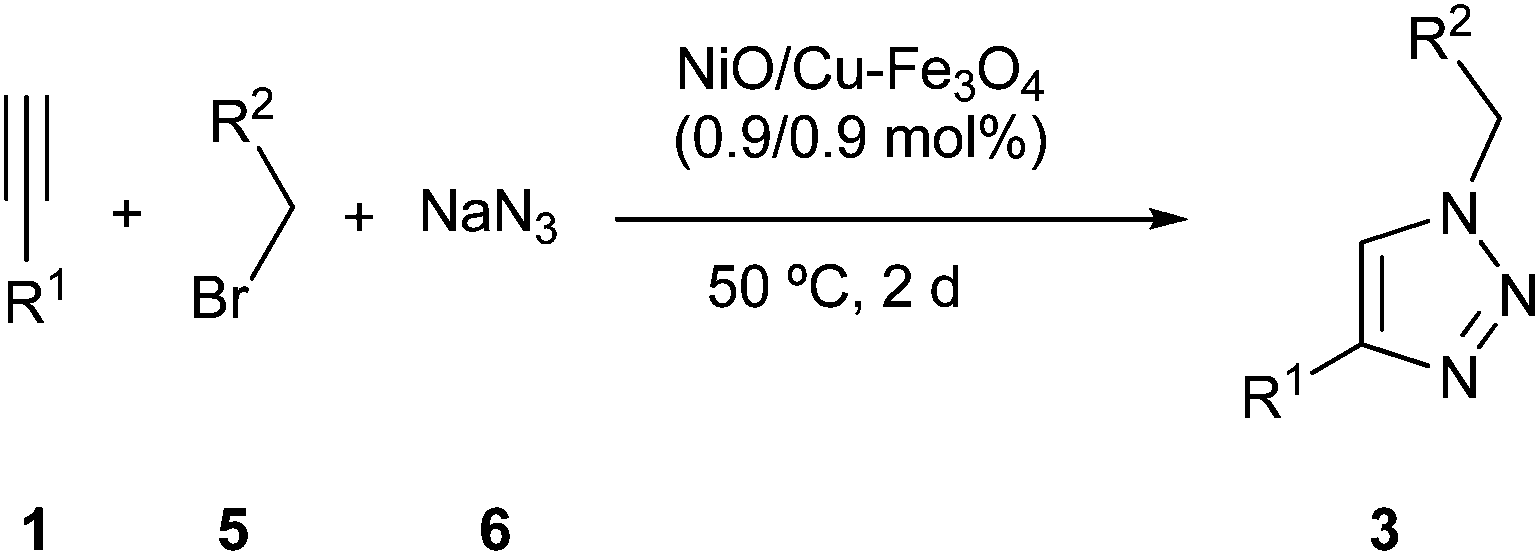
Once the catalytic activity and the recyclability of bimetallic catalyst were proved, the scope of the reaction was tested (Table 4). The reaction gave excellent results independently of substituent position on the aromatic ring of the bromide 5 (entries 2–4). The electronic nature of substituent on the aromatic ring of the bromide 5 seemed not to have influenced on the results (compare entries 1–7), since the results disagrees with the Hammett constants. Also, the reaction was accomplished with akynes 1 with different groups in the aromatic ring, with no clear correlation of the reached yields with the electronic nature of substituents (entries 8–16). The reaction with 2-(bromomethyl)isoindoline-1,3-dione gave the expected compound 3q in a modest yield (entry 17). However, it should be pointed out that the reactions using less electrophilic reagents such as aliphatic bromide (1-bromododecane) or benzyl chloride, failed after seven days under standard conditions, recovering unchanged the starting alkyne, as well as in the case of using either an aliphatic substituted alkyne (oct-1-yne).
Then, the initial source of benzyl azide was tested (Scheme 1). The reaction with benzylic alcohols failed after six days, recovering unchanged the initial alkyne. The reaction also failed using the silyl ether 7b. However, the reaction using benzyl mesylate gave a modest yield (35%) after 2 days reaction time. When the reaction time was increased up to 6 days a reasonable yield was isolated (75%). When the reaction was performed with benzyl tosylate (7d) the result was very modest.
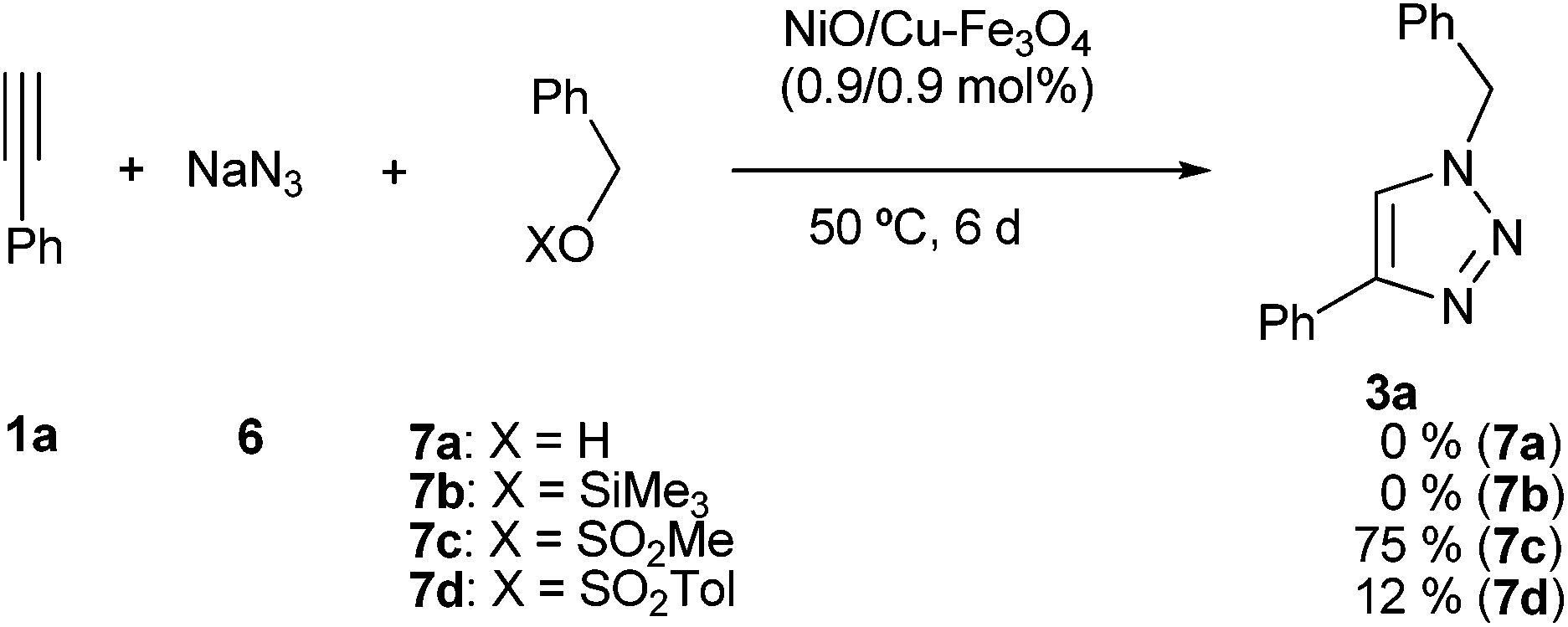 | ||
| Scheme 1 Multicomponent cycloaddition with benzyl derivatives. | ||
The multicomponent reaction with symmetrical internal alkynes 8 gave the expected compound 9 with very modest yield (Scheme 2). This result highlighted the possible selectivity of catalyst, and in order to confirm this, the reaction of benzyl bromide (5a, 2 equiv.), sodium azide (6, 2 equiv.), ethynylbenzene (1a, 1 equiv.) and 1,2-diphenylethyne (8a, 1 equiv.) was performed under standard conditions, finding exclusively the compound 3a (94%) from the analysis of crude mixture.
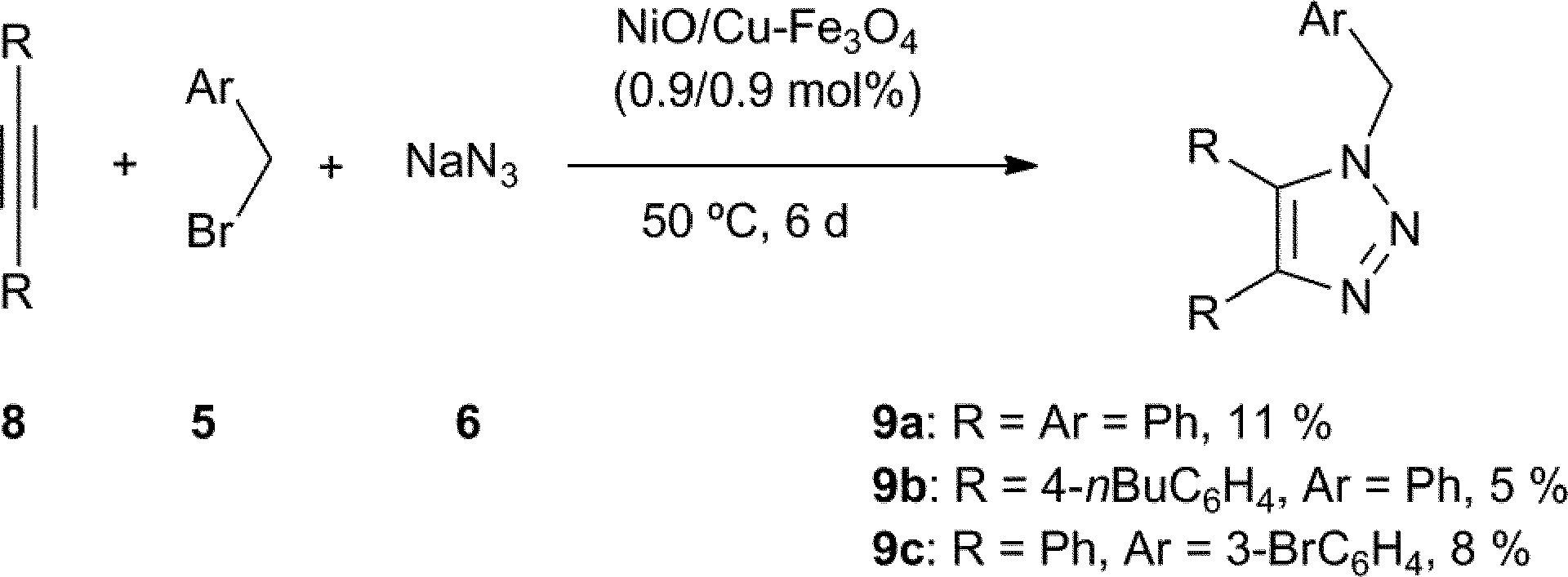 | ||
| Scheme 2 Multicomponent cycloaddition with internal alkynes. | ||
Once the scope of the reaction was studied, we faced the problem of reaction sequentiality. For this proposal, we carried the reaction with the dibromide derivative 10, and a double amount of sodium azide (6), obtaining after six days the azide 11 with a moderate yield (Scheme 3).
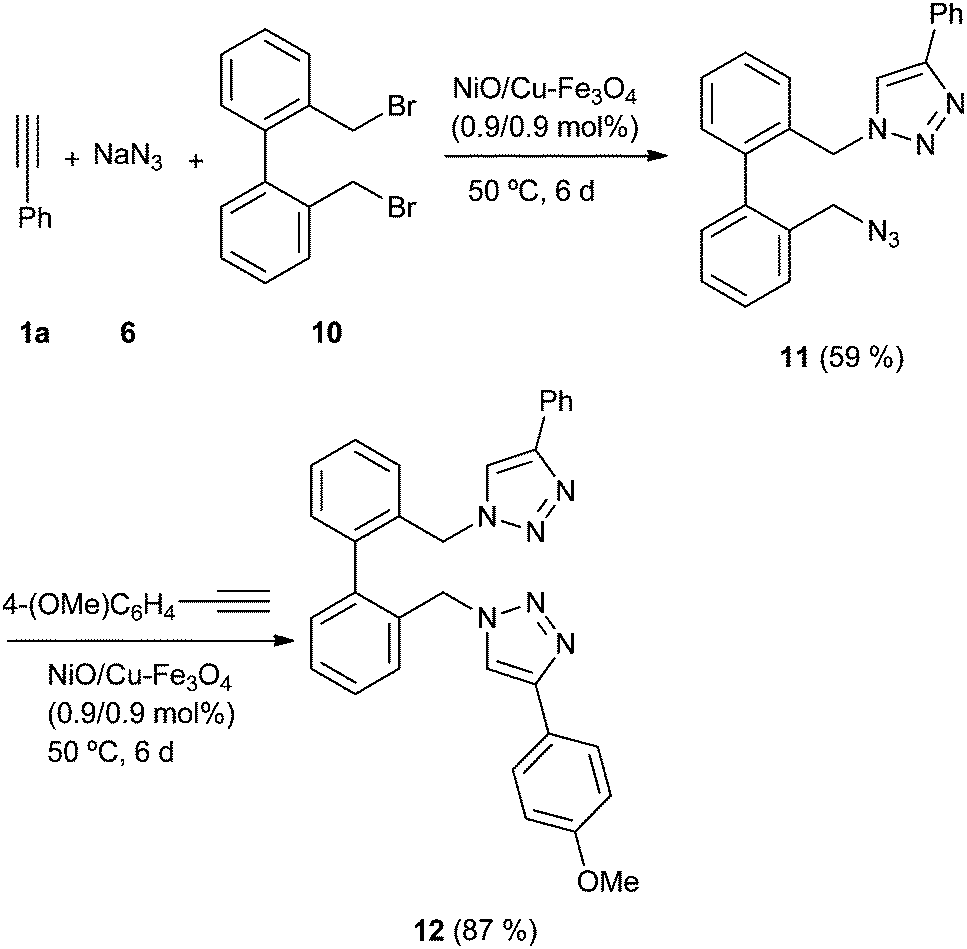 | ||
| Scheme 3 Sequential multicycloaddition process. | ||
The GC-MS analysis of crude mixture did not show the presence of corresponding bis-triazole, with the relate bis-azide derivative being the main by-product. The isolated azide 11 was submitted to another cycloaddition process, yielding the unsymmetrical bis-triazole derivative 12 with good yield. This approach highlights the possibilities of the catalyst in the synthesis of different substituted triazoles.
Conclusions
We have demonstrated that the new bimetallic catalyst derived from nickel and copper supported on magnetite was a good catalyst for the multicomponent reaction of terminal alkynes, sodium azide, and benzyl bromide derivatives. The presence of both metallic species on the surface of magnetite seemed to have a positive and synergetic effect. The catalyst loading was the lowest ever published for a catalyst of copper anchored on any type of iron support, and being in the lowest level for any type of the heterogeneous ones. The catalyst could be easily removed from the reaction media just by magnetic decantation, and it could be reused up to ten times without any negative effect on the initial results.Experimental
General
XPS analyses were carried out on a VG-Microtech Mutilab. XRD analyses were obtained on a BRUKER D-8 ADVANCE diffractometer with Göebel mirror, with a high temperature chamber (up to 900 °C), with a X-ray generator KRISTALLOFLEX K 760-80F (3 kW, 20–60 kV and 5–80 mA). TEM images were obtained on a JEOL, model JEM-2010 equipped with an X-ray detector OXFORD INCA Energy TEM 100 for microanalysis (EDS). XRF analyses were obtained on a PHILIPS MAGIX PRO (PW2400) X-ray spectrometer equipped with a rhodium X-ray tube and a beryllium window. BET isotherms were carried out on a AUTOSORB-6 (Quantachrome), using N2. Melting points were obtained with a Reichert Thermovar apparatus. NMR spectra were recorded on a Bruker AC-300 (300 MHz for 1H and 75 MHz for 13C) using CDCl3 as a solvent and TMS as internal standard for 1H and 13C; chemical shifts are given in δ (parts per million) and coupling constants (J) in Hertz. FT-IR spectra were obtained on a JASCO 4100LE (Pike Miracle ATR) spectrophotometer. Mass spectra (EI) were obtained at 70 eV on a Himazdu QP-5000 spectrometer, giving fragment ions in m/z with relative intensities (%) in parentheses. The chromatographic analyses (GLC) were determined with a Hewlett Packard HP-5890 instrument equipped with a flame ionization detector and 12 m HP-1 capillary column (0.2 mm diam., 0.33 mm film thickness, OV-1 stationary phase), using nitrogen (2 mL min−1) as a carrier gas, Tinjector = 275 °C, Tdetector = 300 °C, Tcolumn = 60 °C (3 min) and 60–270 °C (15 °C min−1), P = 40 kPa. Thin layer chromatography (TLC) was carried out on Schleicher & Schuell F1400/LS 254 plates coated with a 0.2 mm layer of silica gel; detection by UV254 light, staining with phosphomolybdic acid [25 g phosphomolybdic acid, 10 g Ce(SO4)2·4H2O, 60 mL of concentrated H2SO4 and 940 mL H2O]. Column chromatography was performed using silica gel 60 of 40–63 mesh. All reagents were commercially available (Acros, Aldrich, Fluorochem) and were used as received. The ICP-MS analyses were carried out on a Thermo Elemental VGPQ-ExCell spectrometer. The Elemental Analysis was performed on a Elemental Microanalyzer Thermo Finningan Flash 1112 Series.General procedure for the preparation of NiO/Cu–Fe3O4 catalyst
To a stirred solution of CuCl2 (1 mmol, 130 mg) and NiCl2·H2O (1 mmol, 130 mg) in deionized water (120 mL) was added commercially available Fe3O4 (4 g, 17 mmol, powder < 5 μm, BET area: 9.86 m2 g−1). After 10 minutes at room temperature, the mixture was slowly basified with NaOH (1 M) until pH around 13. The mixture was stirred during one day at room temperature in air. After that, the catalyst was filtered and washed several times with deionized water (3 × 10 mL). The solid was dried at 100 °C during 24 h in a standard glassware oven, obtaining thereafter the expected catalyst.General procedures for the preparation of the products
To a stirred solution of sodium azide (6, 2 mmol) and benzyl halide (5, 2 mmol) were added NiO/Cu–Fe3O4 (50 mg, 0.9 mol% of Ni and 0.9 mol% of Cu) and the corresponding alkyne (1 or 8, 1 mmol). The resulting mixture was stirred at 50 °C until the end of the reaction. The catalyst was removed by a magnet and the resulting mixture was quenched with deionized water and extracted with AcOEt (3 × 5 mL). The organic phases were dried over MgSO4, followed by evaporation under reduced pressure to remove the solvent. The product was usually purified by chromatography on silica gel (hexane–ethyl acetate) to give the corresponding products 3 or 9 (see ESI†). The chromatographic analyses were determined with a flame ionization detector and a 30 m capillary column (0.32 mm diam. 0.25 μm film thickness, HP-5 stationary phase), using nitrogen (2 mL min−1) as carrier gas, P = 10 psi, T(injector) = 270 °C and T(detector) = 300 °C; tR values are given under these conditions. Physical and spectroscopic data, only for new compounds, follow:Acknowledgements
This work was supported by the current Spanish Ministerio de Economía y Competitividad (CTQ2011-24151) and University of Alicante. J.M.P. thanks to M.E.C. for a fellowship through the FPI program. We gratefully acknowledge the polishing of our English by Mrs Oriana C. Townley.Notes and references
- A. Michael, J. Prakt. Chem., 1893, 48, 94–95 CrossRef
.
- C. W. Tornøe, C. Christensen and M. Medal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS
-
(a) V. D. Bock, H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2006, 51–68 CrossRef CAS
-
C. Schilling, N. Jung and S. Bräse, in Organic Azides: Syntheses and Applications, ed. S. Bräse and K. Banert, Wiley-VCH, Weinheim, 2010, pp. 269–284 Search PubMed
-
(a) J. McNulty, K. Keskar and R. Vemula, Chem.–Eur. J., 2011, 17, 14727–14730 CrossRef CAS PubMed
-
(a) P. Paul and K. Nag, Inorg. Chem., 1987, 26, 2969–2974 CrossRef CAS
-
(a) F. Alonso, Y. Moglie, G. Radivoy and M. Yus, Tetrahedron Lett., 2009, 50, 2358–2362 CrossRef CAS PubMed
-
(a) J. Y. Kim, J. C. Park, H. Kang, H. Song and K. H. Park, Chem. Commun., 2010, 46, 439–441 RSC
-
(a) B. H. Lipshutz and B. R. Taft, Angew. Chem., Int. Ed., 2006, 45, 8235–8238 CrossRef CAS PubMed
-
(a) U. Sirion, Y. J. Bae, B. S. Lee and D. Y. Chi, Synlett, 2008, 2326–2330 CAS
-
(a) S. Chassaing, M. Kumarraja, A. S. S. Sido, P. Pale and J. Sommer, Org. Lett., 2007, 9, 883–886 CrossRef CAS PubMed
-
(a) S. Kovács, K. Zih-Perényi, Á. Révész and Z. Novák, Synthesis, 2012, 44, 3722–3730 CrossRef PubMed
- B. S. P. A. Kumar, H. V. Reddy, B. Madhav, K. Ramesh and Y. V. D. Nageswar, Tetrahedron Lett., 2012, 53, 4595–4599 CrossRef PubMed
-
(a) A. Megía-Fernández, M. Ortega-Muñoz, J. López-Jaramillo, F. Hernández-Mateo and F. Santoyo-González, Adv. Synth. Catal., 2010, 352, 3306–3320 CrossRef
-
(a) A. K. Feldman, B. Colasson and V. V. Fokin, Org. Lett., 2004, 6, 3897–3899 CrossRef CAS PubMed
-
(a) R. Cano, D. J. Ramón and M. Yus, Tetrahedron, 2011, 67, 5432–5436 CrossRef CAS PubMed
-
(a) R. Martínez, D. J. Ramón and M. Yus, Adv. Synth. Catal., 2008, 350, 1235–1240 CrossRef
-
(a) M. J. Aliaga, D. J. Ramón and M. Yus, Org. Biomol. Chem., 2010, 8, 43–46 RSC
-
(a) B. H. Lipshutz, D. M. Nihan, E. Vinogradova, B. R. Taft and Ž. V. Bošković, Org. Lett., 2008, 10, 4279–4282 CrossRef CAS PubMed
- J. Albdadi, M. Keshavarz, M. Abedini and M. Vafaie-nezhad, Chin. Chem. Lett., 2012, 23, 797–800 CrossRef PubMed
- X. Meng, X. Xu, T. Gao and B. Chen, Eur. J. Org. Chem., 2010, 5409–5414 CrossRef CAS
- D.-R. Hou, T.-C. Kuan, Y.-K. Li, R. Lee and K.-W. Huang, Tetrahedron, 2010, 66, 9415–9420 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Particle size distribution for catalysts, XPS Data and TEM images of new Ni–Cu–Fe3O4 catalyst, as well as copies of 1H and 13C-NMR for all compounds 3, 9, 11 and 12. See DOI: 10.1039/c4ra03149k |
| This journal is © The Royal Society of Chemistry 2014 |