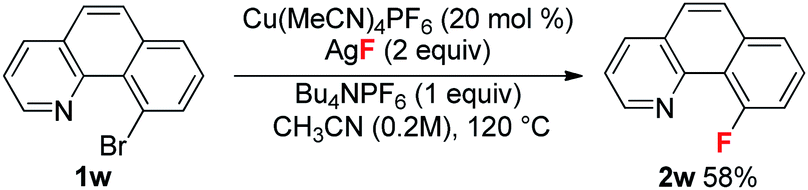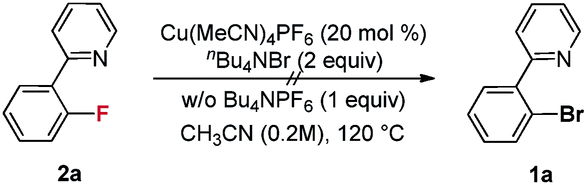DOI:
10.1039/C3SC51876K
(Edge Article)
Chem. Sci., 2014,
5, 275-280
Copper-catalyzed fluorination of 2-pyridyl aryl bromides†
Received
4th July 2013
, Accepted 27th September 2013
First published on 27th September 2013
Abstract
Copper(I)-catalyzed cross-coupling of aryl halides is the subject of extensive interest in synthetic chemistry, but the related catalytic fluorination is unsuccessful. Herein, we have developed a novel copper-catalyzed fluorination of aryl bromides using AgF as the fluorine source. In this transformation a pyridyl directing group is essential for successful catalytic fluorination. A XANES/EXAFS study indicated that the presence of the pyridyl group is essential to stabilize the Cu(I) species and accelerate oxidative addition of the aryl bromide. Further mechanistic studies implicated a Cu(I/III) catalytic cycle in this Cu(I)-catalyzed fluorination, and final aryl C–F bond formation possibly proceeds through an irreversible reductive elimination of the ArCu(III)–F species. This rare report of catalytic fluorination by a copper catalyst provides a valuable foundation for further development of Cu(I)-catalyzed fluorination of aryl halides.
Introduction
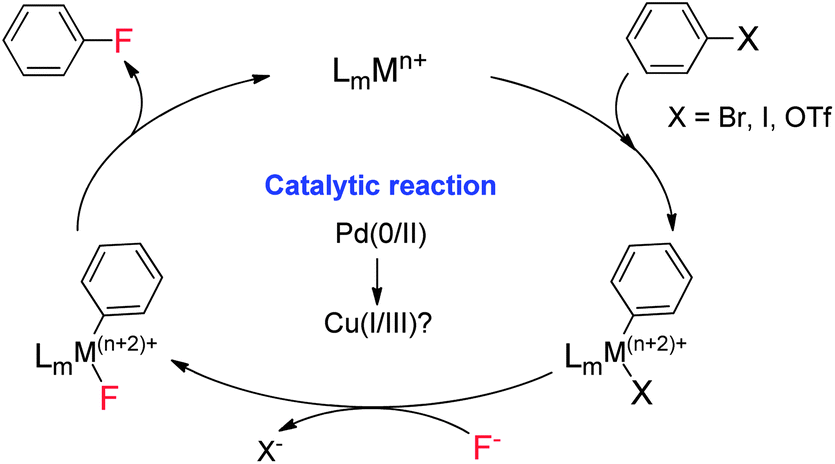
Aryl fluorides as essential moieties have widespread applications in pharmaceuticals, agrochemicals and functional materials. Therefore, the development of an efficient methodology for aryl C–F bond formation has attracted much attention.1 As with many other catalytic organic transformations,2 transition metals have been used to catalyze C–F bond formation.3 Compared to traditional fluorination reactions, such as the Balz–Schiemann reaction and the Halex process,4,5 the catalytic strategy is often highly efficient and encompasses a broad substrate scope.6
As a class of cross-coupling reactions,7 transition metal-catalyzed coupling of aryl halides with inorganic fluorides has attracted much attention for the synthesis of aryl fluorides (Scheme 1). However, this process is highly challenging due to the difficulty in achieving reductive elimination of the ArMF (M = Pd, Rh, etc.) species.8 In 2009, Buchwald and coworkers reported the first example of palladium-catalyzed cross-coupling of an aryl triflate with CsF in the presence of (tBu)BrettPhos.9 Recently, based on studies of well defined Cu(III) complexes and related coupling reactions,10 Ribas11a and Wang11b reported pioneering studies on the reductive elimination of macrocyclic ArylCuIIIF complexes to generate aryl C–F bonds. Thus, we envisioned that this inexpensive and abundant copper catalyst could be applied for catalytic fluorination of aryl halides, which would lead to a new catalytic strategy for late stage synthesis of aryl fluorides.
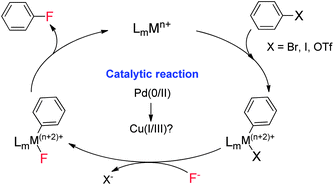 |
| | Scheme 1 Transition metal-catalyzed fluorination of aryl halides. | |
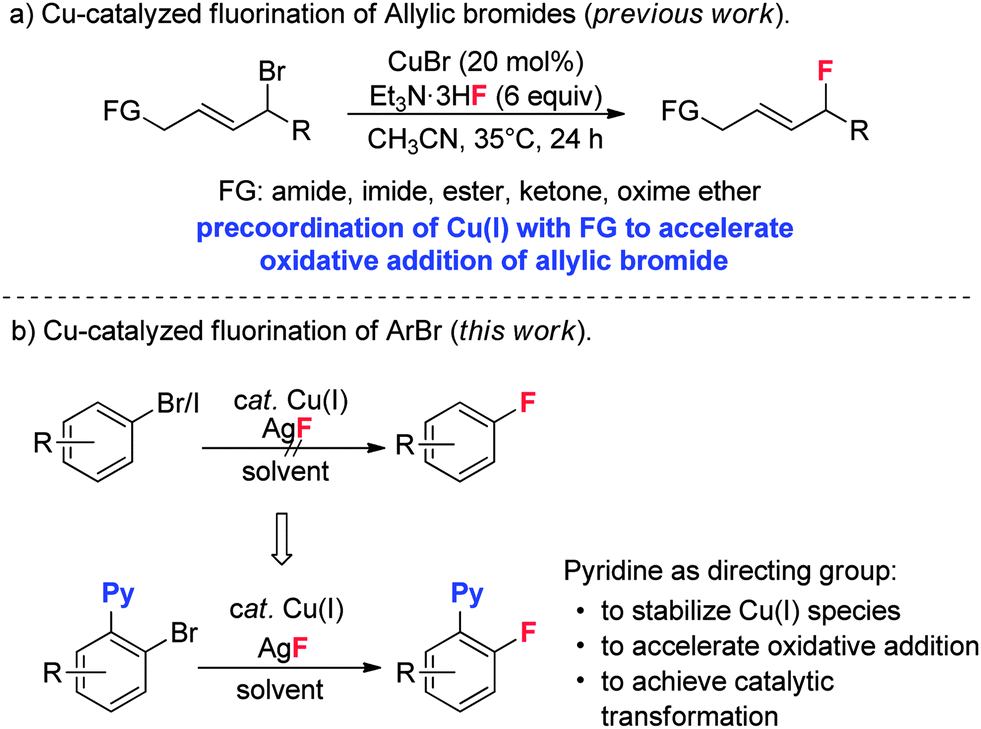
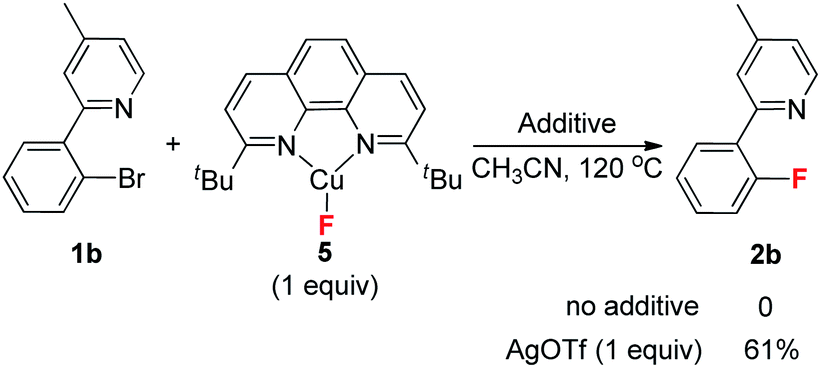
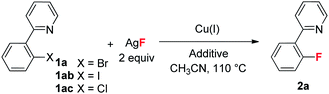
Early studies on the fluorination of aryl halides provided by Grushin demonstrated that CuF2 could be used to achieve fluorination of aryl iodides, but with very low efficiency.12 As part of our efforts to develop transition metal catalyzed fluorinations,13 we also attempted fluorination of ArBr and ArI using catalytic amounts of Cu(I) catalysts. Unfortunately, the desired fluorination product was not detected. Quite recently, Hartwig provided a copper mediated fluorination of aryl iodides using AgF as the fluorine source.14 However, the reaction required a large excess of Cu(I) over AgF, which was tentatively attributed to the competitive oxidation of Cu(I) by AgF, which would form inactive CuF2. In addition, the method was not suitable for aryl bromides. Our group recently discovered a copper-catalyzed fluorination for allylic bromides. In this transformation, the required presence of a coordinating functional group (FG) on the substrate suggests that pre-coordination between the FG and Cu(I) catalyst is crucial to the oxidative addition of the allylic bromide (Scheme 2a).15 Based on this finding, we reasoned that introduction of a directing group into the aryl halides might be helpful to achieve catalytic fluorination. Herein, we report this study, in which pyridine plays dual roles in the catalytic fluorination. Firstly, the ortho-pyridyl group is an effective directing group for accelerating the oxidative addition step. Secondly, the presence of the pyridine could stabilize the Cu(I) species to prevent its oxidation by AgF. These two important factors thus contribute to the successful catalytic transformation (Scheme 2b).
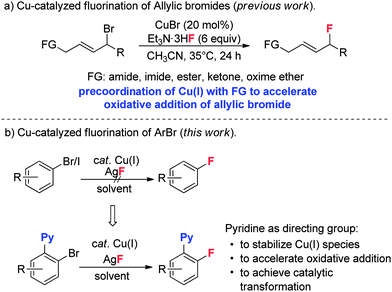 |
| | Scheme 2 Cu(I)-catalyzed fluorination reactions. | |
Results and discussion
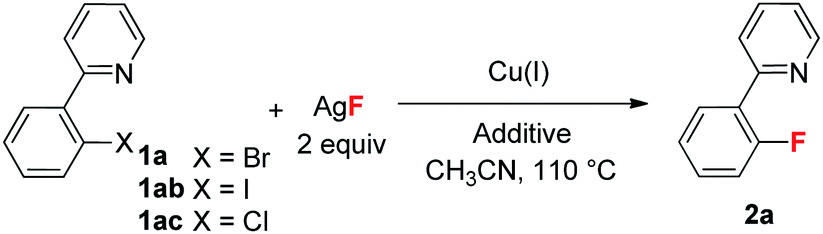
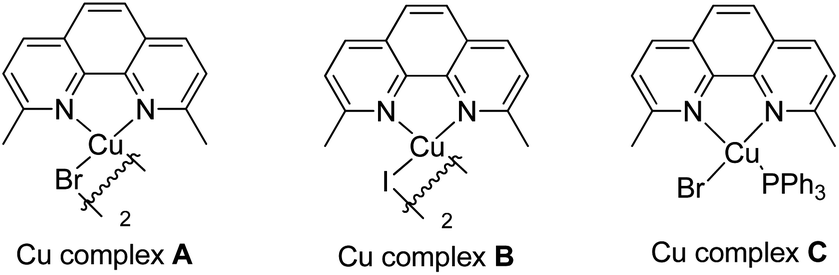
Our initial investigation focused on screening a variety of aryl bromides bearing different ortho-directing groups under various reaction conditions. When these substrates were treated with Cu(CH3CN)4PF6 (1 equiv.) and AgF (2 equiv.) in CH3CN, we were delighted to find that pyridine is a good directing group for the fluorination reaction. In contrast, the other functional groups, such as imine, ester, amide, etc., failed to give the desired products (see the ESI†). Further investigation focused on the optimization of reaction conditions. As shown in Table 1, a stoichiometric amount of Cu(CH3CN)4PF6 was effective, giving 2a in a 55% yield (entry 1). However, other copper salts, such as CuBr, CuI, and [Cu(OTf)]2·C6H6, were ineffective (entries 2–4). Replacement of AgF with combination of silver salts and CsF also provided 2a in similar yields (entries 5–6). Excitingly, the reaction with a catalytic amount of Cu(CH3CN)4PF6 also provided 2a in a 38% yield at 120 °C (entry 7). The reaction was retarded and gave diminished yields when the PF6 anion was replaced with BF4 or OTf (entries 8–9). These results indicate that the counter ion could influence catalytic efficiency. Thus, exogenous hexafluorophosphate salts were added to adjust the reactivity of the copper catalyst. An improved yield was observed in the presence of Bu4NPF6 (entry 12). However, KPF6 and Me4NPF6 gave inferior results (entries 10–11). A higher concentration provided a better yield (entry 13). Compared to the stoichiometric reaction, replacement of AgF with AgOTf/CsF gave a very low yield in the catalytic reaction (entry 5 vs. entry 14). Addition of nitrogen or oxygen containing ligands did not improve the reaction yields (see ESI†). Contrary to the inactive CuBr, surprisingly, copper complex A gave similar results to Cu(CH3CN)4PF6, but copper complexes B and C gave slightly lower yields (entries 15–17 & Fig. 1). No reaction occurred in the absence of copper catalyst. Finally, other 2-pyridyl-phenyl halide reactions were investigated. The reaction of 2-pyridyl-phenyl iodide 1ab exhibited a similar reactivity to 1a, but 2-pyridyl-phenyl chloride 1bc was inactive towards fluorination (entries 18–19).
Table 1 Screening resultsa
|
|
| Entry |
Substrate |
[Cu] (mol%) |
Additive |
Yieldb (%) |
|
Reaction conditions: 1 (0.1 mmol) in 1.0 mL of CH3CN.
Yield determined by 19F-NMR spectroscopy with benzotrifluoride as the internal standard.
AgF was replaced with AgOTf/CsF (2 equiv.).
AgF was replaced with AgBF4/CsF (2 equiv.).
Reaction was carried out at 120 °C.
The concentration was 0.2 M.
|
| 1 |
1a
|
[Cu(MeCN)4]PF6 (100) |
— |
55 |
| 2 |
1a
|
CuBr (100) |
— |
0 |
| 3 |
1a
|
CuI (100) |
— |
0 |
| 4 |
1a
|
[Cu(OTf)]2·C6H6 (100) |
— |
0 |
| 5c |
1a
|
[Cu(MeCN)4]PF6 (100) |
— |
44 |
| 6d |
1a
|
[Cu(MeCN)4]PF6 (100) |
— |
57 |
| 7e |
1a
|
[Cu(MeCN)4]PF6 (20) |
— |
38 |
| 8e |
1a
|
[Cu(MeCN)4]OTf (20) |
— |
31 |
| 9e |
1a
|
[Cu(MeCN)4]BF4 (20) |
— |
29 |
| 10e |
1a
|
[Cu(MeCN)4]PF6 (20) |
KPF6 (1 equiv.) |
10 |
| 11e |
1a
|
[Cu(MeCN)4]PF6 (20) |
Me4NPF6 (1 equiv.) |
38 |
| 12e |
1a
|
[Cu(MeCN)4]PF6 (20) |
n-Bu4NPF6 (1 equiv.) |
53 |
| 13e,f |
1a
|
[Cu(MeCN)4]PF6 (20) |
n-Bu4NPF6 (1 equiv.) |
65 |
| 14c |
1a
|
[Cu(MeCN)4]PF6 (20) |
n-Bu4NPF6 (1 equiv.) |
16 |
| 15e,f |
1a
|
Cu complex A (20) |
— |
63 |
| 16e,f |
1a
|
Cu complex B (20) |
— |
42 |
| 17e,f |
1a
|
Cu complex C (20) |
— |
39 |
| 18e,f |
1ab
|
[Cu(MeCN)4]PF6 (20) |
n-Bu4NPF6 (1 equiv.) |
62 |
| 19e,f |
1ac
|
[Cu(MeCN)4]PF6 (20) |
n-Bu4NPF6 (1 equiv.) |
Trace |
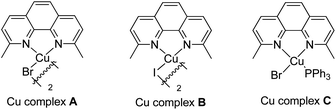 |
| | Fig. 1 The structures of the copper complexes. | |
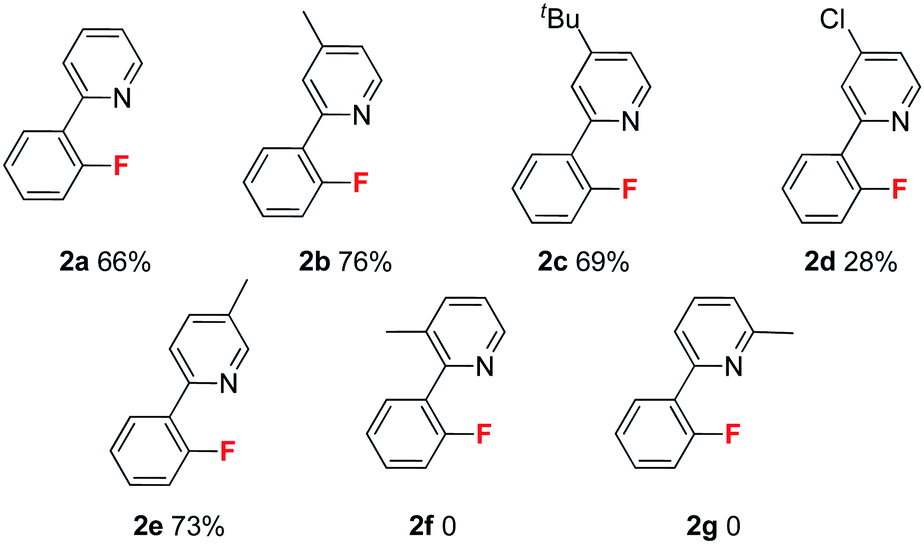
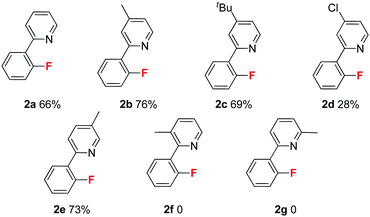
Next, we explored substituent effects on the pyridyl ring (Table 2). The electron donating groups provided superior results than those of the weak electron withdrawing groups. For instance, substrates 1b (4-Me) and 1c (4-t-Bu) gave better results than 1a, but substrate 1d (4-Cl) gave a lower yield. These results suggest that the electron-donating pyridyl group significantly improved the fluorination process. In addition, the positions of the methyl group influenced fluorination. Methyl groups located at the 4° (1b) and 5° (1e) positions provided similar results, but those positioned at the 3° (1f) and 6° (1g) positions could not deliver the corresponding fluorination products.
Table 2 Substituent effects on the pyridine ringa
|
|
|
Reactions were run at 0.2 mmol scale, isolated yield.
|
|
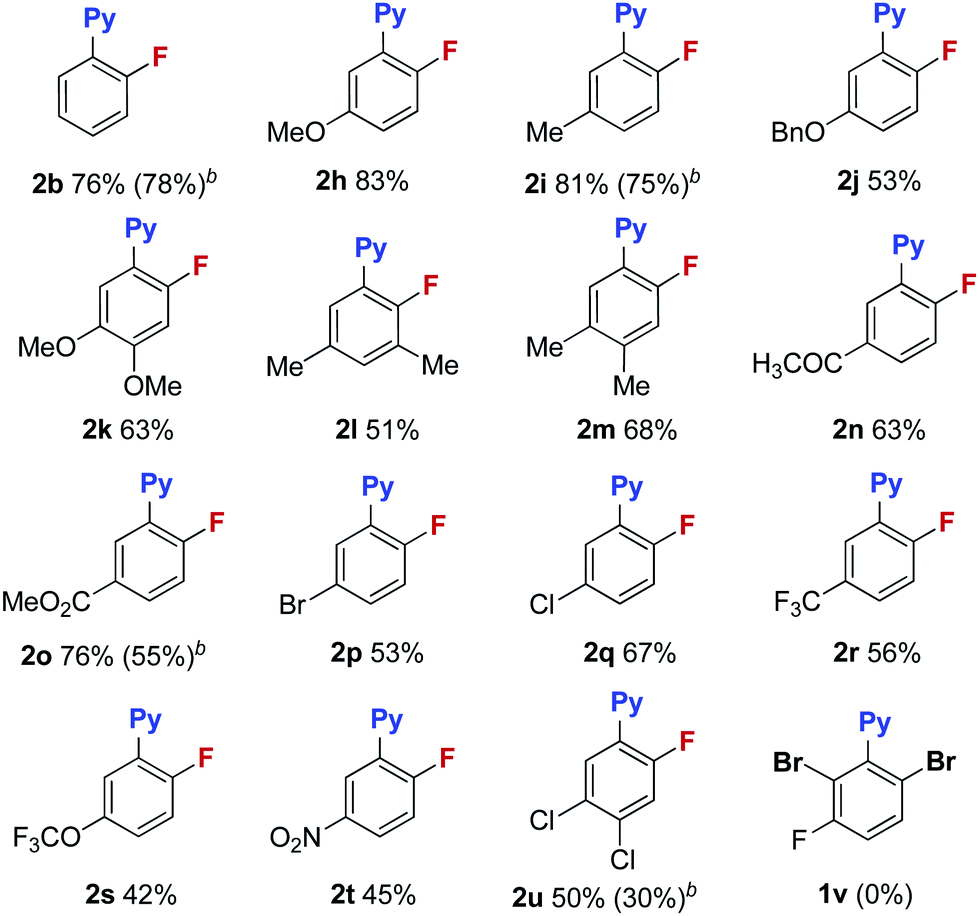
With these optimized reaction conditions in place, the substrate scope with a variety of substituents on the benzene ring was investigated (Table 3). Compared to the Halex process, which is only suitable for electron-poor aryl compounds, electron-rich aryl bromides were compatible with these reaction conditions. For instance, aryl bromides bearing one or two electron donating groups gave moderate to good yields (2b, 2h–2m). Electron poor substrates were also suitable for these reaction conditions giving satisfactory yields. In these reactions, various functional groups, such as a ketone (2n), an ester (2o), halogens (2p–2q) and a trifluoromethyl (2r), survived in this transformation. Substrates bearing trifluoromethoxyl and nitro groups delivered the corresponding products (2s–2t) in slightly low yields (42% and 45%). It is worth noting that, for substrate 1p containing two bromides, only the ortho-bromide was reactive. For the more electron poor substrate 1u, with two chlorides and one bromide, the reaction also proceeded smoothly to afford the fluorination product 2u in a 50% yield without replacement of chlorine by fluorine. These observations again address the vital role of the pyridyl group in promoting the fluorination. Unfortunately, substrate 1v bearing two ortho-bromides failed to give the fluorination product. For electron rich aryl bromides (such as 1b, 1i), it should be pointed out that the Cu complex A showed a similar reactivity to the cationic copper catalyst giving comparable yields, but with slightly lower reactivity towards the electron-poor substrates (1o, 1u). Finally, bromobenzo-[h]quinoline 1w was a good substrate, giving product 2w in a 58% yield (eqn (1)).
| |  | (1) |
Table 3 Substrate scope for fluorination of aryl bromidesa
|
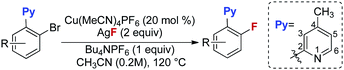
|
|
Reactions were run at 0.2 mmol scale, isolated yield.
The data in parenthesis was obtained with copper complex A (entry 15 in Table 1).
|
|
Mechanism
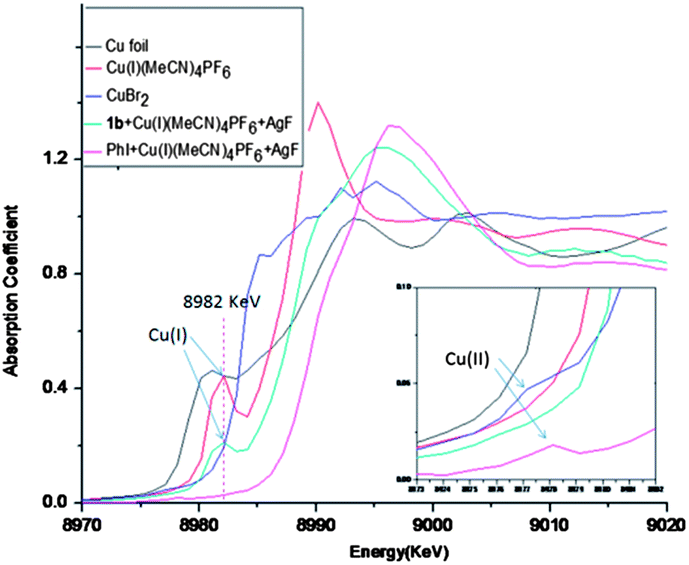
Compared to substrate 1, the standard conditions were not suitable for the fluorination of simple iodobenzene due to the lack of the pyridyl group. It is possible that coordination of the pyridyl with Cu(I) may help to stabilize the Cu(I) species in the excess of AgF and achieve the catalytic fluorination. In order to test this hypothesis, XANES/EXAFS analysis was carried out on the reaction residues of PhI and 1b, providing further insight into the proposed scenario. For the case of PhI, the Cu(I) reagent was fully oxidized to the Cu(II) species, for which the peak at 8982 keV was absent and new signal at 8978 keV appeared.16 In contrast, Cu(I) species still existed in the case of 1b, because oxidation of the Cu(I) had been suppressed (Fig. 2).17 These results revealed that coordination of the pyridyl group is vital for stabilization of the Cu(I) species.
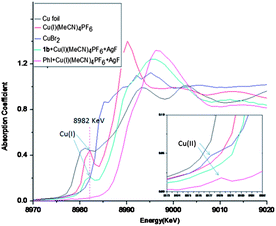 |
| | Fig. 2 XANES/EXAFS analysis of the Cu species. | |
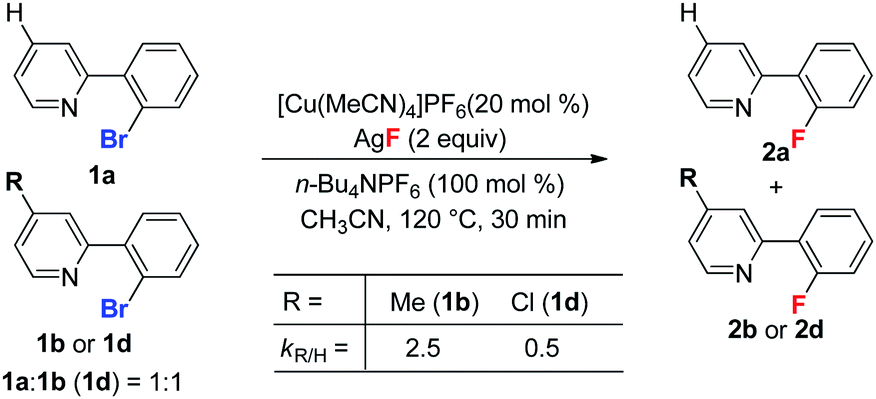
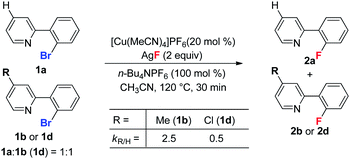
In addition, as shown in eqn (2), substrate 1b bearing an electron donating group on the pyridine ring reacted significantly faster than a substrate with an electron withdrawing group (1d). The possible reason for this is that the electron rich pyridyl group enhances the nucleophilicity of Cu(I) facilitating oxidative addition at the aryl C–Br bond.
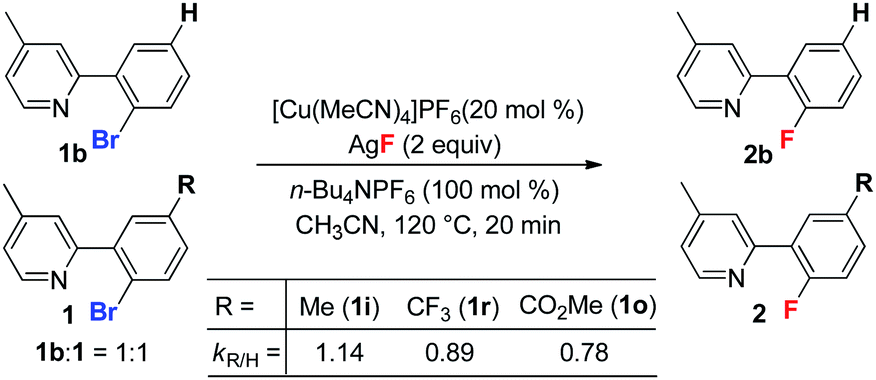
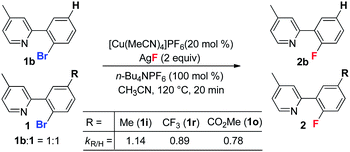
To probe the electronic effect of the aryl bromides, a series of competing reactions were carried out. A mixture of substrates 1b (R = H) and 1i (or 1r, or 1o) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio was subjected to the standard reaction conditions for 20 min to reach a low conversion (less than 20%). As shown in eqn (3), the electron rich substrate (1i) exhibited a faster rate than the electron poor substrates (1r and 1o). This phenomenon is consistent with a Cu(I)/Cu(III) catalytic cycle, which was reported for the copper-catalyzed C–C bond formation reaction.18 Recently, Fu and Peters reported a Cu-catalyzed Ullman C–N coupling involving a single electron transfer (SET) mechanism, in which electron deficient aryl halides react faster than electron donating substrates.19 The observed opposite reactivity indicates that the SET pathway is less likely in the present case.
:1 ratio was subjected to the standard reaction conditions for 20 min to reach a low conversion (less than 20%). As shown in eqn (3), the electron rich substrate (1i) exhibited a faster rate than the electron poor substrates (1r and 1o). This phenomenon is consistent with a Cu(I)/Cu(III) catalytic cycle, which was reported for the copper-catalyzed C–C bond formation reaction.18 Recently, Fu and Peters reported a Cu-catalyzed Ullman C–N coupling involving a single electron transfer (SET) mechanism, in which electron deficient aryl halides react faster than electron donating substrates.19 The observed opposite reactivity indicates that the SET pathway is less likely in the present case.
| |  | (2) |
| |  | (3) |
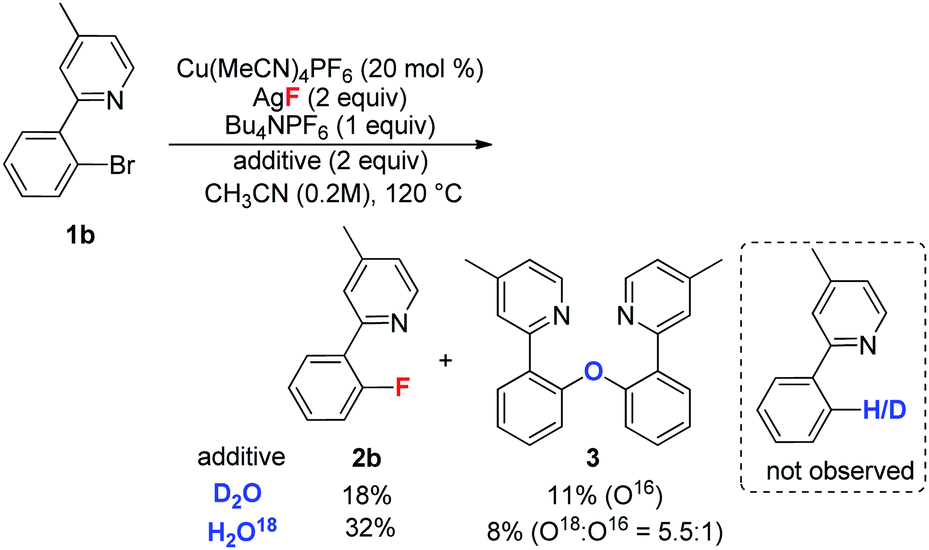
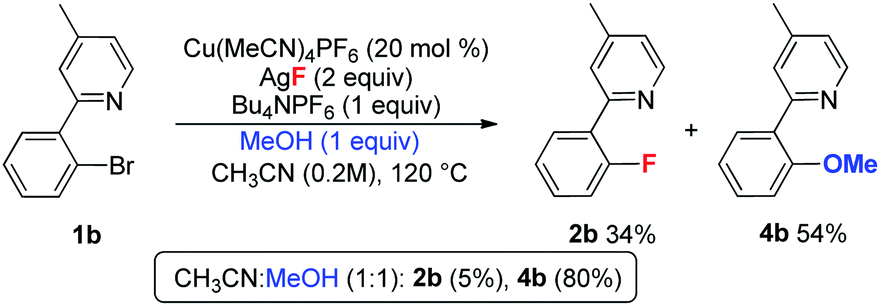
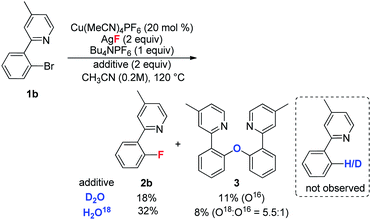
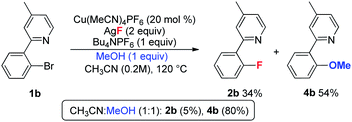
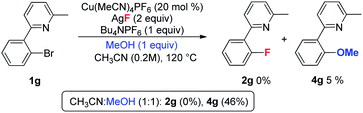
During the optimization of the reaction conditions, a trace amount of side product 3 (less than 5%) was detected. In contrast, the corresponding dehalogenation product was not observed.20 In order to investigate the formation of 3, the reaction of 1b was conducted in the presence of both D2O and H218O. As shown in eqn (4), the yield of 2b was obviously decreased in the presence of water, and 3 was isolated in low yield. In the case of H218O, the incorporation of 18O revealed that product 3 is derived from the reaction of 1b with water.21 When MeOH was used as an additive, the reaction afforded major methoxylation product 4b in 54% yield, along with a 34% yield of the fluorination product 2b. When MeOH was used as a co-solvent, the reaction afforded product 4b in an 80% yield, but with a trace amount of 2b (eqn (5)). Hartwig and coworkers recently reported a copper-mediated fluorination of iodobenzene.14 In this reaction, a significant amount of the dehalogenation side product was observed, which was attributed to the radical pathway.19,22 In contrast, the related dehalogenation reaction did not occur in the current transformation, even in the presence of water (eqn (4)). Combined with the formation of 3 and 4b and other experimental observations we suggest that the current reaction possibly involves a Cu(I/III) pathway.10
| |  | (4) |
| |  | (5) |
| |  | (6) |
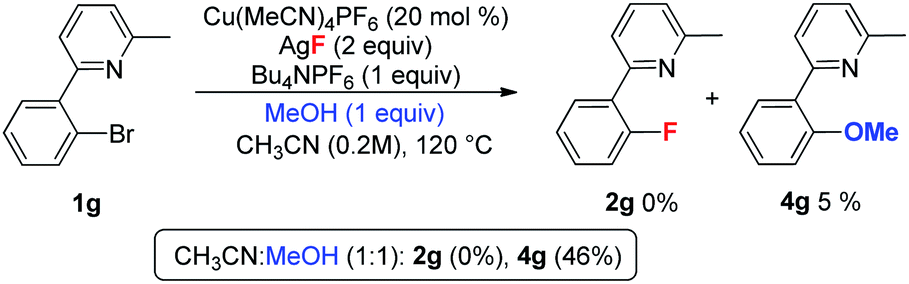
As mentioned in Table 2, substrate 1g was inert towards fluorination due to the steric effect. In contrast, the reaction of 1g delivered product 4g in a 46% yield in the presence of a large excess of MeOH, but provided only a trace amount of 4g with a stoichiometric amount of MeOH (eqn (6)).23 These observations reveal that methoxylation of aryl bromide 1g also exhibits a similar steric effect. However, compared to the above methoxylation, the lack of fluorination in the case of 1g (in Table 2) implied that oxidative addition to the aryl C–Br bond by PyCuIL (Py = 1g, L = F or CH3CN) is slower than that with PyCuI(OMe).24
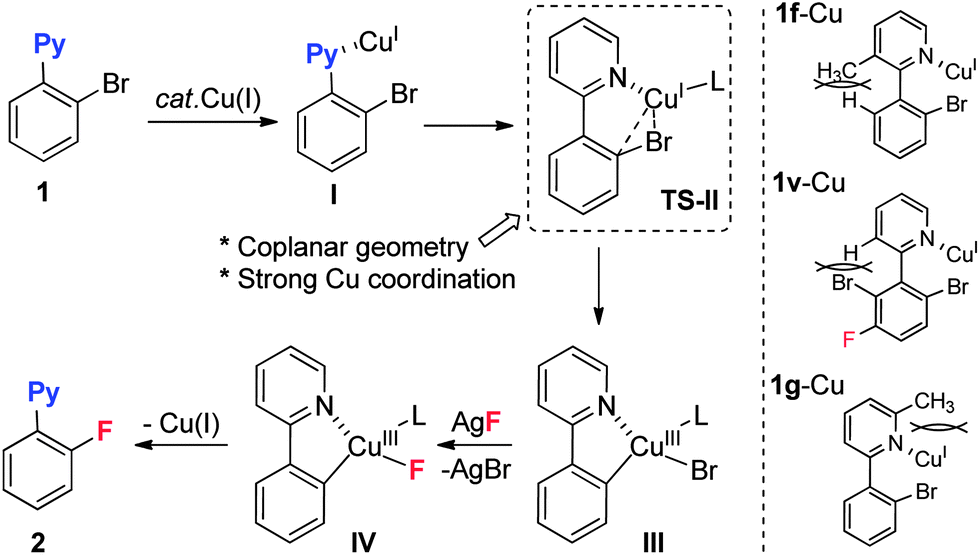
Based on the results above, a mechanism is proposed as shown in Scheme 3. The initial pre-coordination of substrate 1 with Cu(I) promotes an intramolecular oxidative addition to give the Cu(III) intermediate III. Subsequent ligand exchange with AgF and irreversible reductive elimination of IV provides the fluorination product 2.25 In this catalytic cycle, two additional requirements should be mentioned: (1) a coplanar geometry in complex I is required for this reaction. Substrate 1f, which lacks coplanar geometry due to repulsion between the 3-methyl and ortho-H groups (1f-Cu), could not deliver the fluorinated product. A similar effect was observed for substrate 1v. (2) The unsuccessful fluorination of substrate 1g with its 6-methyl group is possibly attributed to weakened coordination due to the steric effect, which also indicated that strong coordination ability in the substrate is crucial for the reaction. The above unfavorable steric effects might destabilize the transition state (TS-II) or the Cu(III) ground state III.
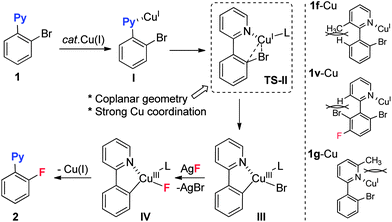 |
| | Scheme 3 Proposed mechanism. | |
Finally, screening of the common fluorine source demonstrated that only AgF could deliver the product. Other inorganic fluorides were ineffective for yielding the fluorination product (see ESI†). Although these results indicate that AgF plays an important role in this transformation, the detailed mechanism is still unknown at the present time. A possible explanation is that the presence of a silver salt helps to activate the C–Br bond and trap the bromide ion in the aryl-Cu(III) complex III generating complex IV, which results in C–F bond formation. For the case of other inorganic fluorides, removal of the bromide ion becomes more difficult. Thus, instead of forming complex IV, complex III could go back to the aryl bromide via reductive eliminations, or generate another side product. In order to provide further insight on this point, the isolated CuF complex 5 (ref. 26) was used in a stoichiometric reaction. Interestingly, no fluorination occurred, but product 2b was obtained in the presence of AgOTf (eqn (7)). These results are consistent with the analysis above.
| | 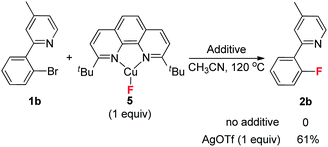 | (7) |
Conclusions
We have developed a novel copper-catalyzed fluorination of aryl bromides with AgF, in which pyridyl groups are important for successful catalytic fluorination. Preliminary mechanistic studies indicated that the coordination of the pyridyl group with Cu(I) could stabilize the Cu(I) species and accelerate oxidative addition of the aryl bromide. Final aryl C–F bond formation possibly follows an irreversible reductive elimination of the ArCu(III)–F species. Due to the current substrate limitation for the pyridyl directing group, further expansion of the substrate scope and application of this chemistry is in progress.
Acknowledgements
We are grateful for financial support from the National Basic Research Program of China (973-2011CB808700), the National Nature Science Foundation of China (nos 20923005, 2125210, 21202185 and 21121062), the Science and Technology Commission of the Shanghai Municipality (11JC1415000), and the CAS/SAFEA International Partnership Program for Creative Research Teams. We also thank Shanghai Synchrotron Radiation Facility for the study of XANES/EXAFS. GSL thank Prof. Zhiqiang Weng for the gift of CuF complex 5. XM thanks Mr Jia-bin Xu for repeating reactions.
Notes and references
-
(a) P. Jeschke, Pest Manage. Sci., 2010, 66, 10 CrossRef CAS PubMed
 ;
(b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC ;
(c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed ;
(d) P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS PubMed .
;
(b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC ;
(c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed ;
(d) P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS PubMed .
-
(a)
Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi, Wiley-Interscience, New York, 2002 Search PubMed ;
(b)
J. F. Hartwig, Organotransition Metal Chemistry, From Bonding to Catalysis, University Science Books, California, 2010 Search PubMed .
- For some recent reviewers on the transition metal-catalyzed fluorination, see:
(a) V. V. Grushin, Acc. Chem. Res., 2010, 43, 160 CrossRef CAS PubMed ;
(b) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed ;
(c) A. Vigalok, Organometallics, 2011, 30, 4802 CrossRef CAS ;
(d) C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929 RSC ;
(e) G. Liu, Org. Biomol. Chem., 2012, 10, 6243 RSC .
- G. A. Olah, J. T. Welch, Y. D. Vankar, M. Nojima, L. Kerekes and J. A. Olah, J. Org. Chem., 1979, 44, 3872 CrossRef CAS .
- D. J. Adams and J. H. Clark, Chem. Soc. Rev., 1999, 28, 225 RSC .
- For some catalytic examples, see:
(a) K. L. Hull, W. Q. Anani and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 7134 CrossRef CAS PubMed ;
(b) X. Wang, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 7520 CrossRef CAS PubMed ;
(c) P. P. Tang, T. Furuya and T. Ritter, J. Am. Chem. Soc., 2010, 132, 12150 CrossRef CAS PubMed ;
(d) T. Truong, K. Klimovica and O. Daugulis, J. Am. Chem. Soc., 2013, 135, 9342 CrossRef CAS PubMed . For some stoichiometric reactions, see;
(e) T. Furuya, A. E. Strom and T. Ritter, J. Am. Chem. Soc., 2009, 131, 1662 CrossRef CAS PubMed ;
(f) Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 4648 CrossRef CAS PubMed ;
(g) P. S. Fier, J. Luo and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 2552 CrossRef CAS PubMed .
-
Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-Interscience, New York, 2nd edn, 2008 Search PubMed .
- For reviews, see ref. 3a and
(a) V. V. Grushin, Chem.–Eur. J., 2002, 8, 1006 CrossRef CAS . For some examples, see;
(b) S. L. Fraser, M. Y. Antipin, V. N. Khroustalyov and V. V. Grushin, J. Am. Chem. Soc., 1997, 119, 4769 CrossRef CAS ;
(c) J. P. Flemming, M. C. Pilon, O. Y. Borbulevitch, M. Y. Antipin and V. V. Grushin, Inorg. Chim. Acta, 1998, 280, 87 CrossRef CAS ;
(d) V. V. Grushin, Organometallics, 2000, 19, 1888 CrossRef CAS ;
(e) V. V. Grushin and W. J. Marshall, Angew. Chem., Int. Ed., 2002, 41, 4476 CrossRef CAS ;
(f) W. J. Marshall and V. V. Grushin, Organometallics, 2004, 23, 3343 CrossRef CAS ;
(g) S. A. Macgregor, D. C. Roe, W. J. Marshall, K. M. Bloch, V. I. Bakhmutov and V. V. Grushin, J. Am. Chem. Soc., 2005, 127, 15304 CrossRef CAS PubMed ;
(h) V. V. Grushin and W. J. Marshall, Organometallics, 2008, 27, 4825 CrossRef CAS ;
(i) V. V. Grushin and W. J. Marshall, J. Am. Chem. Soc., 2009, 131, 918 CrossRef CAS PubMed .
-
(a) D. A. Watson, M. J. Su, G. Teverovskiy, Y. Zhang, J. Garcia-Fortanet, T. Kinzel and S. L. Buchwald, Science, 2009, 325, 1661 CrossRef CAS PubMed ;
(b) T. Noël, T. J. Maimone and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 8900 CrossRef PubMed .
-
(a) X. Ribas, D. A. Jackson, B. Donnadieu, J. Mahia, T. Parella, R. Xifra, B. Hedman, K. O. Hodgson, A. Llobet and T. D. P. Stack, Angew. Chem., Int. Ed., 2002, 41, 2991 CrossRef CAS ;
(b) L. M. Huffman and S. S. Stahl, J. Am. Chem. Soc., 2008, 130, 9196 CrossRef CAS PubMed ;
(c) A. E. King, L. M. Huffman, A. Casitas, M. Costas, X. Ribas and S. S. Stahl, J. Am. Chem. Soc., 2010, 132, 12068 CrossRef CAS PubMed ;
(d) A. Casitas, A. E. King, T. Parella, M. Costas, S. S. Stahl and X. Ribas, Chem. Sci., 2010, 1, 326 RSC ;
(e) A. Casitas, A. Poater, M. Sola, S. S. Stahl, M. Costas and X. Ribas, Dalton Trans., 2010, 39, 10458 RSC ;
(f) L. M. Huffman, A. Casitas, M. Font, M. Canta, M. Costas, X. Ribas and S. S. Stahl, Chem.–Eur. J., 2011, 17, 10643 CrossRef CAS ;
(g) L. M. Huffman and S. S. Stahl, Dalton Trans., 2011, 40, 8959 RSC ;
(h) Z.-L. Wang, L. Zhao and M.-X. Wang, Org. Lett., 2011, 13, 6560 CrossRef CAS PubMed .
-
(a) A. Casitas, M. Canta, M. Solà, M. Costas and X. Ribas, J. Am. Chem. Soc., 2011, 133, 19386 CrossRef CAS PubMed ;
(b) B. Yao, Z.-L. Wang, H. Zhang, D.-X. Wang, L. Zhao and M.-X. Wang, J. Org. Chem., 2012, 77, 3336 CrossRef CAS PubMed .
-
V. V. Grushin and D. E. Hockessin, US20060074261, 2006 .
-
(a) T. Wu, G. Yin and G. Liu, J. Am. Chem. Soc., 2009, 131, 16354 CrossRef CAS PubMed ;
(b) H. Peng and G. Liu, Org. Lett., 2011, 13, 772 CrossRef CAS PubMed ;
(c) T. Xu, S. Qiu and G. Liu, Chin. J. Chem., 2011, 29, 2785 CrossRef CAS ;
(d) T. Xu, X. Mu, H. Peng and G. Liu, Angew. Chem., Int. Ed., 2011, 50, 8176 CrossRef CAS PubMed ;
(e) S. Qiu, T. Xu, J. Zhou, Y. Guo and G. Liu, J. Am. Chem. Soc., 2010, 132, 2856 CrossRef CAS PubMed ;
(f) H. Peng, Z. Yuan, H. Wang, Y. Guo and G. Liu, Chem. Sci., 2013, 4, 3172 RSC ;
(g) H. Zhu and G. Liu, Acta Chim. Sin., 2012, 70, 2404 CrossRef CAS .
- P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 10795 CrossRef CAS PubMed .
- Z. Zhang, F. Wang, X. Mu, P. Chen and G. Liu, Angew. Chem., Int. Ed., 2013, 52, 7549 CrossRef CAS PubMed .
-
(a) A. I. Frenkel, G. V. Korshin and A. L. Ankudinov, Environ. Sci. Technol., 2000, 34, 2138 CrossRef CAS ;
(b) M. Sano, S. Komorita and H. Yamatera, Inorg. Chem., 1992, 31, 459 CrossRef CAS ;
(c) C. He, G. Zhang, J. Ke, H. Zhang, J.-T. Miller, A.-J. Kropf and A. Lei, J. Am. Chem. Soc., 2013, 135, 488 CrossRef CAS PubMed .
- In the case of PhI, the oxidation of Cu(I) by AgF is possible, forming CuF2, which is inactive for this fluorination of 1a.
- Z. Huang and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 1028 CrossRef CAS PubMed .
-
(a) S. E. Creutz, K. J. Lotito, G. C. Fu and J. C. Peters, Science, 2012, 338, 647 CrossRef CAS PubMed ;
(b) J. W. Tye, Z. Weng, A. M. Johns, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 9971 CrossRef CAS PubMed .
- In copper-catalyzed or mediated reactions of aryliodide, dehalogenated ArI was observed as a side product. For details, see ref. 14 and 19a.
-
(a) T. Anis, N. Xia, F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 8725 CrossRef PubMed ;
(b) J. Chen, T. Yuan, W. Hao and M. Cai, Catal. Commun., 2011, 12, 1463 CrossRef CAS PubMed ;
(c) R. Paul, M. A. Ali and T. Punniyamurthy, Synthesis, 2010, 4268 CAS .
- For radical-chain, electron-transfer dehalogenation of arylhalides, see: J. F. Bunnett, Acc. Chem. Res., 1992, 25, 2 CrossRef CAS .
- For copper-catalyzed cross-coupling of arylbromide with alcohols, see:
(a) H. L. Aalten, G. van Koten, D. M. Grove, T. Kuilman and O. G. Piekstra, Tetrahedron, 1989, 45, 5565 CrossRef CAS ;
(b) P. Capdevielle and M. Maumy, Tetrahedron Lett., 1993, 34, 1007 CrossRef CAS .
- In this reaction, AgF acts as a base generating the copper species Cu(OMe).
- When product 2a was treated under standard reaction conditions replacing AgF with Bu4NBr, the reaction did not afford 1a, and 2a was recovered quantitively.
.
-
Y. Liu, C. Chen, H. Li, J. Tan, K.-W. Huang, Y. Yuan, Z. Weng, 2013 DOI:10.1021/om4008967.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedure and spectroscopic characterization data are provided. See DOI: 10.1039/c3sc51876k |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.