Composite supramolecular nanoassemblies with independent stimulus sensitivities†
Conghui
Yuan
ab,
Kishore
Raghupathi
a,
Bhooshan C.
Popere
a,
Judy
Ventura
a,
Lizong
Dai
*b and
S.
Thayumanavan
*a
aDepartment of Chemistry, University of Massachusetts, Amherst, MA 01003, USA. E-mail: thai@chem.umass.edu
bCollege of Materials, Xiamen University, Xiamen, 361005, People's Republic of China. E-mail: lzdai@xmu.edu.cn
First published on 26th September 2013
Abstract
Nanoscale assemblies with stimuli-sensitive features have attracted significant attention due to implications in a variety of areas ranging from materials to biology. Recently, there have been excellent developments in obtaining nanoscale structures that are concurrently sensitive to multiple stimuli. Such nanostructures are primarily focused on a single nanostructure containing an appropriate combination of functional groups within the nanostructure. In this work, we outline a simple approach to bring together two disparate supramolecular assemblies that exhibit very different stimuli-sensitive characteristics. These composite nanostructures comprise a block copolymer micelle core and nanogel shell, both of which can preserve their respective morphology and stimulus sensitivities. The block copolymer is based on poly(2-(diisopropylamino)ethylmethacrylate-b-2-aminoethylmethacrylate hydrochloride), which contains a pH-sensitive hydrophobic block. Similarly, the redox-sensitive nanogel is derived from a poly(oligoethyleneglycolmonomethylethermethacrylate-co-glycidylmethacrylate-co-pyridyldisulfide ethylmethacrylate) based random copolymer. In addition to the independent pH-response of the micellar core and redox-sensitivity of the nanogel shell in the composite nanostructures, the synergy between the micelles and the nanogels have been demonstrated through a robust charge generation in the nanogels during the disassembly of the micelles. The supramolecular assembly and disassembly have been characterized using transmission electron microscopy, dynamic light scattering, zeta potential measurements, fluorescence spectroscopy and cellular uptake.
Polymeric supramolecular assemblies that predictably respond to environmental changes have been of great interest due to their possible use in many applications.1 More recently, assemblies that respond to multiple stimuli have been of interest.2 The key motivation for this interest involves the enhanced selectivity by materials that respond to the concurrent presence of two or more stimuli. Most of the polymers and the resultant assemblies designed for this purpose are based on a single assembly. We have been interested in designing a system, where we bring together two different supramolecular assemblies each of which are independently and synergistically responsive to two different stimuli. To this end, we have developed a new reactive self-assembly route in which we achieve a dynamic composite nanostructure, where a redox sensitive nanogel is coated over a pH sensitive block polymeric micelle (Fig. 1). To fully test the versatility of the approach, we stipulated that: (i) both the micelle and the nanogel should preserve their morphological fidelity in the composite assembly; (ii) the constituent nanoassemblies independently respond to their respective stimuli; and (iii) at least one synergistic feature of the composite nanoassembly is demonstrated.
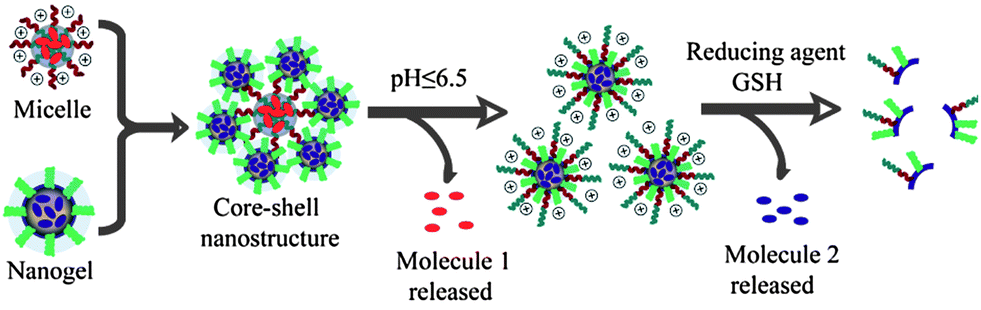 | ||
| Fig. 1 Schematic representation of the composite nanostructure assembly and stimuli-sensitive disassembly. | ||
The composite nanostructure is based on a micellar assembly formed from an amphiphilic block copolymer and a nanogel formed from a self-crosslinking polymer assembly (Scheme 1). Specifically, poly(2-(diisopropylamino)ethylmethacrylate-b-2-aminoethylmethacrylate hydrochloride) (PDPA30-b-PAMA15) is used as the block copolymer, which was synthesized by atom transfer radical polymerization3(ATRP). Similarly, nanogels were prepared from the random copolymer, poly(oligoethyleneglycolmonomethylether methacrylate-co-glycidylmethacrylate-co-pyridyldisulfide ethylmethacrylate) (P(EGMA-GMA-PDSEMA)). The PDSEMA monomer was then used to generate disulfide crosslinks using DL-dithiothreitol (DTT) following the procedures previously reported.4 Here, the EGMA monomer provides a charge-neutral hydrophilic group for self-assembly in the aqueous phase, while the epoxide functionality in the GMA co-monomer provides the functional handle for reactive self-assembly with the block copolymer assembly.
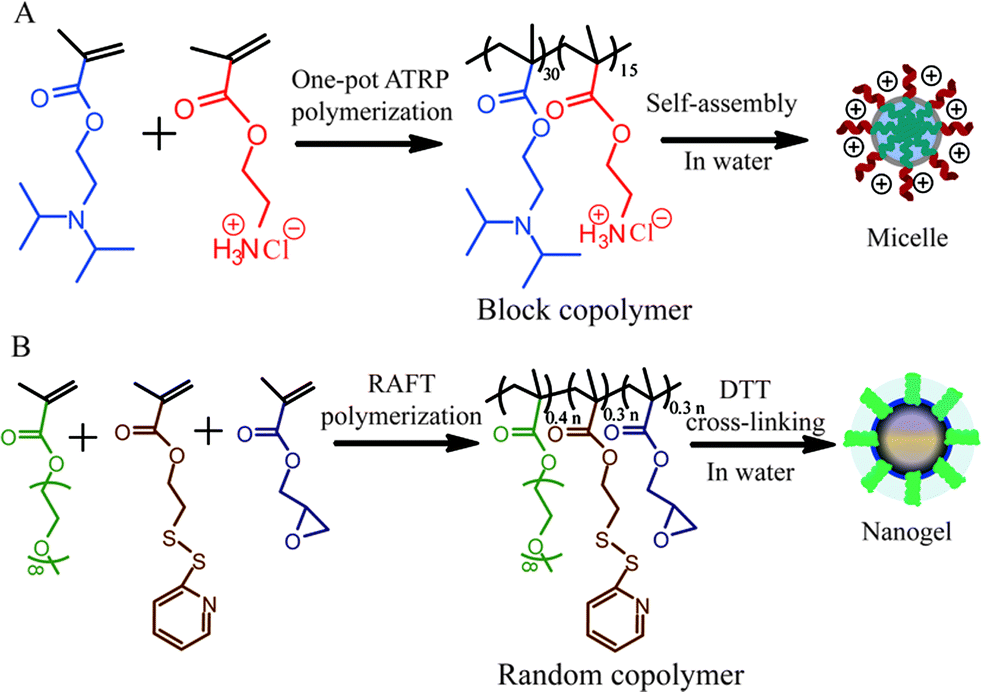 | ||
| Scheme 1 Syntheses of (a) PDPA30-b-PAMA15 block copolymer and (b) P(EGMA-GMA-PDSEMA) random copolymer and the corresponding nanogel. | ||
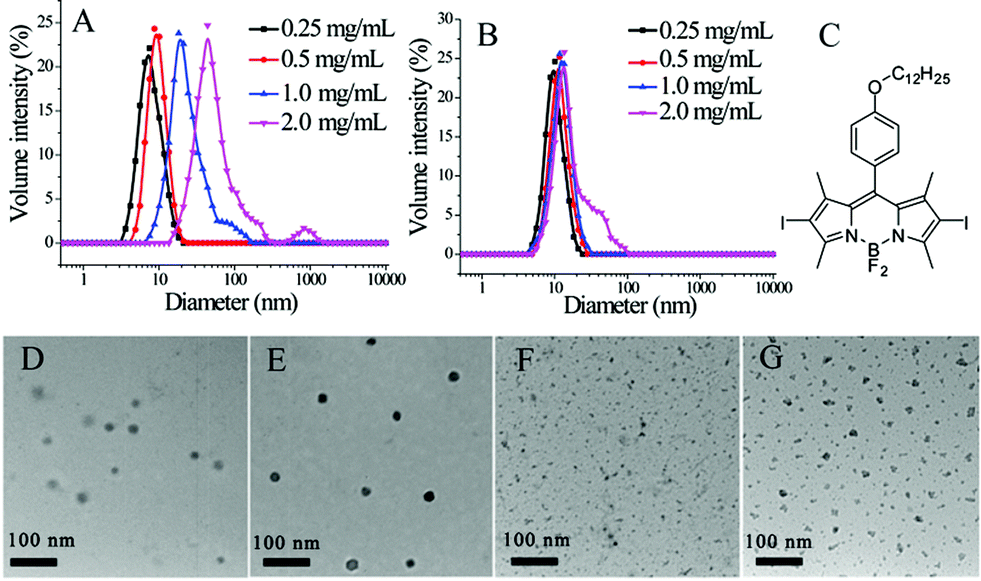 | ||
| Fig. 2 (A) Dynamic light scattering (DLS) of micelles assembled from diblock copolymers at various concentrations; (B) DLS of nanogels at different concentrations; (C) chemical structure of BDP-C12-I2. TEM images of micelles before (D) and after (E) encapsulation of BDP-C12-I2, and nanogels before (F) and after (G) encapsulation of BDP-C12-I2. | ||
Prior to generating the composite nanoassemblies, we characterized the individual supramolecular assemblies generated from the block copolymer and the nanogel using dynamic light scattering (DLS). DLS studies of the block copolymer assembly revealed that the assemblies' diameters range from 10 to 35 nm (Fig. 2A). Since a certain amount of dilution would occur during the formation of these composite assemblies, the block copolymer assemblies were examined before and after dilution. The assemblies had small perturbations, if any, in size after diluting the solution to half concentration (Fig. S3†). Similarly, DLS characterization of the nanogels revealed sizes of ∼10 nm in size (Fig. 2B). Since these nanogels are formed through a chemical crosslinking reaction, we anticipated that the sizes should not decrease upon dilution. Indeed, we found the sizes to be invariant upon dilution of the nanogels (Fig. S3†).
Transmission electron microscopy (TEM) was used to further characterize the block copolymer micelles and the nanogels. As shown in Fig. 2D and F, the sizes of both the block copolymer micellar assembly and the polymeric nanogel observed by TEM are similar to those obtained by DLS. When generating composite nanostructures, it is critical that we distinctly visualize these nanostructures independently. For this purpose, we exploited the capability of both these nanostructures to non-covalently sequester lipophilic guest molecules. We used an iodine-containing hydrophobic dye BDP-C12-I2 and examined their TEM images. As shown in Fig. 2E and G, these structures were indeed darker and clearer. These results also confirm that hydrophobic guest molecules can be incorporated in these assemblies and that this feature can be used to enhance contrast in TEM.
Next, the possibility of obtaining composite nanostructures using the combination of polymer micelles and the nanogels was investigated. Under the reaction conditions, the amine moiety can open the epoxide ring to form the corresponding amino alcohol product. This reaction should covalently attach the micellar assembly and the nanogel. The fidelity of the resultant composite nanostructure was first tested by using micelles and nanogels both with ∼10 nm in size. The reason for this choice is that these two assemblies with such diameters exhibit a narrow polydispersity in particle size. Although the micelle and the nanogel independently are ∼10 nm in size, the composite nanostructure was found to be ∼35 nm (Fig. 3A). The structure of the composite nanostructure was hypothesized to be a simple coating of nanogels on the micelle via covalent bonds.
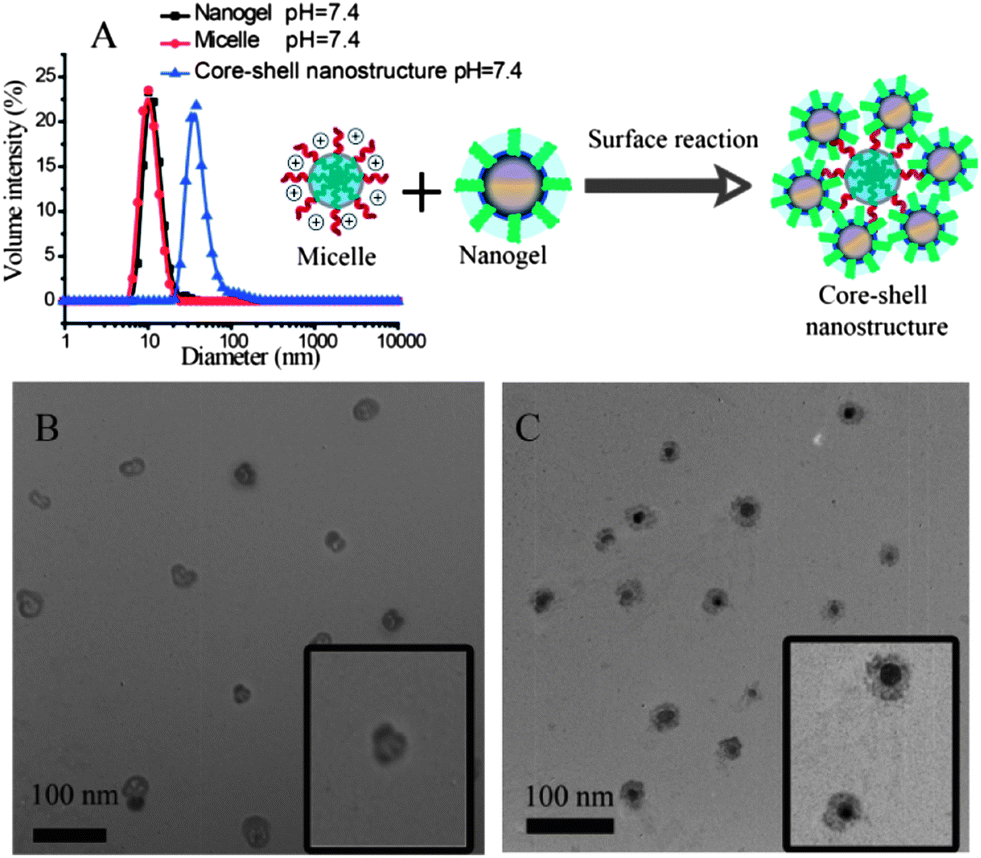 | ||
| Fig. 3 (A) Dynamic light scattering of nanogels (■), micelles (●) and composite nanostructures at pH = 7.4 (▲); the nanogels and the micelles were made from 0.5 mg mL−1 of random copolymer and block copolymer PDPA30-b-PAMA15, respectively; Inset in (A) schematically illustrates the morphology of the composite nanostructure. (B) and (C) TEM images of composite nanostructures formed from the combination of nanogels and micelles; for the sample preparation of (B), the nanogels were loaded with BDP-C12-I2 while the micelles were empty to enhance their contrast; for the sample preparation of (C), the micelles were loaded with BDP-C12-I2 while the nanogels were empty to enhance their contrast. | ||
To test this hypothesis, the composite nanostructures were investigated by TEM. In order to distinguish the micelle from the nanogel, we used: (i) a 25 nm micelle along with the 10 nm nanogel; and (ii) the heavy atom bearing dye molecule, BDP-C12-I2, for it to be non-covalently incorporated into the polymer micelle or the nanogel. The purpose of the latter dye as guest molecule is to conveniently visualize the assembly in which the guest molecule is present. We envisioned that this guest molecule would act as a stain in TEM due to the heavy iodine atoms present in the molecule. If our hypothesis that the combination of the nanogel and the block copolymer assembly forms a composite type nanostructure were correct, then when the dye molecule was incorporated into the nanogel, a darker shell and a hollow core should be observed. On the other hand, when the guest molecule is present in the micelle, a dark core coated with a lighter shell should be present. Indeed, TEM images are consistent with these expectations (Fig. 3B and C), thereby supporting our structural hypothesis. The difference in contrasts in these two structures also show that there is very little guest molecule transfer, if any, from one assembly to the other in the composite nanostructure.
Next, we tested whether the fidelity of the individual nanoassemblies is retained in the composite nanostructures. Since the block copolymer contains poly(2-(diisopropylamino)ethyl methacrylate) as the hydrophobic block, this polymer micellar assembly can be pH-sensitive.5 The pKa of the protonated tertiary amine in this block is expected to be about 6.8. Therefore, while this amine can be unprotonated and hydrophobic at pH 7.4, a significant percentage of this amine moiety is expected to be protonated at pH 6.5.5b This protonation event should decrease the hydrophobicity of the block, thus disassembling the micelle. We independently confirmed that this is indeed the case with the block copolymer micelle used in this study (Fig. S4†). Then, we tested whether this feature is retained in the composite nanostructure. Note that the composite nanostructure prepared from nanogels (∼10 nm) and micelles (∼10 nm) exhibit average diameters of about 35 nm. When the pH of the solution was reduced from 7.4 to 6.5, the composite nanostructure showed a significant decrease in size (Fig. 4A, Fig. S6†). In fact, the final particle sizes of the system at pH 6.5 is nearly the same as those of the nanogel solutions at pH 7.4, suggesting that the pH-induced disassembly features of the block copolymer micelle are indeed retained in the composite nanostructure.
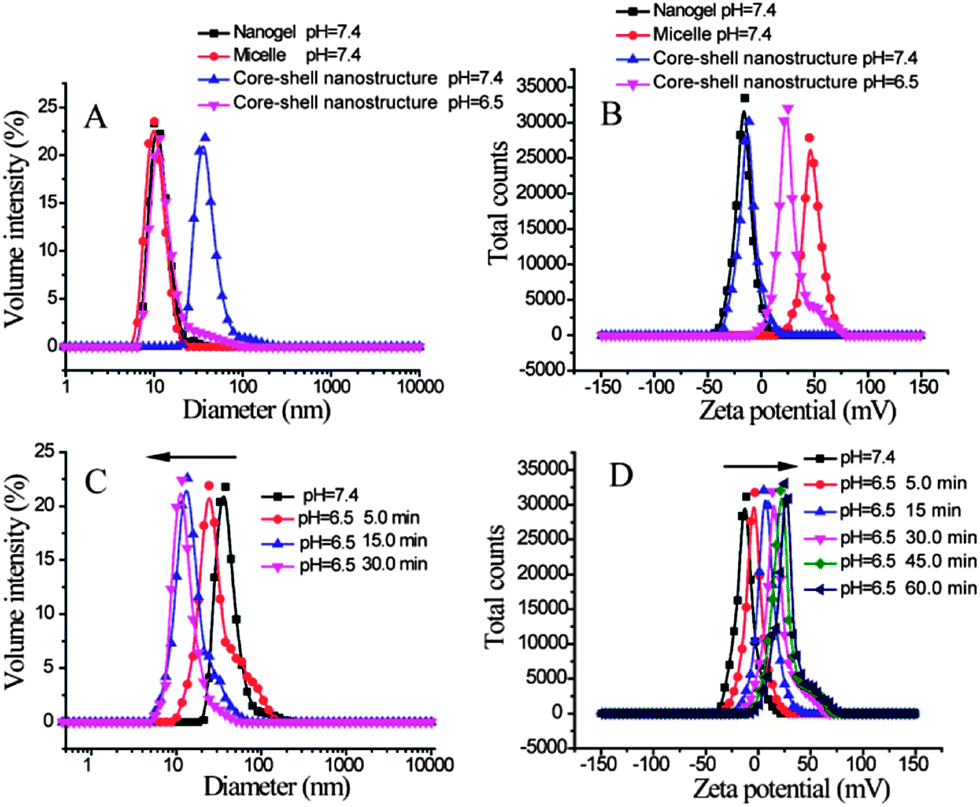 | ||
| Fig. 4 (A) Diameters of composite nanostructures in aqueous solution at pH 7.4 (▲) and 6.5 (▼); to make a comparison, diameters of nanogels (■) and micelles (●) at pH 7.4 are also illustrated. (B) Zeta potentials of nanogels (■), micelles (●) and the composite nanostructures at pH = 7.4 (▲) and pH = 6.5 (▼). (C) and (D) represent the disassembly kinetics and the zeta potential changing trend of the composite nanostructures at pH 6.5. The nanogels and the micelles used in these tests were made from 0.5 mg mL−1 random copolymer and block copolymer PDPA30-b-PAMA15, respectively. | ||
The pH-induced disassembly event is schematically shown as the response to the first stimulus in Fig. 1. Since the nanogel is covalently attached to the block copolymer micelle in the composite nanostructure, a few polymer chains would still be attached to the nanogel after the disassembly. This feature should cause the nanogel to display the protonated amine groups on its surface, which should result in a change in the surface charge of the nanogel. The zeta potentials of the micelles and the nanogels by themselves are positive and negative, respectively (Fig. S3 and S4†). The positively charged surface of the micelle is due to the primary amine moieties on its surface. The reason for the negative surface charge of the oligoethyleneglycol decorated nanogel is not clear; however, this feature has been observed previously with these functional groups.6 The surface charge of the composite nanostructure was negative, as the micelle was densely coated with nanogels. When the pH was reduced from 7.4 to 6.5, the surface charge of the system became positive (Fig. 4B). All these results are consistent with the first step of the disassembly model shown in Fig. 1.
To test the correlation between the composite nanostructure disassembly event and the surface charge change, we compared the time needed to reach saturation in size change with that for the change in surface charge. The size change was saturated within 30 min upon decreasing the solution pH from 7.4 to 6.5 (Fig. 4C). On the other hand, it took about 60 min for the zeta potential change to level off (Fig. 4D). This difference can be attributed to the possibility that disassembly can occur with protonation of a smaller number of amine moieties, as the requisite change in the hydrophilic–lipophilic balance for disassembly is already reached. Also, it is possible that the unreacted epoxide moieties on dissociated nanogels would now react with the water-soluble PDPA-b-PAMA polymer chains, as their reaction with amino groups are facilitated in weakly acidic solutions.
Similarly, the nanogel used in this composite nanostructure is sensitive to the tripeptide, glutathione (GSH). GSH can cleave disulfide bonds through a thiol–disulfide exchange reaction, which results in an uncrosslinking of the nanogel (second step of the disassembly model shown in Fig. 1). DLS and zeta potential measurements are not sufficient to probe this disassembly, because the uncrosslinking has to be quantitative to observe the change in the composite size. Quantitative uncrosslinking is unlikely, since the process is based on an exchange reaction that is under equilibrium. Therefore, we resorted to monitoring GSH-sensitive guest release as the probe to investigate whether the nanogel also preserves its salient features in the composite assembly.
When constructing the composite nanostructure, we incorporated 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) in the nanogels and kept the micelles empty. Fig. 5A shows that no DiI is released from the nanoassembly at pH 7.4 and 6.5, as their emission intensity does not change with time. Since it has been confirmed that the micelle core can disassemble at pH 6.5, it is reasonable that the disassembly of the micelles exhibit no effect on the release of DiI from nanogels. Then, 0.1 mM GSH was added to ascertain whether the DiI, encapsulated within the nanogel, could be released. Indeed, the fluorescence intensity of the DiI decreased gradually with time (Fig. 5A). This decrease is attributed to the GSH-induced disulfide bond cleavage to uncrosslink the nanogels and guest release. This was further confirmed by monitoring the degree of DiI release when the GSH concentration was significantly increased. When the concentration of GSH was increased from 0.1 to 70 mM, the rate of release of the guest molecule was found to be considerably higher (compare Fig. 5A and B; also see Fig. S7† for release in response to 5 mM GSH).
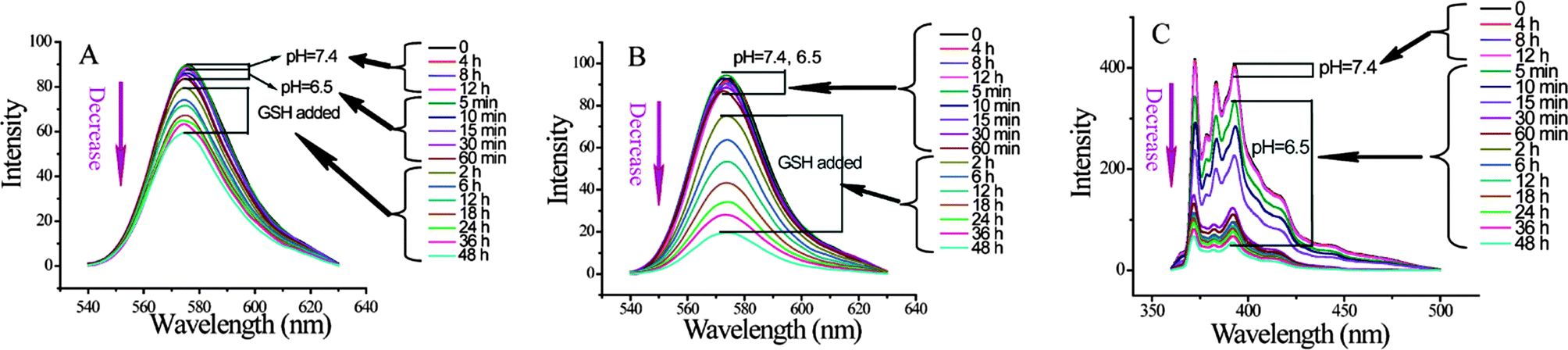 | ||
| Fig. 5 Fluorescence spectra trace the release of DiI from the composite nanostructures in response to GSH with different concentrations, (A) 0.1 mM and (B) 70 mM; and release of pyrene in response to pH (C). The composite nanostructures were constructed by copolymer nanogels (0.5 mg mL−1) and PDPA30-b-PAMA15 micelles (0.5 mg mL−1). All nanogels used in these studies were 40% cross-linked. DiI was encapsulated in nanogels. | ||
Similarly, we were also interested in testing whether the guest molecule trapped within the block copolymer micelle can be independently released, since this assembly is sensitive to reduced pH. For this purpose, we sequestered pyrene within the hydrophobic interior of the block copolymer micelle. Reducing the pH to 6.5 resulted in significant loss of pyrene over time (see Fig. 5C). Note that the change in pH did not result in any significant loss in DiI, which was sequestered within the redox-sensitive nanogels. These results suggest that the composite nanostructures can independently release the incorporated hydrophobic molecules in response to redox or pH change.
Finally, we demonstrate the possibility of synergy in combining two nanostructures using this system as an example. It is well-known for many nanoscale architectures that those with positively charged surfaces are capable of being taken up by cells faster than anionic or charge-neutral assemblies.7 Note that the surface charge of the composite nanostructure is very similar to that of the nanogel itself. Therefore at pH 7.4, the composite nanoassembly should not have significant cellular uptake. However, when the pH is reduced to 6.5, the micelle at the core disassembles leaving behind the positively charged protonated tertiary amine block on the surface of the nanogel, which renders the nanogel positively charged (see Fig. 1 for an illustration). Therefore, at this pH the nanocomposite should exhibit a significant cellular uptake. We performed cellular uptake experiments with HeLa cells for 30 min (Fig. 6A and B). No cellular uptake was observed at pH 7.4, whereas significant uptake was observed at pH 6.5. To insure that the nanogels themselves do not have any pH-dependent uptake, we carried out the control experiments with the nanogels themselves at both pHs. There was no discernible cellular uptake of the nanogels under either of these conditions (see Fig. S8†). Strategies for pH-induced surface charge changes have been previously reported.8 The strategy outlined here, based on composite nanoassemblies, is based on a fundamentally different approach.
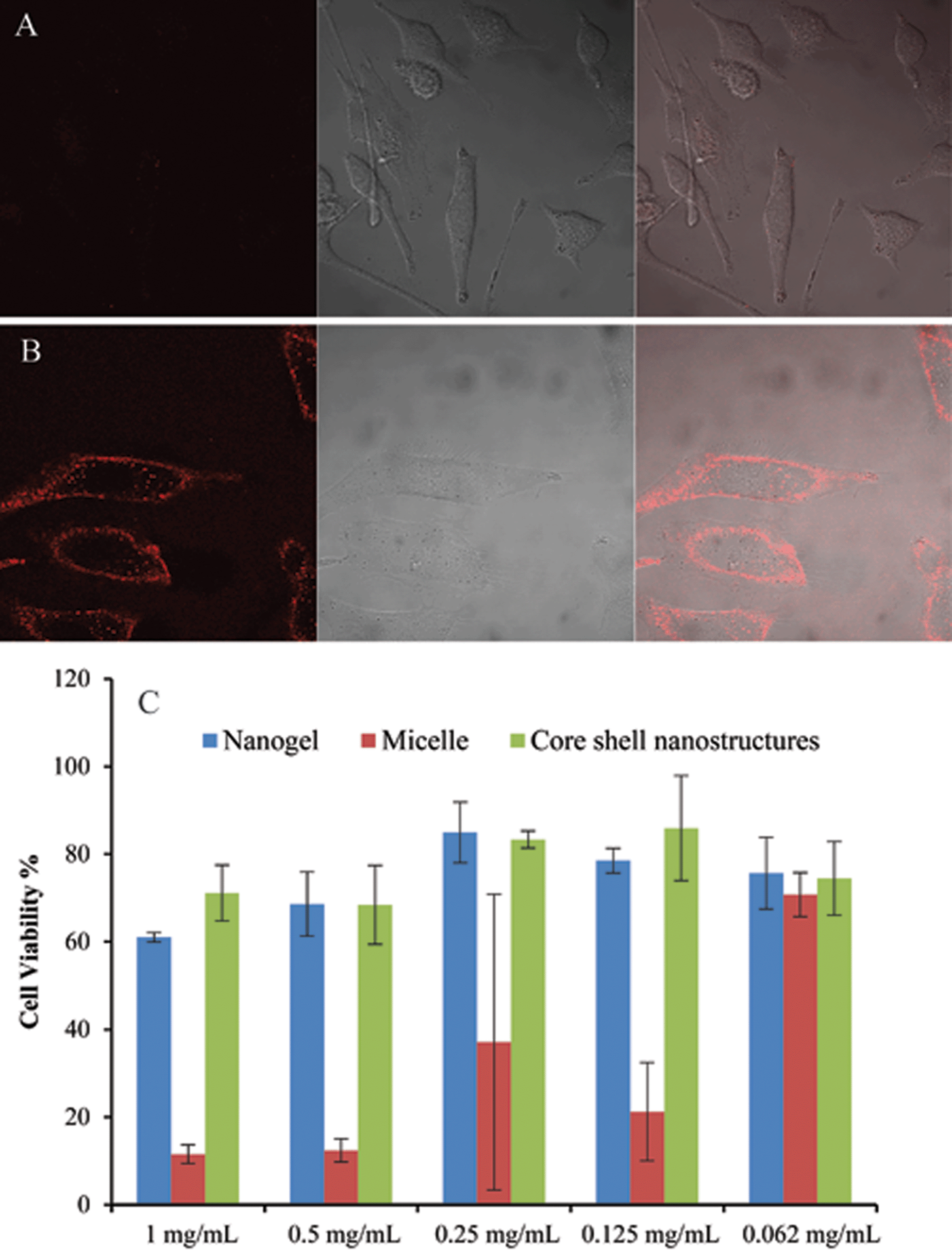 | ||
Fig. 6 Cell uptake of composite nanoassemblies at (A) pH 7.4 and (B) pH 6.5 after incubation with cells for 30 min. The composite nanoassemblies used in the dye release testing and cell uptake experiment were made from 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of copolymer 1 nanogels (40% crosslinked, 0.5 mg mL−1) and PDPA30-b-PAMA15 micelles (0.5 mg mL−1). Among each set of confocal images: the left panel corresponds to DiI emission, the middle panel corresponds to DIC image and right panel shows an overlap of both. (C) Cytotoxicity of nanogels, micelles and composite nanostructures by Alamar blue assay. :1 ratio of copolymer 1 nanogels (40% crosslinked, 0.5 mg mL−1) and PDPA30-b-PAMA15 micelles (0.5 mg mL−1). Among each set of confocal images: the left panel corresponds to DiI emission, the middle panel corresponds to DIC image and right panel shows an overlap of both. (C) Cytotoxicity of nanogels, micelles and composite nanostructures by Alamar blue assay. | ||
To further illustrate the synergy in combining two nanostructures, we also tested the toxicity of the composite and the individual nanostructures. It is well-known with many nanoscale architectures that surfaces with cationic functionality exhibit high cytotoxicity, while with PEG functionality exhibit a low cellular toxicity.9 If the surface functional groups are primary determinants of the cellular toxicity, we should see that the composite nanoassemblies are significantly less toxic compared to those of the block copolymers. Fig. 6C shows the cell viability of nanogels, micelles and composite nanostructure. Indeed, the cationic block copolymer micelles exhibit higher cytotoxicity than the charge-neutral nanogels. Interestingly, the composite nanostructure with nanogel coating on the surface of micelle, exhibits almost the same lower cytotoxicity as the nanogels alone. These results support the synergistic impact of the nanogels upon the micelles in the composite nanostructure. The synergy in cytotoxicity exists because the positively charged micelle is effectively shielded by the nanogel shell.
Conclusions
In summary, we have shown that two different polymeric nanostructures, viz. a pH-sensitive block copolymer micelle and a redox-sensitive crosslinked polymeric nanogel, can be brought together to form a composite nanoassembly using a reactive self-assembly approach. We have shown that: (i) the composite nanostructure is formed with the block copolymer assembly as the core and the nanogel as the shell; (ii) the relative placement of these assemblies can be ascertained by encapsulating a heavy-atom containing guest molecule selectively in one of the nanostructures as a stain for TEM imaging; (iii) the stimuli-sensitive host–guest properties of the two assemblies are retained in the composite nanoassembly; (iv) the presence of nanogel as the shell around the block copolymer assembly causes a change in the overall surface charge; (v) the surface charge change results in reduced cytotoxicity of the composite nanostructure, relative to the block copolymer assembly; and (vi) the pH-sensitive disassembly of the block copolymer changes the surface of the nanogel to be cationic, which results in an enhanced cellular uptake of the nanoassembly at lower pH. The possibility of charge generation and guest molecular release in response to two different triggers, in the current system, highlights the possible utility of this type of composite nanoassemblies in areas such as theranostics and dual delivery.10Acknowledgements
This work was partially supported by the U.S. Army Research Office. The Materials Research Science and Engineering Centre (MRSEC) at UMass Amherst, funded by the National Science Foundation, also supported this work. C. Y.'s work at UMass Amherst was partially supported by the Chinese National Science Foundation (51173135). Contributions from BCP (synthesis and utility of the BODIPY dye molecule) were partially supported by the PHaSE Energy Frontier Research Centre, funded by the Basic Energy Sciences of the U.S. Department of Energy. J. V. was partially supported by the NSF-IGERT program.Notes and references
- For an introduction to a collection of reviews on stimuli-sensitive materials, see: (a) P. Theato, B. S. Sumerlin, R. K. O'Reilly and T. H. Epps III, Chem. Soc. Rev., 2013, 42, 7055–7056 RSC; for a selected recent reviews on stimuli-sensitive solution phase assemblies, see: (b) E. G. Kelley, J. N. L. Albert, M. O. Sullivan and T. H. Epps III, Chem. Soc. Rev., 2013, 42, 7057–7071 RSC; (c) J. Thévenot, H. Oliveira, O. Sandre and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 7099–7116 RSC; (d) J.-F. Gohy and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7117–7129 RSC; (e) K. M. Wiggins, J. N. Brantley and C. W. Bielawski, Chem. Soc. Rev., 2013, 42, 7130–7147 RSC; (f) Z. Chu, C. A. Dreiss and Y. Feng, Chem. Soc. Rev., 2013, 42, 7174–7203 RSC; (g) M. I. Gibson and R. K. O'Reilly, Chem. Soc. Rev., 2013, 42, 7204–7213 RSC; (h) D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC; (i) Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289–7325 RSC; (j) J. Huang and A. Heise, Chem. Soc. Rev., 2013, 42, 7373–7390 RSC; (k) R. B. Grubbs and Z. Sun, Chem. Soc. Rev., 2013, 42, 7436–7445 RSC; (l) F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- J. Zhuang, M. Gordon, J. Ventura, L. Li and S. Thayumanavan, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- (a) K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS; (b) D. J. Siegwart, J. K. Oh and K. Matyjaszewski, Prog. Polym. Sci., 2012, 37, 18–37 CrossRef CAS PubMed.
- J.-H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227–17235 CrossRef CAS PubMed.
- (a) J. Du and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 12800–12801 CrossRef CAS PubMed; (b) J. Du, Y. Tang, A. L. Lewis and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 17982–17983 CrossRef CAS PubMed; (c) for another seminal example of a pH-sensitive block copolymer assembly, see: J. Rodríguez-Hernández and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026–2027 CrossRef PubMed.
- J. Zhuang, S. Jiwpanich, V. D. Deepak and S. Thayumanavan, ACS Macro Lett., 2012, 1, 175–179 CrossRef CAS.
- (a) J.-Z. Du, T.-M. Sun, W.-J. Song, J. Wu and J. Wang, Angew. Chem., 2010, 122, 3703–3708 CrossRef; (b) C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS PubMed; (c) L. Li, K. Raghupathi, C. Yuan and S. Thayumanavan, Chem. Sci., 2013, 4, 3654–3660 RSC.
- (a) Y. Lee, T. Ishii, H. Cabral, H. J. Kim, J. H. Seo, N. Nishiyama, H. Oshima, K. Osada and K. Kataoka, Angew. Chem., Int. Ed., 2009, 48, 5309–5312 CrossRef CAS PubMed; (b) C. M. LaManna, H. Lusic, M. Camplo, T. J. McIntosh, P. Barthélémy and M. W. Grinstaff, Acc. Chem. Res., 2012, 45, 1026–1038 CrossRef CAS PubMed.
- (a) M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed; (b) H. Summers, Nat. Nanotechnol., 2011, 7, 9–10 CrossRef PubMed; (c) D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS PubMed.
- (a) X. Chen, S. S. Gambhir and J. Cheon, Acc. Chem. Res., 2011, 44, 841 CrossRef CAS PubMed; (b) S. Sengupta, D. Eavarone, I. Capila, G. Zhao, N. Watson, T. Kiziltepe and R. Sasisekharan, Nature, 2005, 436, 568–572 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and characterization details, 1H NMR spectra, GPC, TEM images and DLS results of the resultant products. See DOI: 10.1039/c3sc52347k |
| This journal is © The Royal Society of Chemistry 2014 |