 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot three-component sulfone synthesis exploiting palladium-catalysed aryl halide aminosulfonylation†
Charlotte S.
Richards-Taylor
a,
David C.
Blakemore
b and
Michael C.
Willis
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: michael.willis@chem.ox.ac.uk; Fax: +44 (0)1865 285002; Tel: +44 (0)1865 285126
bNeusentis Chemistry, Pfizer Worldwide Research and Development, The Portway Building, Granta Park, Cambridge, CB21 6GS, UK
First published on 22nd October 2013
Abstract
A palladium-catalysed aminosulfonylation process is used as the key-step in a one-pot, three-component sulfone synthesis. The process combines aryl-, heteroaryl- and alkenyl iodides with a sulfonyl unit and an electrophilic coupling fragment. The sulfonyl unit is delivered in the form of an aminosulfonamide, which then serves as a masked sulfinate. The sulfinate is combined, in situ, with an electrophilic coupling partner, such as a benzylic, allylic or alkyl halide, an electron-poor arene, or a cyclic epoxide, to provide the corresponding sulfone products in good to excellent yields. The mild reaction conditions and use of commercially available reaction components allows the easy preparation of a broad range of sulfones featuring a variety of functional groups. The process obviates the need to employ thiol starting materials, and oxidative operations.
Introduction
The distinctive structural and electronic features present in sulfones have resulted in these unique functional groups being imbedded in a number of important pharmaceutical and agrochemical molecules. For example, molecules used against medical indications as diverse as migraine and prostate cancer, or as the herbicides mesotrione and cafenstrole, all feature aryl sulfone units (Scheme 1).1 The variety of chemical reactivity possible with a sulfone group means that they also serve as versatile intermediates in many synthetic routes.2 This combination of significant biological activity and compelling synthetic utility has resulted in the development of a number of methods for sulfone preparation. They are most commonly synthesised either by oxidation of the corresponding sulfide or sulfoxide,3 or by alkylation of a sulfinate salt.4 Both methods suffer drawbacks: for oxidative methods, the substrate scope is limited to molecules devoid of oxidation-sensitive functional groups. In addition, the ultimate starting material is often a thiol, many of which are foul-smelling, and there is limited commercial availability for alkenyl- or heteroaryl variants. Sulfinate salts have very limited commercial availability, and are usually prepared from the corresponding sulfonyl chloride.5 However, sulfonyl chlorides themselves can require multi-step procedures, which often feature harsh reaction conditions.6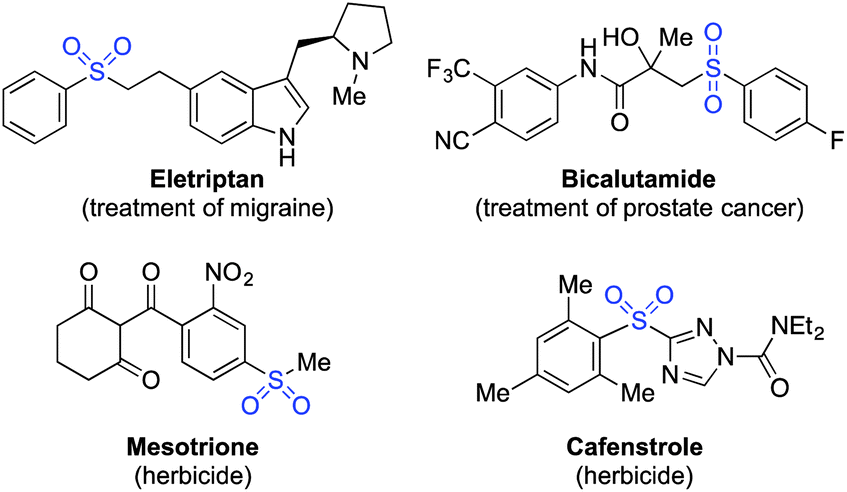 | ||
| Scheme 1 Representative biologically active aryl sulfones. | ||
Despite the limitations presented above, sulfone synthesis based on the combination of a sulfinate anion with a carbon-centered electrophile (for example, 1 → 2, Scheme 2) remains a potentially attractive route to these valuable molecules, due in particular to the wide range of electrophiles that can be employed successfully.4,7 We believed the main challenge in delivering a more useful variant of these transformations was to develop a sulfinate based route that avoids the necessity of a sulfonyl chloride intermediate, and the use of harsh reaction conditions.
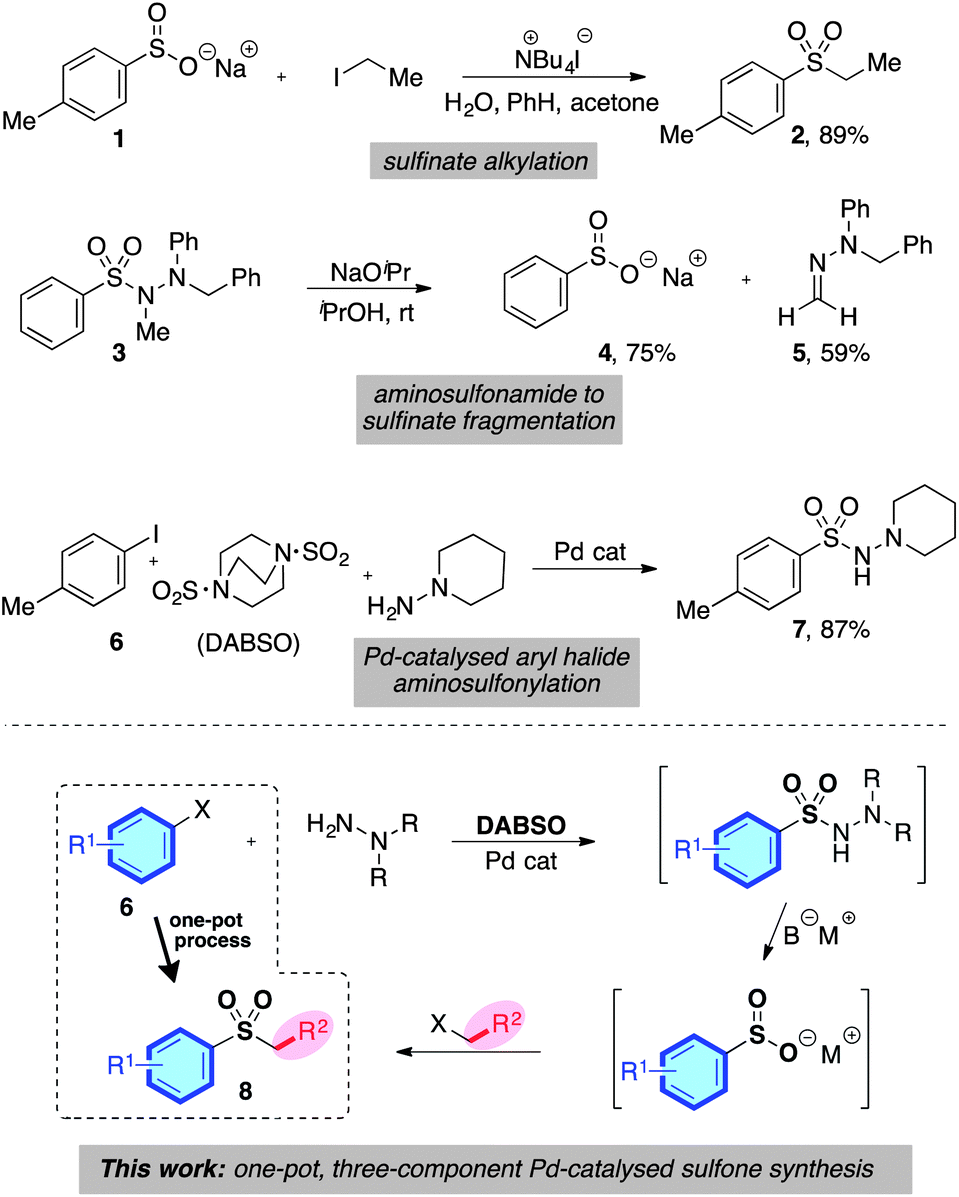 | ||
| Scheme 2 The origin of a one-pot, three-component sulfone synthesis. | ||
We were aware of a number of reports that had demonstrated the effective formation of metal sulfinates from a variety of N-aminosulfonamide derivatives; for example, Dornow and Bartsch had shown that N,N′,N′-trialkyl aminosulfonamides could be converted into sodium sulfinates under the action of sodium isopropoxide at ambient temperature (3 → 4, Scheme 2).8 In addition, various N′,N′-dialkyl aminosulfonamides have also been converted into the corresponding sodium sulfinates under the action of base,9 as have unsubstituted sulfonylhydrazides.10 Our laboratory has recently reported the palladium-catalysed formation of N′,N′-dialkyl aminosulfonamides from the combination of aryl halides, an SO2-surrogate (DABSO), and N′,N′-dialkylhydrazines (for example, 6 → 7, Scheme 2).11 This process was also effective for heteroaryl- and alkenyl halide substrates. Given the mild reaction conditions employed in the three key transformations – catalytic aminosulfonamide synthesis, aminosulfonamide to sulfinate degradation, and sulfinate alkylation – combined with the excellent availability of aryl halide substrates, we were attracted to the possibility of developing a one-pot sulfone synthesis based on the merger of these three transformations (6 → 8, Scheme 2).
Results and discussion
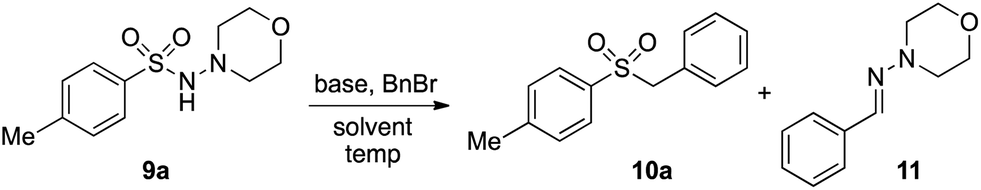
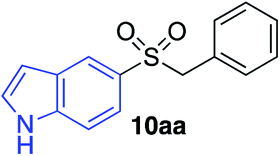
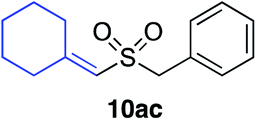
In developing the proposed route to sulfones we first elected to explore the conditions necessary for sulfinate generation and electrophile-trapping using pre-formed aminosulfonamides (Table 1). Literature precedent suggested that degradation of trialkyl aminosulfonamides could be achieved more readily and under milder reaction conditions than decomposition of the corresponding dialkyl-derivatives.8,9 Accordingly, we explored the alkylation/degradation of dialkyl aminosulfonamide 9a using a variety of weak bases and solvents, and employing benzyl bromide as the electrophile; the alkylating reagent was present from the start of the reaction with the expectation that the trialkylated aminosulfonamide would be generated in situ. As can be seen from Table 1, after evaluating a variety of solvent, base and temperature combinations, efficient formation of sulfone 10a could be achieved using either Cs2CO3 or K2CO3 as a base in 1,4-dioxane at 100 °C, in combination with two equivalents of benzyl bromide. The by-product from these reactions was hydrazone 11; supporting the proposed in situ formation of a trialkyl aminosulfonamide intermediate.12 Control experiments established that conversion of dialkyl aminosulfonamide 9a to the corresponding sulfinate in the absence of an alkylating reagent was less efficient. For example, after 1 h reaction at 100 °C with two equivalents of K2CO3, only unreacted aminosulfonamide was returned, and after 8 h a 40% conversion to the sulfinate salt was observed by 1H NMR spectroscopy. Complete conversion was observed after 16 h. In comparison, reaction with two equivalents of benzyl bromide present in the flask allowed full conversion to the sulfone to be achieved after only 30 minutes reaction.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Base equiv. | BnBr equiv. | Temp. (°C) | Yieldb (%) | |
| 10 | 11 | |||||
| a Reaction conditions: (i) N-aminosulfonamide (1 equiv.), BnBr, base, 16 h, solvent [0.3 M]. b Determined by 1H NMR spectroscopy. c Isolated yield. d 30 min reaction time. | ||||||
| 1 | Methanol | Cs2CO3 (2) | 6 | 70 | 55c | 55c |
| 2 | Ethanol | Cs2CO3 (2) | 2 | 70 | 85 | 87 |
| 3 | Ethanol | Cs2CO3 (2) | 2 | 90 | 99 | 98 |
| 4 | Toluene | Cs2CO3 (2) | 2 | 110 | 95 | 96 |
| 5 | Dioxane | Cs2CO3 (2) | 2 | 100 | 98 | 94 |
| 6 | Dioxane | K2CO3 (2) | 2 | 100 | 98d | 95d |
| 7 | Dioxane | K2CO3 (1) | 2 | 100 | 74 | 67 |
| 8 | Dioxane | K2CO3 (2) | 1 | 100 | 42 | 38 |
| 9 | Dioxane | — | 2 | 100 | 0 | 0 |
Using the reaction conditions developed in Table 1, a range of alternative electrophiles were combined with a small selection of N-aminosulfonamides to deliver the corresponding sulfones in good to excellent yields (Method I, Table 2). Despite the sulfinate anion being an ambident nucleophile, sulfinate ester products (10′), resulting from alkylation at an O-atom as opposed to the desired S-atom, were only observed for the alkyl iodide examples (entries 2, 3 and 8) and even in these cases in less than 10% yield.13
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2-X | Product | Yieldb,c | |
| Method I | Method II | ||||
| a Reaction conditions: Method I; N-aminosulfonamide (1 equiv.), R2X (2 equiv.), K2CO3 (2 equiv.), 100 °C, 1,4-dioxane [0.3 M], 1–16 h. Method II; N-aminosulfonamide (1 equiv.), BnBr (0.95 equiv.), K2CO3 (2 equiv.), 50 °C, 1,4-dioxane [0.3 M], 1 h, then R2X (1.5 equiv.), 100 °C, 15 h. b Isolated yields. c Number in parentheses corresponds to the sulfinate ester. d 6 equiv. of MeI. e Conversion determined by 1H NMR spectroscopy. f Et3N (1 equiv.) added after 1 h. | |||||
| 1 | Me | Me-I |
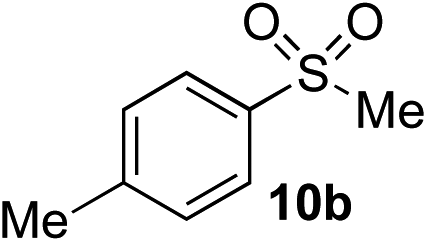
|
77%d | — |
| 2 | Me | Pr-I |
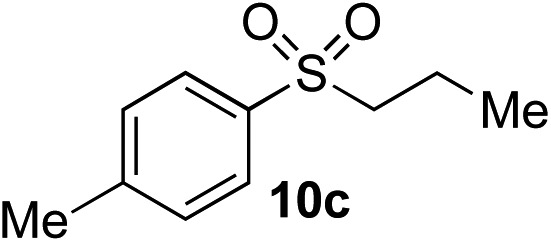
|
61% (5%)e | — |
| 3 | Me | Hex-I |
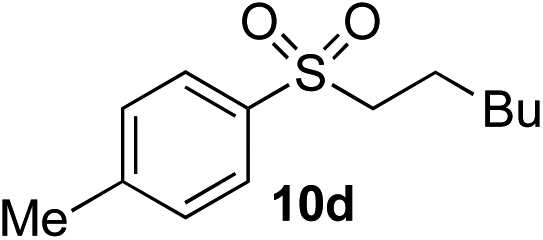
|
67% (8%) | — |
| 4 | Me | Bn-Br |
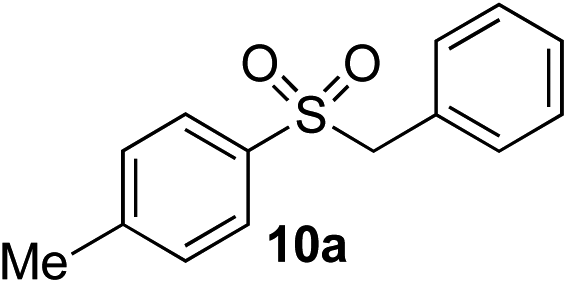
|
92% | — |
| 5 | Me | Bn-Cl |
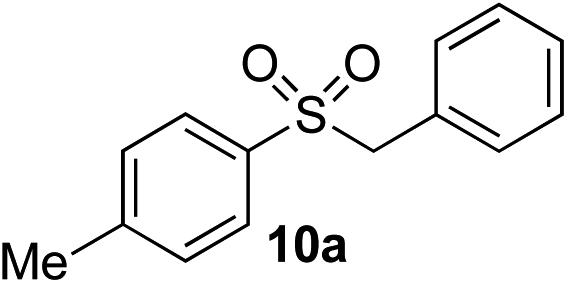
|
69% | — |
| 6 | OEt | Bn-Br |
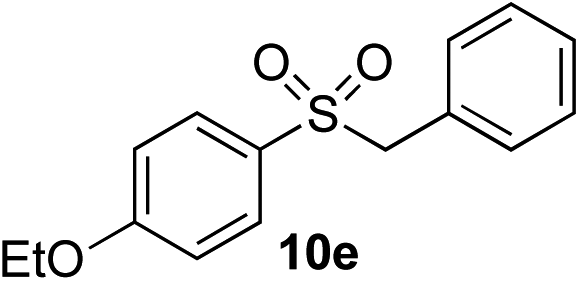
|
94% | — |
| 7 | CF3 | Bn-Br |
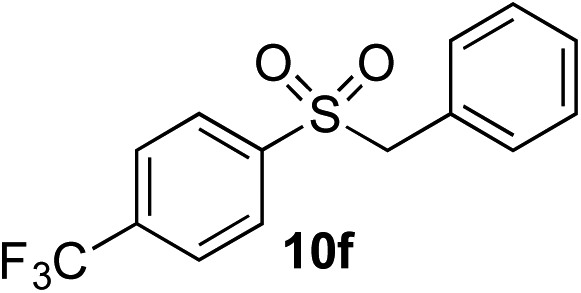
|
70% | — |
| 8 | Me | i Pr-I |
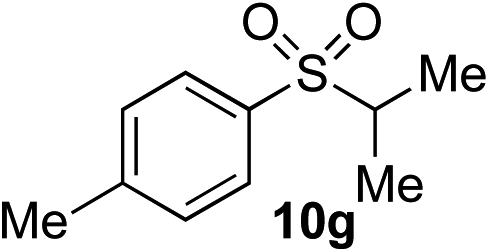
|
47% (7%) | 44% (5%) |
| 9 | Me |
|
|
51% | 58% |
| 10 | Me |
|
|
18%f | 53%f |
| 11 | Me |
|
|
0% | 40% |
| 12 | Me |
|
|
20% | 59% |
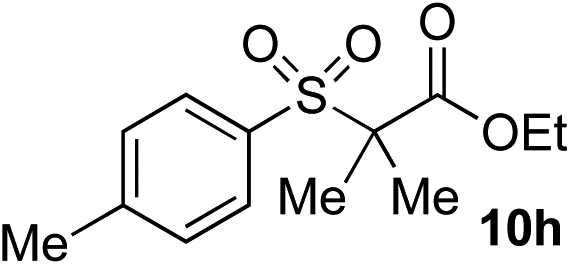
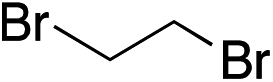
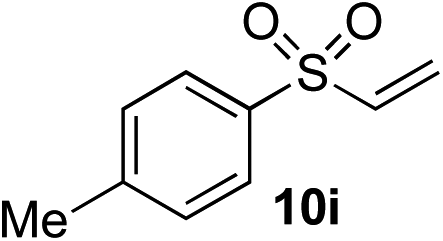
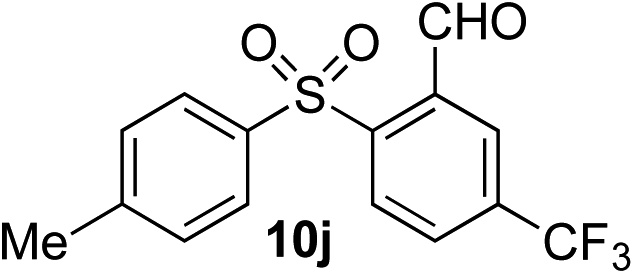
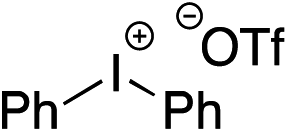
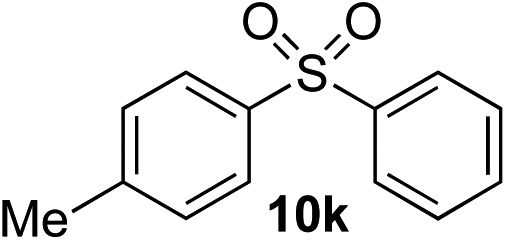
Given the requirement to form a trialkyl aminosulfonamide in situ, the methodology as presented is limited to electrophiles that can undergo the necessary N-alkylation reaction. For example, generation of a diaryl sulfone either by an SNAr reaction with an aryl fluoride, or by reaction with an iodonium salt, was expected to be challenging. As the decomposition of dialkyl aminosulfonamides by K2CO3 alone is slow, a procedure was sought that would provide faster generation of the sulfinate salt that could then react with the desired electrophile. We found that a solution was to employ benzyl bromide as a sacrificial electrophile that could be added to give controlled generation of the sulfinate salt, and then a second, different electrophile could be added to the reaction mixture to be incorporated into the sulfone product. In practice, after 1 h reaction at 50 °C with 0.95 equivalents of benzyl bromide and two equivalents of base, analysis of the crude reaction mixture showed formation of the sulfinate salt and hydrazone by-product. A second electrophile was then added and the temperature increased to 100 °C to give solely the sulfone incorporating the second electrophile. This method (Method II, Table 2) was explored with a small number of electrophiles that had previously given lower yields using Method I. Although little or no improvement was observed for entries 8 or 9, in which secondary and tertiary alkyl iodides were employed, Method II gave a much cleaner reaction and a significant improvement in yield for the synthesis of an α,β-unsaturated sulfone (entry 10) and the aryl sulfones (entry 11 and 12) formed by reaction with an electron-poor aryl fluoride7b and an iodonium salt,7c,d respectively. For entries 8–12, in which only moderate yields of the sulfones were obtained, the remainder of the mass balance was predominantly un-reacted sulfinate anion, presumably due to the lower reactivity of this series of electrophiles.
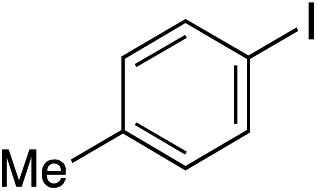
Having established two complementary methods for the degradation and functionalization of dialkyl aminosulfonamides, the next task was the development of one-pot reaction conditions to synthesise the sulfone directly from the aryl-, heteroaryl- or alkenyl iodides. 4-Iodotoluene was selected as the test substrate, and was employed in the palladium-catalysed aminosulfonylation reaction, and after 16 h, K2CO3 and benzyl bromide were added. The highest yield of sulfone achieved using this method was 57% (entry 1, Table 3), despite a large excess (4 equiv.) of benzyl bromide and base being used. Sulfinate salts are known to have a low solubility in organic solvents, and as such, literature precedent suggests that polyethylene glycol, DMSO and DMF are good solvents for sulfone formation from the sulfinate salt.7g,14 Unfortunately, addition of these solvents for the alkylation step resulted in a significant drop in conversion to the desired sulfone (entries 2–6). Pleasingly, the addition of water,15 together with a slight modification to the first, palladium-catalysed step, in which the amount of aminomorpholine was slightly reduced (to 1.2 equiv.), allowed a 91% isolated yield of the sulfone (10a) to be realised (entry 9). These optimised reaction conditions employed 2.5 equivalents of both benzyl bromide and base (entry 9), and the total reaction time for this one-pot process was 21 hours. Reduction of the reaction temperature to 90 °C was necessary to prevent hydrolysis of benzyl bromide which occurred at 100 °C.
|
|
||||
|---|---|---|---|---|
| Entry | K2CO3 equiv. | BnBr equiv. | Co-solventb | Yieldc (%) |
| a Reaction conditions: 4-iodotoluene (1 equiv.), Pd(OAc)2 (10 mol%), PtBu3·HBF4 (20 mol%), 4-aminomorpholine (1.5 equiv.), DABSO (0.6 equiv.), DABCO (0.5 equiv.), 70 °C, 16 h, 1,4-dioxane, [0.3 M]; then BnBr, K2CO3, 20 h, 100 °C. b 0.5 mL of added solvent. c Isolated yield. d Determined by 1H NMR spectroscopy. e Second step; 1.2 equiv. of N-aminomorpholine employed and heated to 90 °C. f 5 h for second step. | ||||
| 1 | 4 | 4 | — | 57 |
| 2 | 4 | 4 | EtOH | 10d |
| 3 | 2.5 | 2.5 | PEG-400 | 0d |
| 4 | 2.5 | 2.5 | Diglyme | 0d |
| 5 | 2.5 | 2.5 | DMSO | 15d |
| 6 | 2.5 | 2.5 | DMF | 15d |
| 7 | 4 | 4 | H2O | 80 |
| 8 | 2 | 2 | H2O | 87e |
| 9 | 2.5 | 2.5 | H2O | 91e,f |
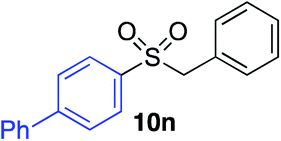
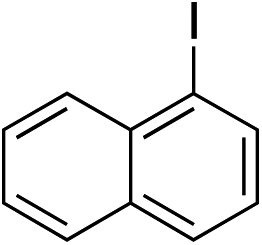
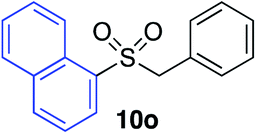
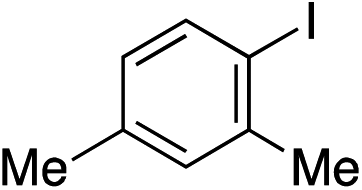
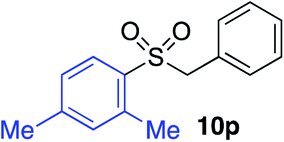
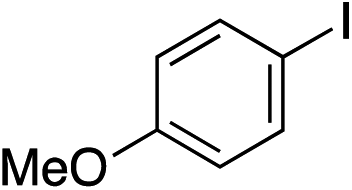
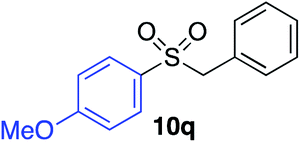
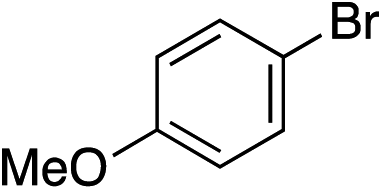
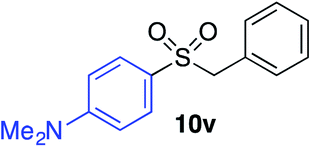
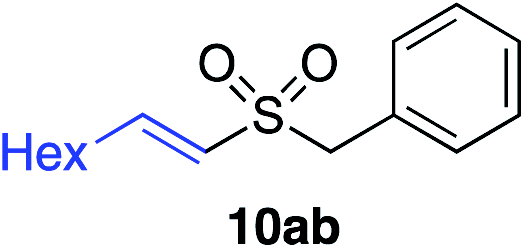
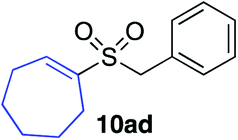
With the optimised conditions in hand, benzyl bromide was reacted with a range of halide coupling partners (Table 4). As described in previous reports from our laboratory, slower reacting substrates, such as those with electron-withdrawing groups, were found to give improved yields when extra DABSO (1.1 equiv. total) was employed.11a,c Aryl iodides with neutral and electron-donating substituents gave excellent yields of the desired sulfones (entries 1–14, Table 4); however, when an aryl bromide was used as the coupling partner, in place of the corresponding iodide, a reduced yield was obtained and reflects the lower reactivity of aryl bromides in the Pd-catalysed aminosulfonylation step (entry 8).11a,c Substrates with ortho-substituents were well tolerated (entries 6 and 11). Aryl iodides with electron-withdrawing groups (entries 15 and 16) gave lower yields; we attributed this to stabilisation of the sulfinate salt consequently reducing their reactivity towards the electrophile. Entry 16 demonstrates that an aryl chloride substituent remains intact during the transformation and so can potentially be used as a handle for subsequent functionalization of the product. Heteroaryl iodides gave moderate yields, which is in agreement with the yields obtained in the parent N-aminosulfonamide forming reactions (entries 17–20).11a,c Pleasingly, alkenyl iodides gave the corresponding sulfones in moderate to good yields (entries 21–23). That sulfide (entry 12), amine (entries 13 and 14) and olefin (entries 21–23) functionalities were employed without issue, highlights the tolerance of the process to oxidation-sensitive functional groups. 4-Aminomorpholine was employed as the standard hydrazine component in all of the examples discussed above; however, alternative hydrazines are also viable, for example, entry 9 was also performed using 1-aminopiperidine with almost identical results.
|
|
|||
|---|---|---|---|
| Entry | Aryl halide | Product | Yieldb |
| a Reaction conditions: 4-iodotoluene (1 equiv.), Pd(OAc)2 (10 mol%), PtBu3·HBF4 (20 mol%), 4-aminomorpholine (1.2 equiv.), DABSO (0.6 equiv.), DABCO (0.5 equiv.), 70 °C, 16 h, 1,4-dioxane, [0.3 M]; then K2CO3 (2.5 equiv.), BnBr (2.5 equiv.), water (0.5 mL), 90 °C, 5–20 h. b Isolated yield. c DABSO (1.1 equiv.) used; no DABCO. d BnCl (2.5 equiv.) used in place of BnBr. e 1-Aminopiperidine (1.2 equiv.) used in place of 4-aminomorpholine. | |||
| 1 |
|
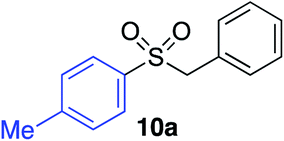
|
91% |
| 2 |
|
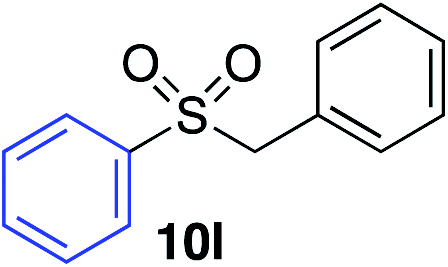
|
65%c |
| 3 |
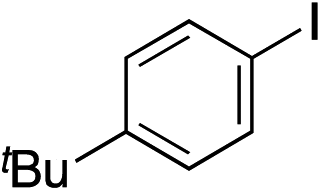
|
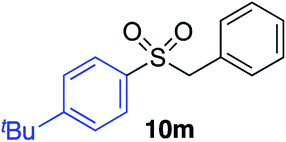
|
87% |
| 4 |
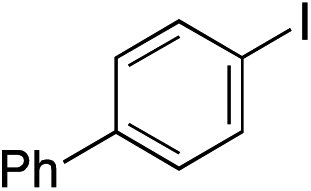
|
|
78%c |
| 5 |
|
|
72%c |
| 6 |
|
|
88% |
| 7 |
|
|
87% |
| 8 |
|
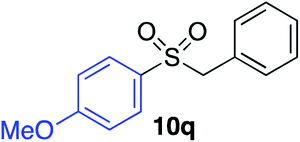
|
46% |
| 9 |
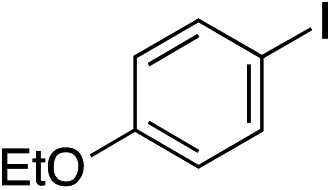
|
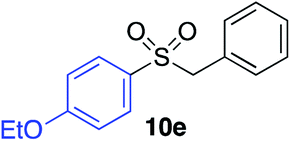
|
90% (75%)d (88%)e |
| 10 |
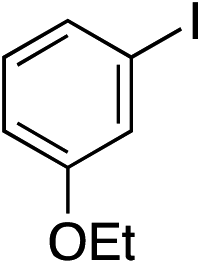
|
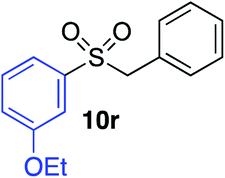
|
87% |
| 11 |
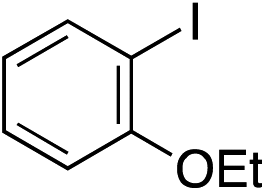
|
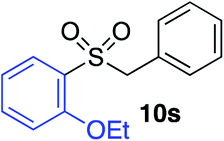
|
75%c |
| 12 |
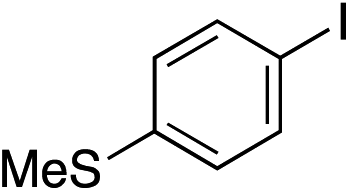
|
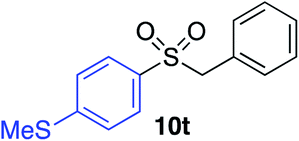
|
89% |
| 13 |
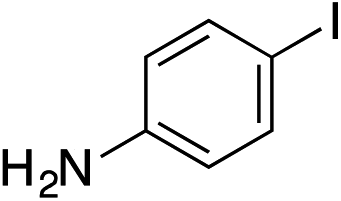
|
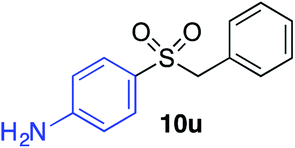
|
57% |
| 14 |
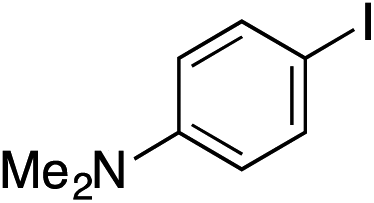
|
|
74% |
| 15 |
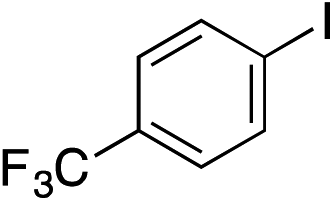
|
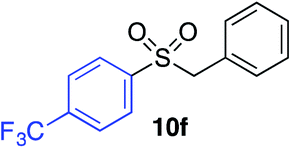
|
42%c |
| 16 |
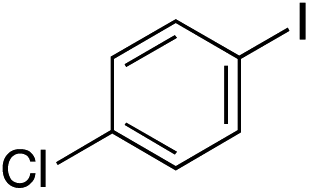
|
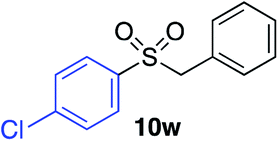
|
67%c |
| 17 |
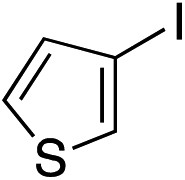
|
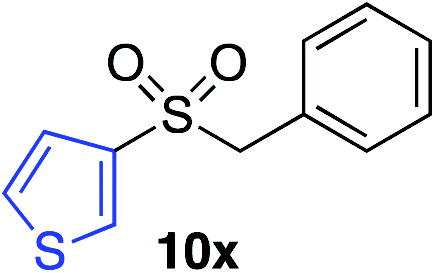
|
62%c |
| 18 |
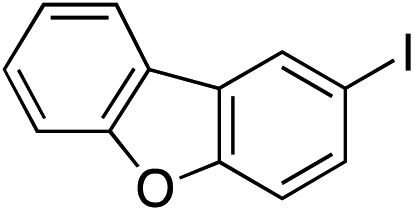
|
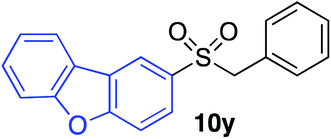
|
64%c |
| 19 |
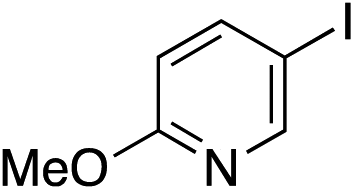
|
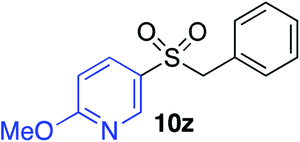
|
70%c |
| 20 |
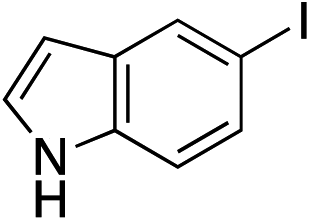
|
|
36%c |
| 21 |
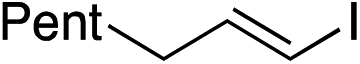
|
|
46%c |
| 22 |
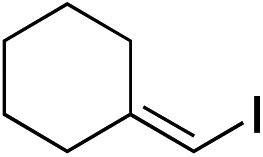
|
|
87%c |
| 23 |
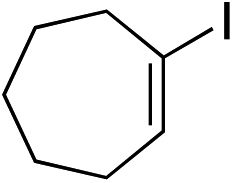
|
|
81%c |
We next investigated the scope of the electrophilic component. 1-Ethoxy-4-iodotoluene was used as the standard aryl halide, and Method I or II (see Table 2) was employed, as appropriate. A range of benzylic bromides could all be incorporated using Method I (entries 1–3). Allylic bromides delivered higher yields of the desired sulfones when Method II was utilized (entries 4 and 5), as did cyclohexene oxide (entry 6).7f,g Alkyl iodides (entries 7 and 8) gave reasonable yields, although interestingly side-products corresponding to the derived sulfinate esters were not observed (cf.Table 2). Under the mild aqueous basic conditions, the ester group incorporated in entry 9 was tolerated well. Implementing Method II allowed an electron-poor aryl fluoride to be employed as the electrophile, delivering a diaryl sulfone product, albeit in low yield (Table 5, entry 10).7b,16
|
|
||||
|---|---|---|---|---|
| Entry | Electrophile | Product | Yieldb | |
| Method I | Method II | |||
| a Reaction conditions: 1-ethoxy-4-iodobenzene (1 equiv.), Pd(OAc)2 (10 mol%), PtBu3·HBF4 (20 mol%), 4-aminomorpholine (1.2 equiv.), DABSO (0.6 equiv.), DABCO (0.5 equiv.), 70 °C, 16 h, 1,4-dioxane, [0.3 M]; then Method I: K2CO3 (2.5 equiv.), RX (2.5 equiv.), water (0.5 mL), 90 °C, 5–20 h; or Method II: K2CO3 (2.5 equiv.), BnBr (0.95 equiv.), 90 °C, 1 h, water (0.5 mL), then RX (2 equiv.), 19 h. b Isolated yield. | ||||
| 1 |
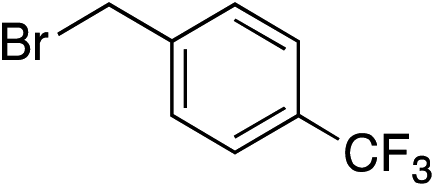
|
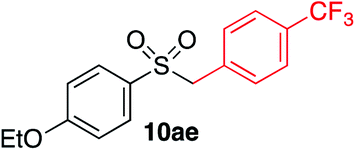
|
91% | — |
| 2 |
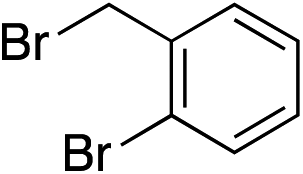
|
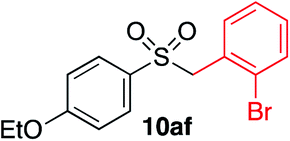
|
90% | — |
| 3 |
|
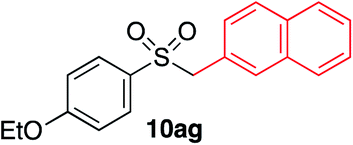
|
93% | — |
| 4 |
|
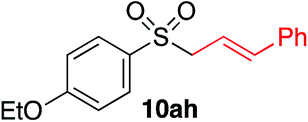
|
51% | 83% |
| 5 |
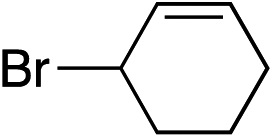
|
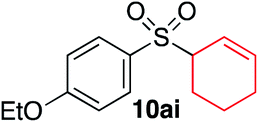
|
— | 55% |
| 6 |
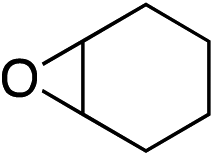
|
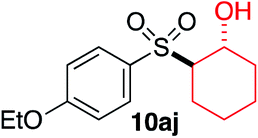
|
35% | 75% |
| 7 |
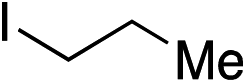
|
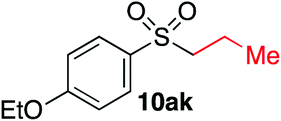
|
55% | — |
| 8 |
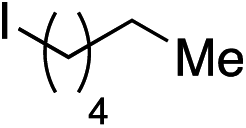
|
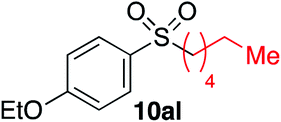
|
63% | — |
| 9 |
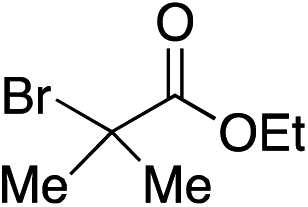
|
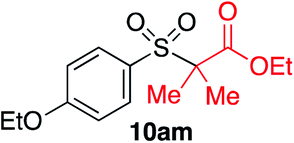
|
57% | — |
| 10 |
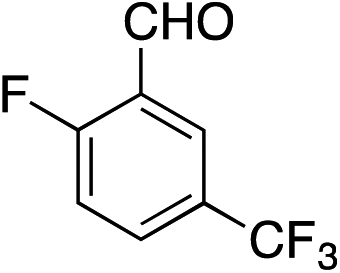
|
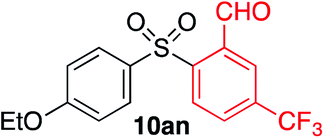
|
— | 35% |
Palladium-catalysed syntheses of N-aminosulfonamides have also been achieved from aryl halides by employing a potassium metabisulfite/tetrabutylammonium bromide combination in place of DABSO,17a and from aryl boronic acids and DABSO under oxidative conditions;17b both of these protocols were also amenable to the developed one-pot sulfone synthesis.18
To demonstrate the utility of our aryl–alkyl sulfone methodology, an intermediate to Eletriptan (trade name Relpax, marketed and manufactured by Pfizer) was synthesised by the coupling of an appropriately 5-substituted indole (12), commercially available iodobenzene and DABSO (Scheme 3). Eletriptan can be prepared from sulfone 13 using two established transformations.19
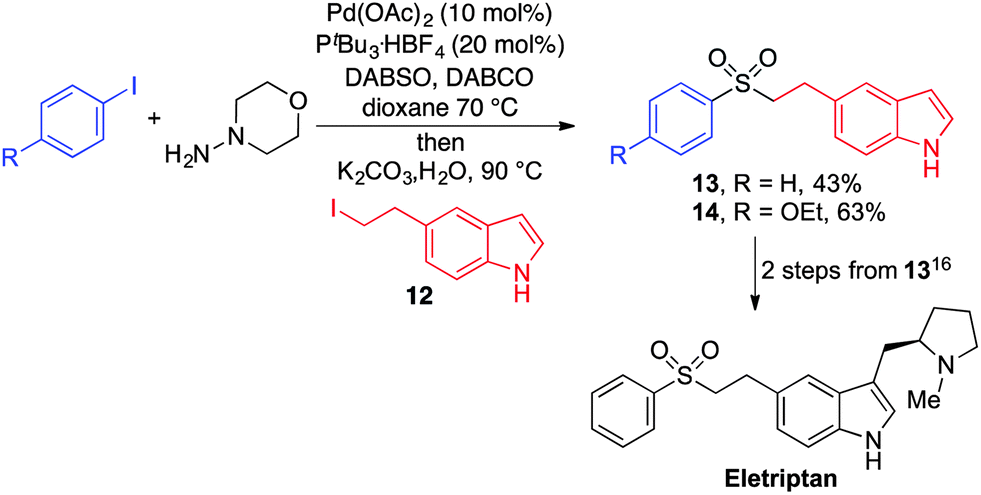 | ||
| Scheme 3 Synthesis of an intermediate towards the preparation of Eletriptan. | ||
Conclusions
We have shown that a wide range of sulfones can be successfully synthesised using a one-pot three-component system that combines aryl-, heteroaryl- or alkenyl iodides with a SO2-surrogate (DABSO) and a range of C-electrophiles. The key sulfonyl unit is introduced using a Pd-catalysed aminosulfonylation process, delivering an N-aminosulfonamide intermediate, which is primed for degradation to the corresponding sulfinate salt; subsequent electrophilic trapping delivers the required sulfones. The methodology tolerates a broad range of functionality on both coupling partners, including oxidation-sensitive functional groups. This new methodology avoids the use of thiol starting materials and oxidative transformations.Acknowledgements
The authors gratefully acknowledge the EPSRC and Pfizer for funding of this work.Notes and references
- (a) P. Tfelt-Hansen, P. De Vries and P. R. Saxena, Drugs, 2000, 60, 1259 CrossRef CAS PubMed; (b) P. F. Schnellhammer, Expert Opin. Pharmacother., 2002, 3, 1313 CrossRef PubMed; (c) G. Mitchell, D. W. Bartlett, T. E. M. Fraser, T. R. Hawkes, D. C. Holt, J. K. Townson and R. A. Wichert, Pest Manage. Sci., 2001, 57, 120 CrossRef CAS; (d) P. Boger, J. Pestic. Sci., 2003, 28, 324 CrossRef CAS.
- N. S. Simpkins, Sulfones in Organic Synthesis, Pergamon Press, Oxford, 1993 Search PubMed.
- (a) B. M. Trost and R. Braslau, J. Org. Chem., 1988, 53, 532 CrossRef CAS; (b) K. Sato, M. Hyodo, M. Aoki, X.-Q. Zheng and R. Noyori, Tetrahedron, 2001, 57, 2469 CrossRef CAS.
- (a) J. K. Crandall and C. Pradat, J. Org. Chem., 1985, 50, 1327 CrossRef CAS; (b) Y. Ju, D. Kumar and R. S. Varma, J. Org. Chem., 2006, 71, 6697 CrossRef CAS PubMed.
- W. E. Truce and A. M. Murphy, Chem. Rev., 1951, 48, 69 CrossRef CAS.
- (a) S. Fujita, Synthesis, 1982, 423 CrossRef CAS; (b) A. Barco, S. Benetti, G. P. Pollini and R. Taddia, Synthesis, 1974, 877 CrossRef CAS.
- (a) B. Das, M. Lingaiah, K. Damodar and N. Bhunia, Synthesis, 2011, 2941 CrossRef CAS PubMed; (b) A. Ulman and E. Urankar, J. Org. Chem., 1989, 54, 4691 CrossRef CAS; (c) N. Umierski and G. Manolikakes, Org. Lett., 2013, 15, 188 CrossRef CAS PubMed; (d) F.-Y. Wang, Z.-C. Chen and Q.-G. Zheng, J. Chem. Res., Synop., 2003, 10, 620 CrossRef; (e) N. Suryakiran, T. Srikanth Reddy, K. Ashalatha, M. Lakshman and Y. Venkateswarlu, Tetrahedron Lett., 2006, 47, 3853 CrossRef CAS PubMed; (f) S. Narayana Murthy, B. Madhav, V. Prakash Reddy, K. Rama Rao and Y. V. D. Nageswar, Tetrahedron Lett., 2009, 50, 5009 CrossRef CAS PubMed; (g) A. K. Maiti and P. Bhattacharyya, Tetrahedron, 1994, 50, 10483 CrossRef CAS.
- A. Dornow and W. Bartsch, Liebigs Ann. Chem., 1957, 602, 23 CrossRef CAS.
- (a) B. F. Powell, C. G. Overberger and J. P. Anselme, J. Heterocycl. Chem., 1983, 20, 121 CrossRef CAS; (b) P. Carter and T. S. Stevens, J. Chem. Soc., 1961, 1743 RSC; (c) D. M. Lemal, T. W. Rave and S. D. McGregor, J. Am. Chem. Soc., 1963, 85, 1944 CrossRef CAS; (d) D. M. Lemal, F. Menger and E. Coats, J. Am. Chem. Soc., 1964, 86, 2395 CrossRef CAS; (e) L. A. Carpino, Chem. Ind., 1957, 172 CAS; (f) L. A. Carpino, J. Am. Chem. Soc., 1957, 79, 4427 CrossRef CAS; (g) W. Baker, J. F. W. McOmie and D. R. Preston, J. Chem. Soc., 1961, 2971 RSC.
- R. Ballini, E. Marcantoni and M. Petrini, Tetrahedron, 1989, 45, 6791 CrossRef CAS.
- (a) B. Nguyen, E. J. Emmett and M. C. Willis, J. Am. Chem. Soc., 2010, 132, 16372 CrossRef CAS PubMed; (b) H. Woolven, C. González-Rodríguez, I. Marco, A. L. Thompson and M. C. Willis, Org. Lett., 2011, 13, 4876 CrossRef CAS PubMed; (c) E. J. Emmett, C. S. Richards-Taylor, B. Nguyen, A. Garcia-Rubia, B. R. Hayter and M. C. Willis, Org. Biomol. Chem., 2012, 10, 4007 RSC.
- Experiments performed at lower temperatures (40–50 °C) allowed the presumed trialkyl aminosulfonamide intermediate to be observed in a crude 1H NMR spectrum (see ESI† for details).
- J. S. Meek and J. S. Fowler, J. Org. Chem., 1968, 33, 3422 CrossRef CAS.
- (a) K. Sukata, Bull. Chem. Soc. Jpn., 1984, 57, 613 CrossRef CAS; (b) Z.-H. Guan, W. Zuo, L.-B. Zhao, Z.-H. Ren and Y.-M. Liang, Synthesis, 2007, 1465 CAS; (c) C. Caupène, C. Martin, M. Lemarié, S. Perrio and P. Metzner, J. Sulfur Chem., 2009, 30, 338 CrossRef.
- N. Chumachenko and P. Sampson, Tetrahedron, 2006, 62, 4540 CrossRef CAS PubMed.
- For recent examples of reactions of sulfinate anions with electron-poor arenes, see ref. 7b and; (a) J. Zou, F. Li and F. G. Tao, Chin. Chem. Lett., 2009, 20, 17 CrossRef CAS PubMed; (b) I. Bruce, M. Akhlaq, G. C. Bloomfield, E. Budd, B. Cox, B. Cuenoud, P. Finan, P. Gedeck, J. Hatto, J. F. Hayler, D. Head, T. Keller, L. Kirman, C. Leblanc, D. Le Grand, C. McCarthy, D. O'Connor, C. Owen, M. S. Oza, G. Pilgrim, N. E. Press, L. Sviridenko and L. Whitehead, Bioorg. Med. Chem. Lett., 2012, 22, 5445 CrossRef CAS PubMed.
- (a) S. Ye and J. Wu, Chem. Commun., 2012, 48, 10037 RSC; (b) S. Ye and J. Wu, Chem. Commun., 2012, 48, 7753 RSC.
- See ESI for details.†.
- M. R. Pullagurla, J. B. Rangisetty, N. Naidu, N. Maddela, R. Nagarapu and P. R. Polagani, P. R. PCT Int. Appl., 2010049952, 2010.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and accompanying spectra. See DOI: 10.1039/c3sc52332b |
| This journal is © The Royal Society of Chemistry 2014 |