
Terahertz gas-phase spectroscopy: chemometrics for security and medical applications
P. F.-X.
Neumaier
*a,
K.
Schmalz
b,
J.
Borngräber
b,
R.
Wylde
cd and
H.-W.
Hübers
ae
aGerman Aerospace Center, Institute of Optical Sensor Systems, Rutherfordstr. 2, 12489 Berlin, Germany. E-mail: Philipp.Neumaier@dlr.de; Heinz-Wilhelm.Huebers@dlr.de
bIHP Microelectronics, Leibniz-Institute on Innovative Microelectronics, Im Technologiepark 25, 15236 Frankfurt (Oder), Germany. E-mail: Schmalz@ihp-microelectronics.com; Borngraeber@ihp-microelectronics.com
cThomas Keating Ltd, Station Mills Billingshurst, West Sussex RH14 9SH, UK. E-mail: R.Wylde@terahertz.co.uk
dSchool of Physics and Astronomy, University of St. Andrews, North Haugh, St Andrews, KY16 9SS, Scotland, UK
eHumboldt-Universität zu Berlin, Institute of Physics, Newtonstraße 15, 12489 Berlin, Germany. E-mail: Heinz-Wilhelm.Huebers@physik.hu-berlin.de
First published on 7th November 2014
Abstract
We describe a spectrometer consisting of a vector network analyzer, a gas absorption cell, and a quasi-optical bench that acquires terahertz spectra of gaseous substances and mixtures. We tested volatile organic compounds that are medical biomarkers or chemicals which can be found on the US Environment Protection Agency list of harmful substances. Absorption spectra at gas pressures between 10 Pa and 5000 Pa were recorded. A subsequent multivariate data analysis demonstrated excellent qualitative and quantitative identification of pure substances and complex mixtures. The applied multivariate algorithms are principal components analysis, partial least square regression and soft independent modelling of class analogy.
I. Introduction
The qualitative and quantitative analysis of molecules in the gas phase is of great importance for many analytical tasks. Since spectroscopy in the terahertz (THz) region is a powerful tool for chemical sciences,1 there are increasing research efforts in the development of spectroscopic THz gas sensors2 for applications in medicine and health care,3 chemistry/pharmacy4–7 and security and defense.8,9The analysis of biomarkers in human breath, which is among the least invasive diagnostic methods, is gaining increasing importance. The human breath contains more than 1000 volatile organic compounds (VOCs) associated with core metabolic processes, diseases, drug consume or exposure to environmental pollutants.10,11 Lung cancer, for instance, can be diagnosed by monitoring certain respiratory gases such as acetaldehyde, acetone, isopropyl alcohol and ethanol.12–16 Since sample collection is easy17 breath analysis has distinct advantages for patients and medical doctors, in terms of a pain-free, non-invasive, and safe method. This method is inexpensive compared to common imaging devices such as computer tomography (CT), which in addition have the risk of radiation exposure.18
There is also a need for environmental monitoring of gaseous toxic industrial chemicals (TICs) which are often used in industry. Minute amounts of TICs as well as automotive exhausts affect the urban air quality. Many TICs as well as major air pollutants19 such as NO2, SO2, and CO have absorption lines in the THz region.20 Since TICs might be used in terrorist attacks or may be freed by terrorist attacks on chemical production sites, their sensitive and specific detection is of prime importance.
Since many molecules – in particular small ones – and larger hydrocarbons have maximum absorption at THz frequencies21,22 spectroscopy in this region is well suited to detect even small amounts of VOCs, TICs, biomarkers, air pollutants, process gases or drugs.23 Despite the dense spectra there is virtually no overlap of lines in the Doppler limit and at moderately low pressure. This makes the spectra redundant and suitable for substance identification even if only a small spectral band width is available, provided the spectrometer has sufficient spectral resolution. In addition the line widths contain information about the substances' concentration, since the broadening mechanisms are pressure dependent.
Unfortunately at increasing pressure the high spectral density of lines becomes an issue, because spectral information is lost due to the line broadening and spectral overlap. With gas mixtures this is a particular problem as the line density is even higher and the spectral fingerprint gets blurred at even lower pressure. Along with the pressure, further disturbances – for example standing waves and receiver noise – can complicate or prevent gas detection and identification.
Multivariate data analysis (MVA) is applied to many measurement techniques.24 The objective of this study is to assess for the first time the potential of absorption spectroscopy around 245 GHz in combination with MVA for qualitative and quantitative substance and mixture identification. The measurements are performed at pressures near and above the Doppler limit. The strengths of MVA lie in pattern recognition25 which reveals hidden structures allowing reduction of data volumes to their minimum, and making the system less prone to error.26 Additionally a future integrated system would be easy to handle and can be used by laymen through automation. The MVA methods applied in this study are principal component analysis (PCA), partial least square regression (PLS), principal components regression (PCR) and soft independent modeling of class analogy (SIMCA).
With future security and medical applications in mind, we selected chemicals and VOCs, which are on the list of the US Environmental Protection Agency (EPA)27 and/or are known as biomarker in human breath.12–16 Our substance selection purposely covers a wide range in terms of absorption line strengths and line densities. In particular the qualitative and quantitative determination of a mixtures composition regardless their exact chemical behaviour or reaction is of central importance.
We have chosen a group of seven VOCs based upon their relevance for security and medical diagnostics. All of the substances are used in industrial, chemical or pharmacological synthesis processes, while four chemicals are on the EPA list of harmful substances and five are well known as biomarkers for certain diseases. The medical biomarkers acetaldehyde,16 acetone,14–16 and isopropyl alcohol14,15 are tracers for lung cancer, while acetaldehyde additionally is found to be a tracer for alcoholism and liver related diseases.12 Acetone indicates furthermore dietary fat losses, congestive heart failures, and diabetes.12,14 Methanol is a biomarker for a nervous system disorder, and ethanol is a tracer for the production of gut bacteria.12
The detection of acetaldehyde and methanol, with annual production quantities of approximately 1.3 million28 respectively 40 million29 metric tons, is important for security applications. While acetaldehyde and methanol are used for the chemical synthesis of acetic acid, methanol is used additionally for formaldehyde and MTBE (methyl tert-butyl ether; gasoline additive) synthesis that are important source materials for many products. In organisms they cause the degeneration of olfactory epithelium or the growth of extra cervical ribs. They can be incorporated by skin contact and via breathing. In addition acetone is a widely used substance for example as diluter or in enamel remover and it is, in common with acetonitrile, on the EPA list of harmful substances.27
Ideally any spectrometer for such applications has to be capable of detecting several substances simultaneously, since there is a mixture of VOCs in a patient's breath,13 or when monitoring a chemical accident one has to deal with many escaping gases and possibly their reaction products. In this paper we investigate the potential of THz gas-phase spectroscopy in combination with chemometric methods for these applications.
II. Experimental
A. Samples
For our investigations we have chosen a set of seven pure substances and nine mixtures of these (see Table 1). Acetaldehyde, methanol, isopropyl alcohol and ethanol were purchased from Merck® with a purity ≥ 99.5%. Deuterated methanol, has a purity of 99.0% and was obtained from Cambridge Isotope Laboratories, Inc. Acetone and acetonitrile were bought from VWR International® with a purity ≥ 99.0%. Table 1 lists the measured substances and mixtures, along with some physical, chemical and toxicological key properties. For preparation of the mixtures the liquid chemicals were mixed first and the degassing vapour was spectroscopically analysed.| Chemical substance CAS# | Mol. mass (g mol−1) | Chemical formula | p s (20 °C) | EPA | Biomarker | LD50 oral | LC50 inhal. | |
|---|---|---|---|---|---|---|---|---|
| a List of measured pure substances and mixtures. Top: chemical and toxicological properties together with a classification in terms of harmful molecules according the EPA list27 and medical breath biomarkers.12–16ps (20 °C) is the vapor pressure at 20 °C. The lethal doses (oral intake and inhalation) and concentrations concern a rat metabolism. Bottom: mixtures made from the above mentioned pure chemicals (no. 1–7) stated in mass fraction. | ||||||||
| Pure substance | ||||||||
| 1 | Acetaldehyde 75-07-0 | 44.05 | (CH3)CHO | 1006 | √ | √ | 664 mg kg−1 | 24 mg l−1 kg−1 |
| 2 | Methanol 67-56-1 | 32.04 | (CH3)OH | 129 | √ | √ | 5630 mg kg−1 | 83.9 mg l−1 4 h−1 |
| 3 | Deut. methanol 1455-13-6 | 33.05 | (CH3)OD | 129 | √ | √ | 5630 mg kg−1 | 83.9 mg l−1 4 h−1 |
| 4 | Acetone 67-64-1 | 58.08 | (CH3)CO(CH3) | 246 | √ | √ | 5800 mg kg−1 | |
| 5 | Isopropyl alcohol 67-63-0 | 60.10 | (CH3)(CH)(CH3)OH | 43 | √ | 5050 mg kg−1 | ||
| 6 | Acetonitrile 75-05-8 | 41.05 | (CH3)CN | 94 | √ | 2460 mg kg−1 | ||
| 7 | Ethanol 64-17-5 | 46.07 | (CH3)(CH2)OH | 58 | √ | 7060 mg kg−1 | ||
| Acetaldehyde | Methanol | Deuterated methanol | Acetone | Isopropyl alcohol | Acetonitrile | Ethanol | |
|---|---|---|---|---|---|---|---|
| Mixtures | |||||||
| 8 | 0.654 | 0.346 | |||||
| 9 | 0.309 | 0.691 | |||||
| 10 | 0.088 | 0.912 | |||||
| 11 | 0.5 | 0.5 | |||||
| 12 | 0.052 | 0.405 | 0.543 | ||||
| 13 | 0.191 | 0.809 | |||||
| 14 | 0.038 | 0.297 | 0.665 | ||||
| 15 | 0.091 | 0.522 | 0.387 | ||||
| 16 | 0.218 | 0.293 | 0.489 | ||||
B. Experimental setup
For this study a Rohde & Schwarz® vector network analyzer (VNA) type ZVA 24 was used to generate and receive the THz radiation. The internal local oscillator provides 10 MHz to 24 GHz and two subsequent extender modules (ZVA-Z325 Converter WR03) are multiplying the signal to a frequency between 220 and 325 GHz. The band width in the experiment was set to 238–252 GHz with a frequency resolution of 500 kHz and an internal integration band width of 1 kHz that results in a sweeping time of approximately 60 s per spectrum.Electroformed corrugated feedhorns (Thomas Keating Ltd) are attached to the extender modules. The horns generate and receive axially symmetric low sidelobe Gaussian beams with a waist radius of 7.5 mm. The transmitted beam is propagated through a gas absorption cell using a Quasi-Optical (QO) circuit, built using four ellipsoidal focusing mirrors. In addition, two free standing wire grids for a 45° beam polarization with respect to the optical table (Fig. 1). The overall power loss is 6 dB, coming from the orientation of the polarizing grids, and used to dampen down standing waves.
 | ||
| Fig. 1 Left: photograph of the setup. The gas cell is in the foreground, with the gas injection pipework. The cell is supported on the QO bench, with the S1 and S2 feedhorns attached to the VNA extender modules and the VNA hardware in the rear. Right: schematic picture of the setup. | ||
The circuit is designed to be frequency independent and can be used at other frequencies. The cell is 12 cm in diameter with a length of 56 cm and HDPE windows are mounted in Brewster angle, to minimize standing waves, a common problem in low loss QO circuits. The gas pressure in the cell is controlled by two gas-independent pressure gauges (1 × 10−4 to 1 × 102 hPa measurement range), a dosing valve and a turbo molecular pump.
The VNA is used to measure the S-parameters that are the bi-directional signal transmission (S12, S21) and signal reflection-characteristics (S11, S22) of the THz beam. For spectroscopy the S21 and S12 signals are measured since these contain the absorption spectra.
First a series of measurements at different pressures from 10 to 5000 Pa was performed. One spectrum per pressure was measured. The second measurement series was done with 16 substances (Table 1) at 30, 100 and 300 Pa, obtaining 10 spectra at each pressure and S-parameter, thus recording 960 spectra. Prior to each measurement the absorption cell was evacuated to a pressure below 1 × 10−3 hPa and the VNA was recalibrated.
C. Data processing
The spectra obtained with the VNA have a resolution of 0.5 MHz which gave 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 data points in the measured spectral range between 238 and 252 GHz. Each spectrum was stored in one ASCII file that was subsequently merged into a single data matrix (28000 × 960) for further processing with the commercial software The Unscrambler X (CAMO Software AS). This matrix contains 960 rows, which are the S-parameters S21 and S12 of 16 substances measured ten times at three different pressures, and 28000 columns containing the measured frequencies.
000 data points in the measured spectral range between 238 and 252 GHz. Each spectrum was stored in one ASCII file that was subsequently merged into a single data matrix (28000 × 960) for further processing with the commercial software The Unscrambler X (CAMO Software AS). This matrix contains 960 rows, which are the S-parameters S21 and S12 of 16 substances measured ten times at three different pressures, and 28000 columns containing the measured frequencies.
MVA was used to analyse the spectra with a set of methods: PCA, PCR, PLS and SIMCA.
PCA is an unsupervised method whose algorithm is using the raw spectral information without any additional information like class affiliation or substance composition. Mathematically the PCA is transferring measured values in an eigenvalue-problem and projecting them into an appropriate low-dimensional subspace by calculating new eigenvectors (so called Loadings).
The Non-Linear Iterative Partial Least Squares (NIPALS) method is used to find the maximum variance in a hub centred data matrix that is constructed from the initial variables (in this case, the spectra). Considering this maximum variance the NIPALS algorithm iteratively and successively calculates the loadings that constitute the new orthogonal bases respectively the new axes (Principal Components: PCs) into which the eigenvalues (scores) are transformed. Because of the successive calculation in terms of descending variance, the first Principal Component (PC-1) determine most of the information in the measured data, while the following PCs explain progressively less and less information. Thus, a large and complex data set is reduced by taking only the first few PCs that in an ideal case explain the whole information in the data/spectrum.
PCR is a multiple linear regression (MLR) technique that uses the scores from the PCA to find the functional correlation between many independent measured original values and one dependent target value. While the ordinary MLR needs more measured data sets (spectra) than variables (spectral points), the PCR uses the PCA scores of the first PCs instead of the original values that contain almost the whole information. This reduction makes this technique applicable to high resolution spectra, while an analysis of the regression coefficients points out important spectral regions concerning a target value, such as class affiliation like the qualitative and quantitative identification of a chemical.
The PLS is a regression technique that finds the functional correlation between a measured spectrum and a variety of dependent values (target matrix). This is similar to the PCR but with the advantage of a correlation between the scores of the data matrix and the target matrix. Both PCAs are affecting each other such that less PCs (factors) are necessary for a prediction.
The SIMCA method is another supervised technique that combines several independent PCA models, each created from a data set with known class affiliation. For an assignment, SIMCA projects the spectra with unknown affiliation to each class-subspace individually, while a short distance to a certain subspace means a high probability of class membership. Further information on all of these methods can be found in ref. 22 and 23. For the model generation, the full cross validation was always used, which uses every spectral data twice, for calibration and validation.
Based on the abstract mathematical algorithm the PCA is well suited for pattern and structure recognition, while data pre-processing sometimes can have a positive effect on information separation, data modeling and model predictions. It was found that FT filtering, absorption line pre-selection and tuning the frequency resolution by averaging had no influence in the PCA model quality in the selected rage of variation. This fact and the strong relationship between a substance, its quantity and its spectrum, makes over-fitting unlikely.
Thus the raw data was used for all calculations and for the reduction of explanatory values and computation time the spectral resolution was lowered by averaging five adjacent spectral points thus reducing the data matrix to the dimension of 960 × 5600. With the spectral band width of 14 GHz this means a spectral resolution of 2.5 MHz, which is in the order or less than the pressure broadening of the investigated molecules.
III. Results
A. Absorption spectra
Absorption spectra of the sixteen substances were measured at 30, 100 and 300 Pa between 238 and 252 GHz. In that frequency range the Doppler broadening, full width at half maximum (FWHM), of the substances is between 380 and 530 kHz. In all measurements the pressure broadening is dominant with line widths between 5 and 50 MHz. Fig. 2 shows absorption spectra of acetaldehyde and acetronitrile with absorptions up to 60% while other substances such as methanol, acetone and isopropyl alcohol have weak absorptions around 1%. Fig. 3 shows the spectrum of a mixture of methanol and deuterated methanol.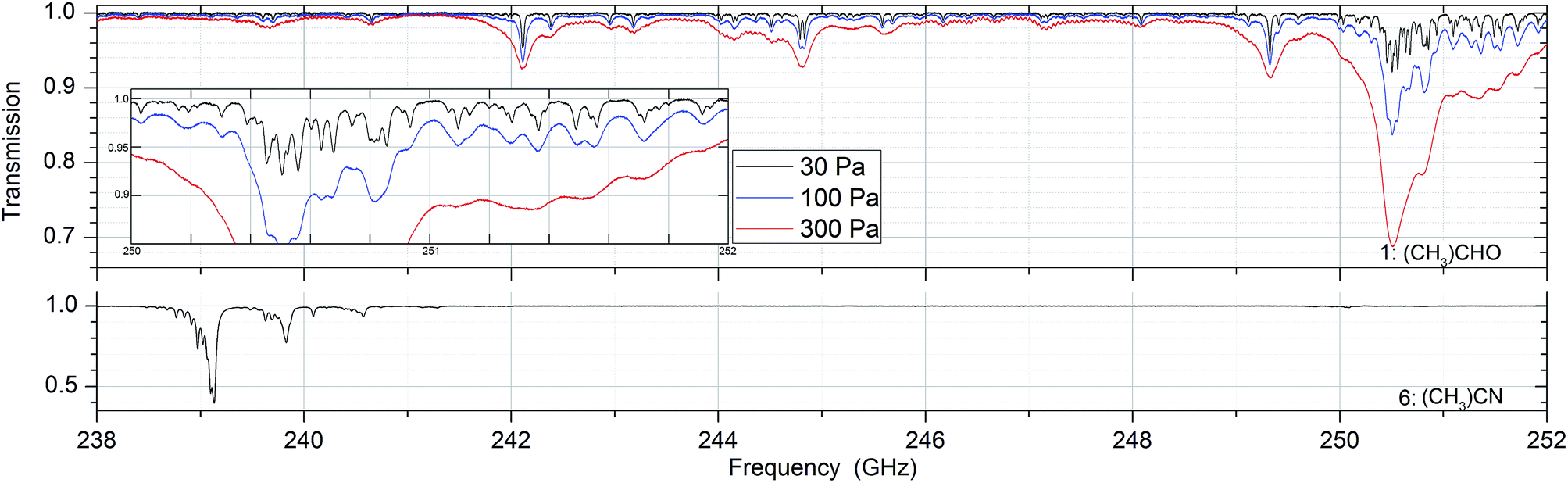 | ||
| Fig. 2 Selected absorption spectra. Acetaldehyde ((CH3)CHO, substance no. 1) and acetonitrile ((CH3)CN, no. 6) at pressures of 30, 100 and 300 Pa. It underlines the difference of the samples in line strength, number and distribution of lines and shows the pressure broadening. | ||
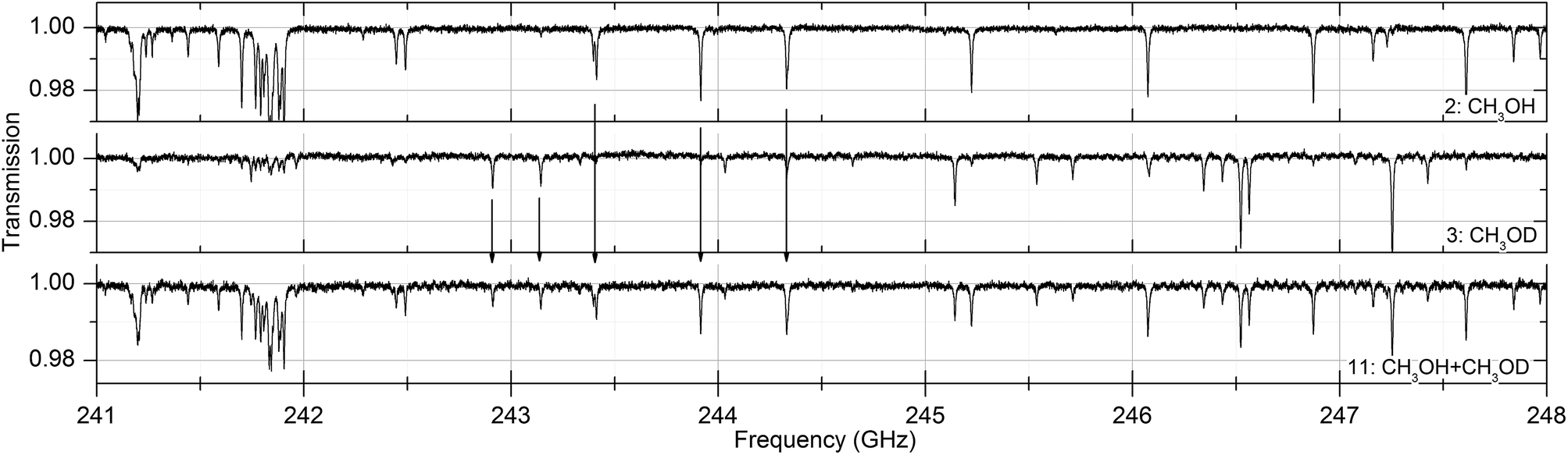 | ||
| Fig. 3 Absorption spectra of pure methanol ((CH3)OH, substance no. 2), deuterated methanol ((CH3)OD, no. 3) and a mixture of both (no. 11) as described in Table 1 at a pressure of 30 Pa. As can be seen, the spectrum of the mixture contains the spectral information of both two pure spectra. | ||
Since there is no chemical reaction between both substances and no suppression of evaporation due to intermolecular interaction between the different molecules, the spectrum of the mixture contains the spectral features of both pure substances.
With a spectral resolution of 500 kHz, all measured absorption lines are resolved with at least 10 pts per line. Since the integration band width was set to 1 kHz, the overall sweeping time was approximately 60 s and the standard deviation of the baseline is σnoise = 0.0008.
For the calculation of the PCA, PCR, PLS and SIMCA, all spectra were smoothed by adjacent averaging of five data points. This reduced the resolution to 2.5 MHz, which is equivalent to a sweeping time of 12 s per spectrum. A benefit of the smoothing is that the dimension of the data matrix is lowered, thus reducing the computation time and the probability of over-fitting.
B. Principal components analysis
The PCAs were performed separately for each gas pressure. Fig. 4 and 5 are showing two-dimensional score-plots for measured absorption spectra based on S21. The data set consists of 10 spectra for each of the 16 substances at a pressure of 300 Pa. Each point in the scores plot represents a spectrum. Evidently the PCA delivers excellent results with respect to substance identification, since all of the substance classes are forming clusters. Most of them are emerging already in the first two PCs.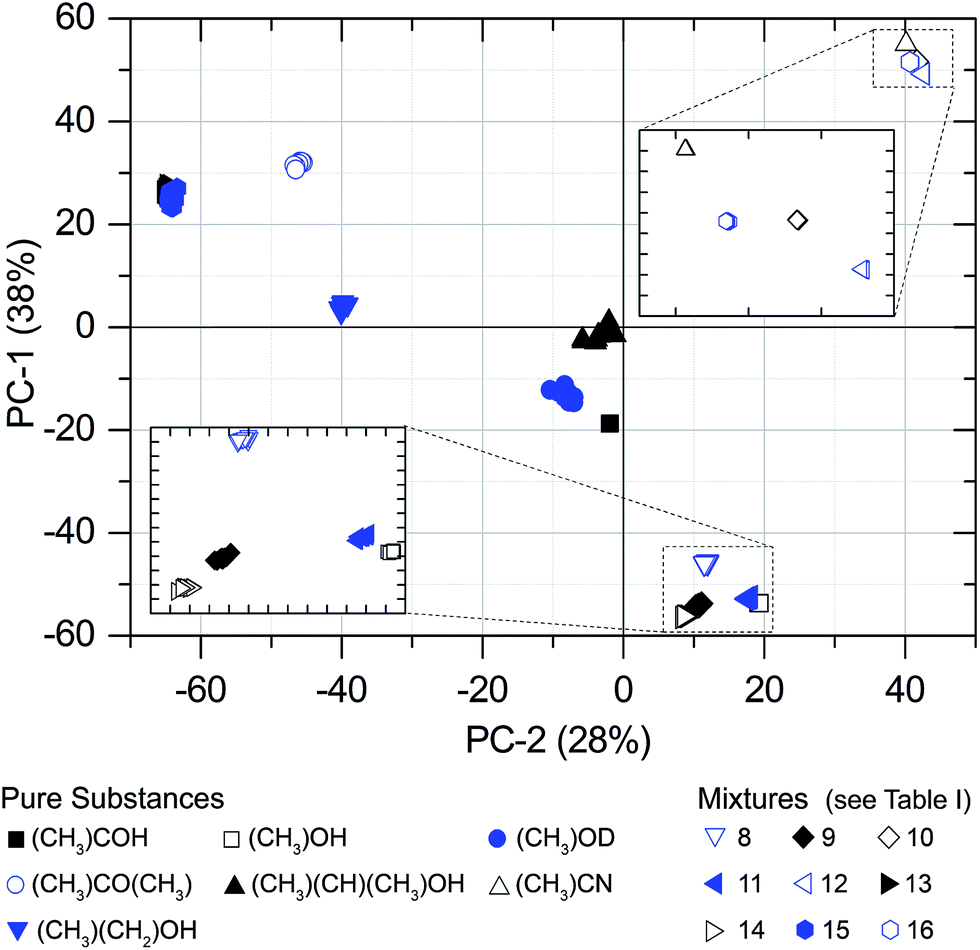 | ||
| Fig. 4 PCA scores. 16 substances at a pressure of 300 Pa (S21 spectra). 14 substances are clustering without overlap. Mixtures no. 13 and 15 are overlapping, but are separated in PC-3 (Fig. 5). | ||
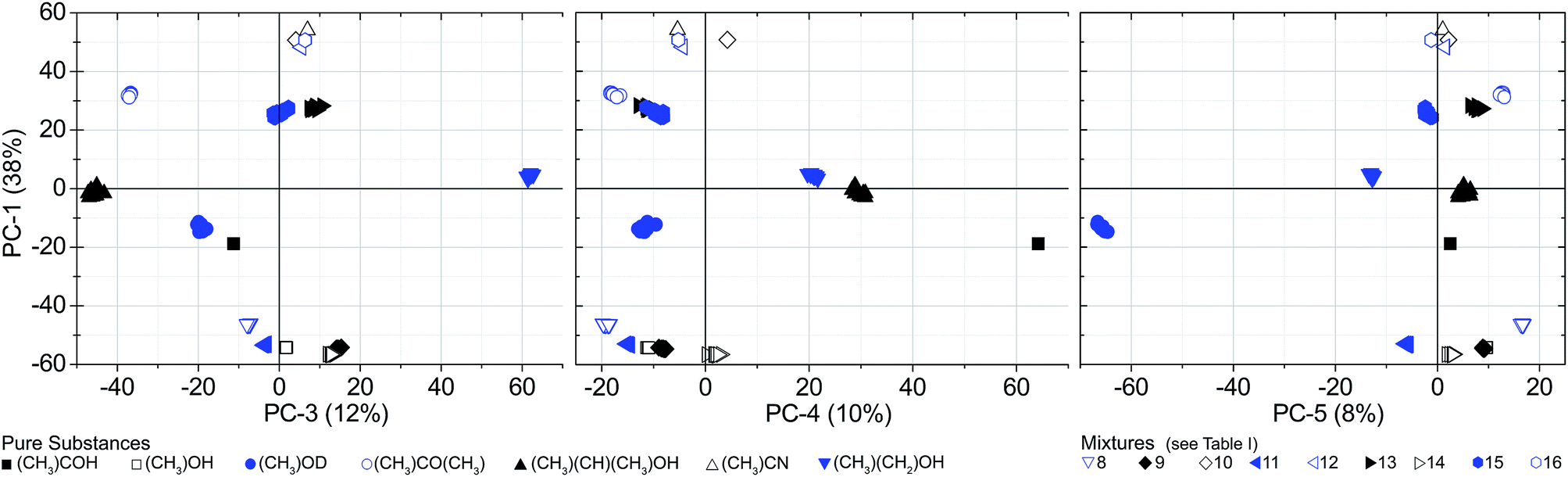 | ||
| Fig. 5 Scores of the PCA. 16 substances/mixtures at a pressure of 300 Pa (S21 spectra). Mixtures no. 13 and 15 are clustering without overlap in PC-3. | ||
As mentioned in section II, each PC contributes a certain amount to the explanation of the data and enables to distinguish between spectra by cluster formation. With N = 6 PCs the model describes the original data to 99.9% meaning that the remaining difference between the original spectrum and the PCA is only 0.1%.
To quantify and compare the model qualities, we determine the barycentre of each cluster and apply a Euclidean metric to quantify the samples' spreading S and the clusters' mutual distances D. To obtain a scalar measure we averaged the Euclidean distances over N PCs and the number of recorded substance spectra n. Eqn (1) gives the spreading S of a clusters' spectra around its respective barycentre ỹj, while j is the number of the PCs' dimension and yj,i is the score of a certain measured spectrum i. The lower S the more compact the cluster and the better the substance differentiation. S represents the similarity between the spectra of one certain substance, hence it is a figure of merit for the reproducibility and the signal-to-noise ratio of the measurement.
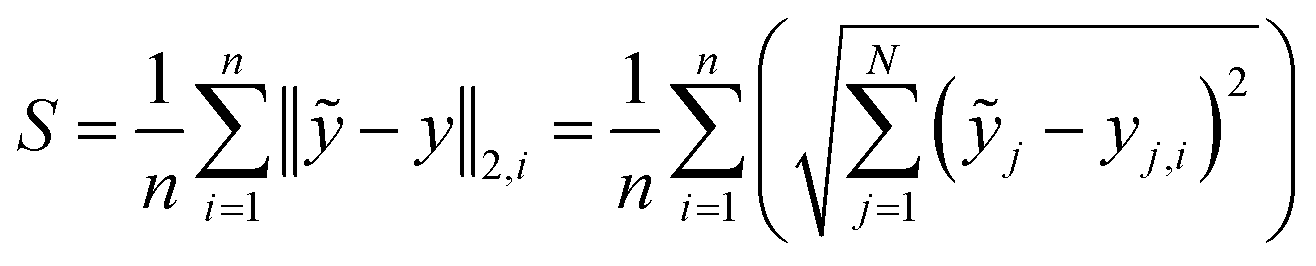 | (1) |
Along with the spreading S, the Euclidean distance D between barycentre of a substance ỹ and its nearest neighbour ỹ′ is another figure of merit (eqn (2)). The larger D, the better the substance differentiation and the better the PCA model. D represents the difference between the spectra of different substances. Hence it is an indication whether substances can be separated and whether they can be described by the same model.
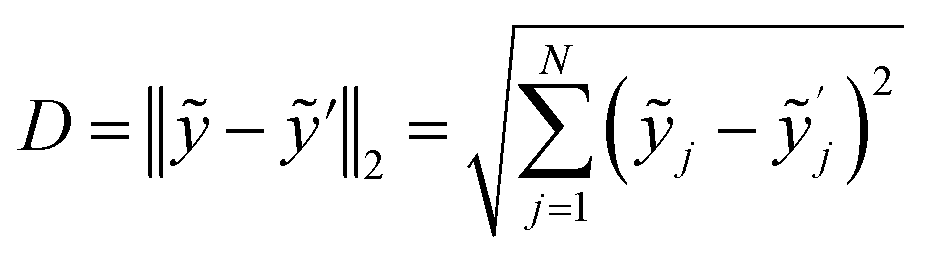 | (2) |
We purposely define the clusters quality q as the fraction q = D/S. q > 1 means a successful delimitation and cluster formation. Table 2 lists the barycentre of all samples in the respective PC.
| PC-1 (38%) | PC-2 (28%) | PC-3 (12%) | PC-4 (10%) | PC-5 (8%) | PC-6 (4%) | Spreading S | Distance D | Quality q | |
|---|---|---|---|---|---|---|---|---|---|
| a Results for 300 Pa (S21). | |||||||||
| Pure substance | |||||||||
| 1 | −18.74 | −1.90 | −11.48 | 64.13 | 2.29 | −29.60 | 0.08 | 38.67 | 483.38 |
| 2 | −53.69 | 19.29 | 1.94 | −11.31 | 9.73 | 0.53 | 0.23 | 2.10 | 9.13 |
| 3 | −13.04 | −8.28 | −18.21 | −12.70 | −65.91 | −5.33 | 1.76 | 54.39 | 30.90 |
| 4 | 31.71 | −46.10 | −36.54 | −18.02 | 13.03 | −15.12 | 0.85 | 16.22 | 19.08 |
| 5 | −1.61 | −3.44 | −46.10 | 29.00 | 3.64 | 41.89 | 2.97 | 30.05 | 10.12 |
| 6 | 55.06 | 40.14 | 7.13 | −5.34 | 1.14 | −0.05 | 0.03 | 3.55 | 118.28 |
| 7 | 4.01 | −39.84 | 62.09 | 21.00 | −12.48 | 15.20 | 0.95 | 50.82 | 53.50 |
|
|||||||||
| Mixtures | |||||||||
| 8 | −45.80 | 11.75 | −7.38 | −19.27 | 16.86 | −2.90 | 0.61 | 9.02 | 14.78 |
| 9 | −54.13 | 10.65 | 14.54 | −8.27 | 9.41 | 3.31 | 0.52 | 2.38 | 4.58 |
| 10 | 51.65 | 41.67 | 4.06 | 4.29 | 2.26 | −4.70 | 0.04 | 3.02 | 75.40 |
| 11 | −52.64 | 17.87 | −2.96 | −15.25 | −5.72 | −0.72 | 0.49 | 7.79 | 15.89 |
| 12 | 49.25 | 42.57 | 5.49 | −4.40 | 1.63 | 0.11 | 0.04 | 2.88 | 72.10 |
| 13 | 26.77 | −64.73 | 9.15 | −11.17 | 8.27 | −4.41 | 1.33 | 2.74 | 2.06 |
| 14 | −56.24 | 8.78 | 13.05 | 1.81 | 7.62 | −2.42 | 0.87 | 4.12 | 4.73 |
| 15 | 25.15 | −64.02 | 0.56 | −9.51 | 8.69 | 2.68 | 2.15 | 2.68 | 1.24 |
| 16 | 51.59 | 40.71 | 6.50 | −5.25 | 0.46 | 1.30 | 0.06 | 1.34 | 22.32 |
The value Q, which is the arithmetic mean of all q-values, indicates the overall model quality. In this case it is Q = 59.
Since the PCA method is quite robust, the cluster formation and the resulting substance identification are possible even for challenging candidates like isopropyl alcohol (sample no. 5). It has a comparatively high S-value that is caused by very weak absorption lines in the observed spectral range. Despite that, the large distance to adjacent substances in two dimensions (PC-1 and PC-2) yields a good differentiation. As well for sample no. 13 and 15 the spreading S is comparatively high and the barycenter distance to each other in PC-1 and PC-2 is small. But in PC-3 the space between the barycenter is sufficiently large to distinguish between both mixtures. Fig. 6 shows the associated loadings with some assigned spectral features, marked with the number of the substance.
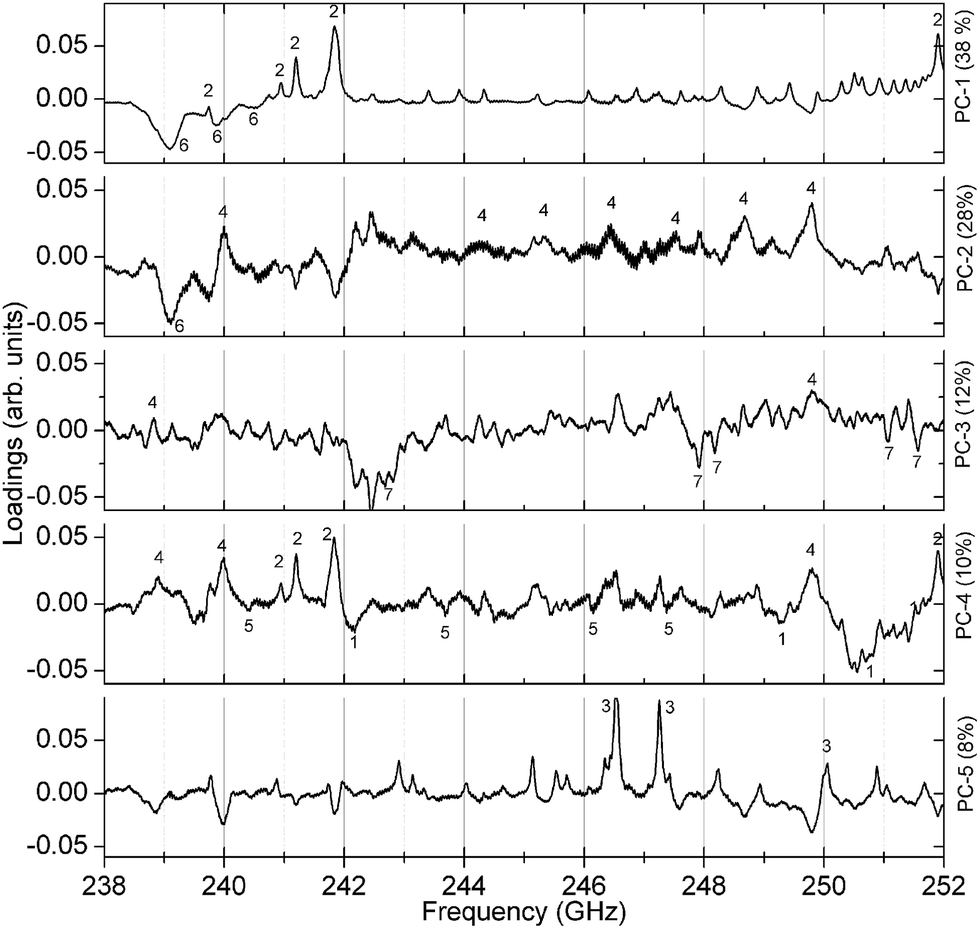 | ||
| Fig. 6 The first five loadings calculated with the PCA. Features of all seven substances can be found as peaks and dips. The numbers are referring to a particular substance number (compare Table 1 and spectra in Fig. 2 and 3). | ||
Furthermore, PCA models with absorption spectra measured at 30 Pa and 100 Pa with 16 substances were calculated. All have well separated clusters with a comparatively high model quality. For N = 6, the PCA model for 30 Pa shows Q = 16 and the model for 100 Pa Q = 24. This indicates an increasing model quality between 30 Pa and 300 Pa. Thus the MVA enables substance identification even at high pressures where pressure broadening dominates.
Another measurement with a series of gas pressures from 10 to 5000 Pa shows the suitability of a qualitative substance-class-differentiation and a quantitative amount-of-substance-analysis. The scores of spectra of a substance at different pressures are arranged in increasing order along a spline in the PC-3 direction (Fig. 7). This shows the feasibility of identifying gases and gas mixtures even at pressures where the absorption lines are strongly pressure broadened.
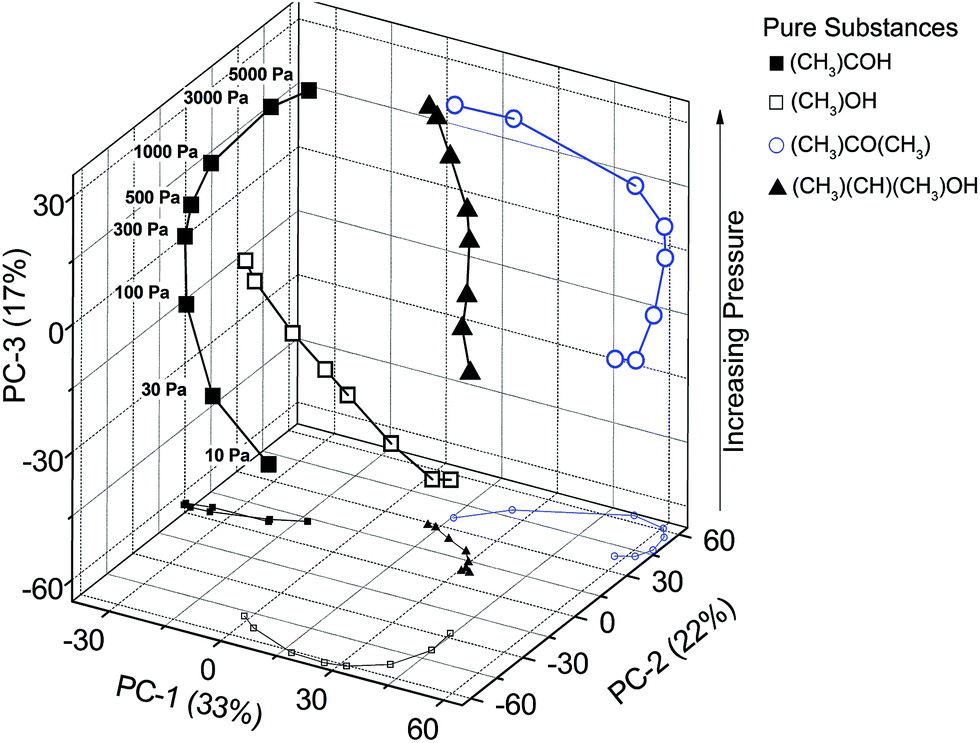 | ||
| Fig. 7 PCA scores of four substances at eight gas pressures. The PCA is separating all spectra and PC-3 contains most of the pressure information. | ||
C. Partial least square regression (PLS)
Half of the 10 spectra were used to calculate a PLS model, while the remaining half was treated as unknown and used for a prediction. Two types of predictions have been calculated.First, the PLS model was created by using the exact amount of substance in a mixture as target value (model hereinafter denoted as PLSE). The second model used discrete value, where 0 means that a substance is not included in a mixture and 1 means a substance is present (model hereinafter denoted as PLSD). It should be emphasized, that the second (discrete) model PLSD has to be capable of assigning a substance to a mixture even if it comprises only a small amount. Reference is made to some mixtures with sub-quantities of less than 4% (see Table 1). The following calculations and results are referring to the 300 Pa spectra (S21) of the samples no. 1 to 15. Mixture no. 16 was not included. Instead it was used for an independent test of the model in order to check whether an unknown mixture can be correctly associated.
Fig. 8 (left) shows the results of the PLSE prediction of the exact amount of a substance in a mixture. The blue straight line is the actual mass fraction value which is expected from the mixing ratio, while the black squares are the prediction based on the spectra treated as unknown. σsub is the mean deviation between the real and predicted values of the five unknown spectra concerning a certain mixture under test. 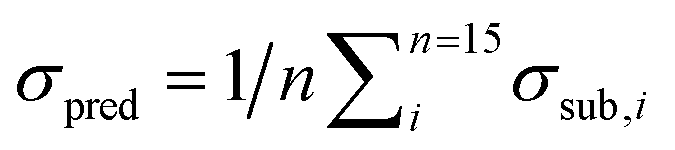 is the mean deviation for a substance prediction in all measured mixtures.
is the mean deviation for a substance prediction in all measured mixtures.
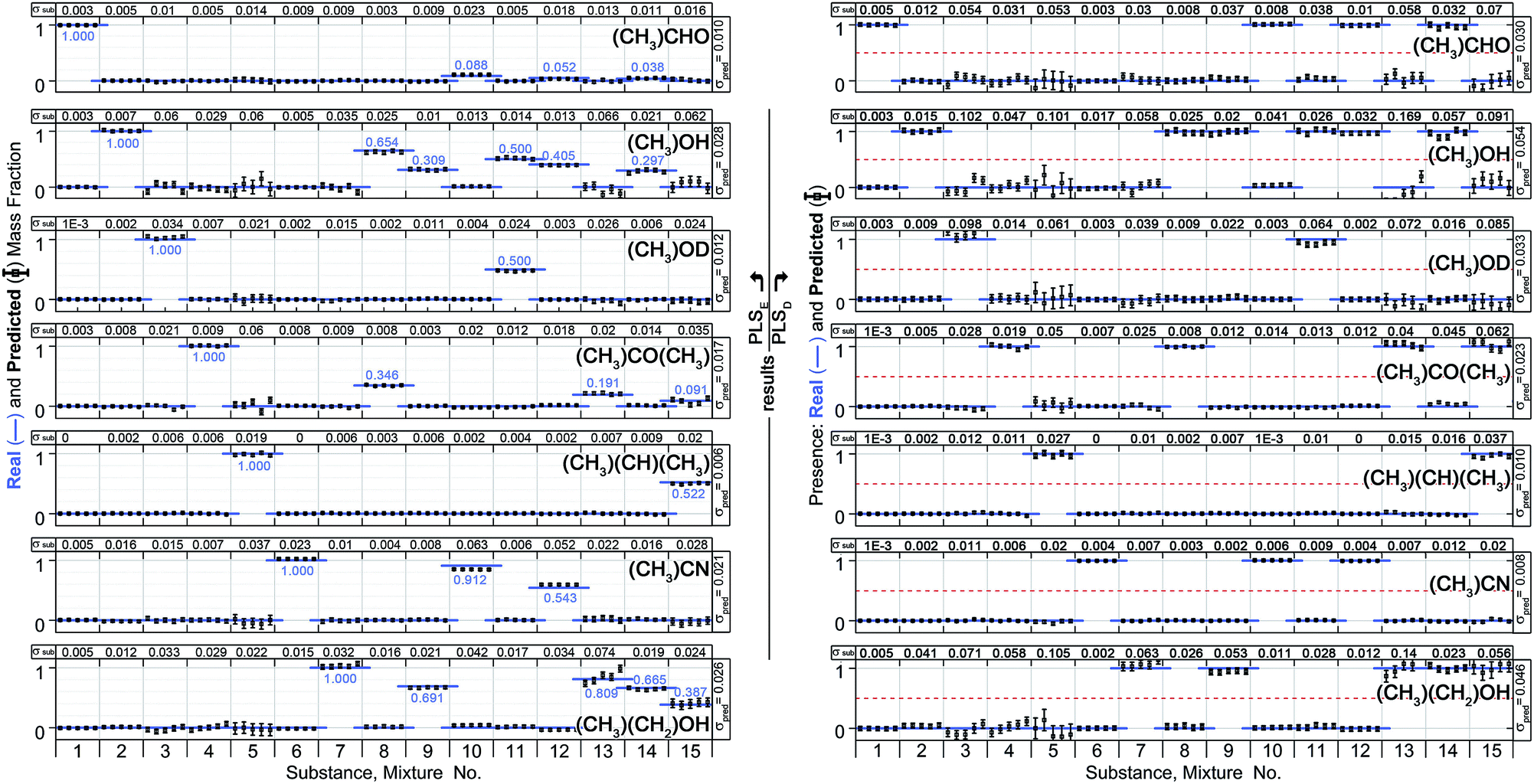 | ||
Fig. 8 Left: results of PLSE analysis with exact mass fraction prediction. Right: results of the PLSD analysis with discrete substance prediction (substance present = 1, not present = 0); the blue lines are the real values, black symbols are the predicted values based on the respective PLS model. σsub is the mean deviation between the real and predicted values of the five unknown spectra concerning a certain substance under test. 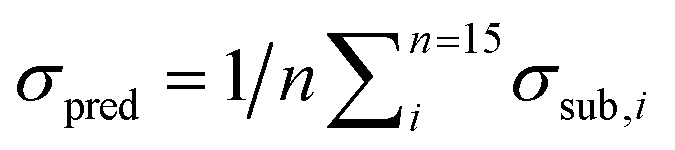 is the prediction error of a particular substance for all measured spectra. The overall error of prediction for the PLSE model is is the prediction error of a particular substance for all measured spectra. The overall error of prediction for the PLSE model is 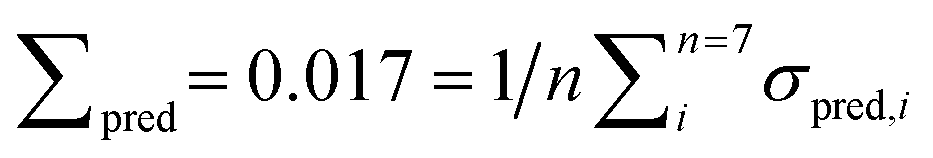 . For the PLSD model it is Σpred = 0.029. The qualitative and quantitative substance identification even of small amounts of a substance in a complex mixture is excellent. Reference is made to the mixtures no. 10, 12 and 14 with single shares of acetaldehyde down to below 4%. . For the PLSD model it is Σpred = 0.029. The qualitative and quantitative substance identification even of small amounts of a substance in a complex mixture is excellent. Reference is made to the mixtures no. 10, 12 and 14 with single shares of acetaldehyde down to below 4%. | ||
As can be seen the model gives an excellent quantitative analysis with an overall error of prediction Σpred = 0.017, i.e. the absolute deviation between predicted and real amount averaged over all predictions in the entire model is approximately 2%.
Also the challenging prediction using the discrete model PLSD provides good results (Fig. 8, right). In this model a threshold was set to 0.5 meaning that above this threshold the substance is in the mixture while below it is not. This model has a prediction error of Σpred = 0.029. Even the very little amount of acetaldehyde in the mixtures 10, 12 and 14 was correctly identified. Applying the model on mixture no. 16 all ingredients have been characterized correctly without false positive, except of an error for ethanol.
The accuracy of the PLSE model with respect to the determination of an amount of substance in a mixture is shown in Fig. 9 for ethanol. The comparison between reference and prediction shows a slope of the regression line close to one which is almost ideal. The standard error of the residuals (for calibration; SEC) gives a determination accuracy for the mass fraction of ethanol of 2.4 mass%. The BIAS is a measure of the systematic uncertainty and is very close to zero, which indicates a good calibration. Accordingly the coefficient of determination is very high (99.6%).
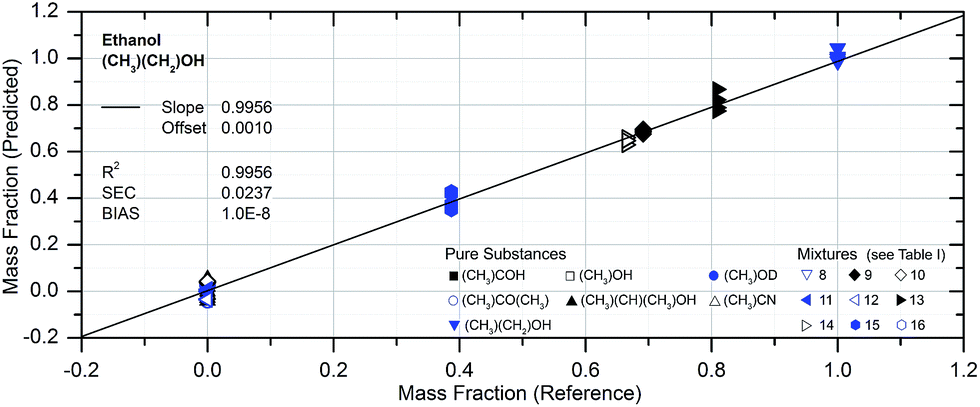 | ||
| Fig. 9 Results of the predicted mass fraction of ethanol obtained from PLSE regression. The slope is close to the ideal value of 1 while the standard deviation of residuals (SEC) is 2.4 mass%. The coefficient of determination R2 shows that 99.6% of the total variance in the amount of substance is explained by the spectra. | ||
The scaling of a PLS model depends on the required prediction precision and can be derived from the root mean square error RMSEV (full validation). It is calculated according to:
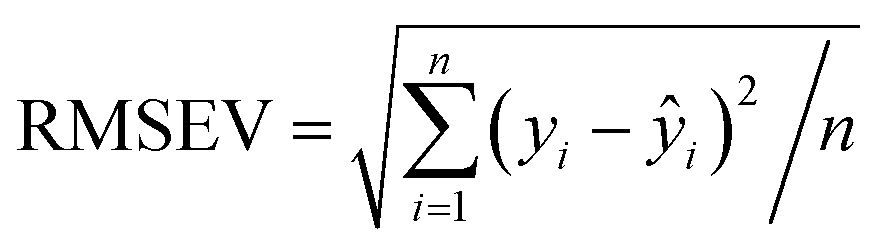 | (3) |
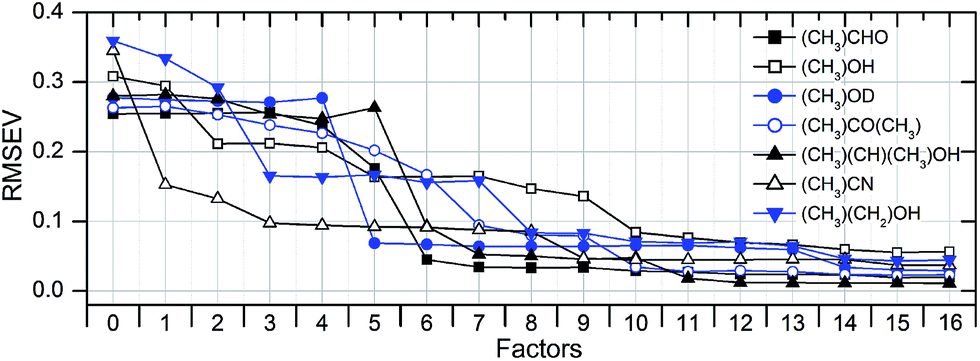 | ||
| Fig. 10 Successive reduction of the root mean square error (RMSEV) with increasing number of used factors (PLSE model; full validation). With ten and more factors the validation error is well below 10%. Since the RMSEV is not increasing, a PLSE model with 16 factors is still not over-determined. | ||
The cluster analysis for PLSE showed a model quality of Q = 78 for N = 6 factors. This enhancement in comparison to the PCA (sec. III B), is due to the integration of the target matrix as considered in sec. II C.
The prediction with the PLSE model showed a good stability against noise in the unknown spectra. This was simulated by adding noise to the measured spectra.30 The PLSE model proves to be robust, because a 25 times increased noise resulted in a prediction error Σpred below 0.5. Acetaldehyde, for example, has a prediction error σpred < 0.05.
D. Principal components regression (PCR)
For the PCR, the same spectra are used as in sec. III. The PCR uses individually calculated PCAs for every target value. The PCR can be useful to create a calibration model, if there is only information on one certain target substance.Twelve individual models were created, including two models for each of the six substances with a discrete affiliation and the exact amount of substance as discussed in sec. III C. The good quality of the PCR in terms of substance identification is similar to the PLS regression. Concerning the exact amount of substance, the prediction showed a mean error of Σpred = 0.04, being twice the error of the PLSE model. The discrete prediction has a mean error of Σpred = 0.08 with a differentiation-threshold at 0.44.
E. Soft independent modeling of class analogy (SIMCA)
The SIMCA analysis was realized by creating 96 PCA models, one for each of the 16 substances and mixtures at three pressures (30, 100 and 300 Pa) and for both measurement directions (S21 and S12). The model calibration was performed with every second spectrum, while the other half represented the unknown spectra for the prediction.All 480 spectra were assigned correctly to their classes. Not only substance membership, but also the pressure regardless the measurement direction was associated correctly. Only propyl alcohol had false positive events, where a set of five measurements at 100 Pa was wrongly associated with 30 Pa.
IV. Summary and conclusion
Absorption spectra of seven gaseous VOCs and mixtures there of have been measured at frequencies between 238 GHz and 252 GHz. The investigated substances represent both medical biomarkers and security relevant substances. Several MVA techniques have been applied to the analysis of the spectra. These techniques were investigated with respect to their performance in terms of substance detection and identification.PCA models yield score-plots with a well-defined cluster formation for all 16 substances. The clusters are well localized with large barycentre distances. The associated loadings show distinctive features of the involved substances.
It was found that the PCA model quality for spectra taken at 300 Pa was four times better than for spectra at 30 Pa. Measurement and PCA analyses at different pressures, showed the capability of the PCA to identify substances and mixtures at pressures between 10 Pa and 5000 Pa.
The qualitative and quantitative substance identification with the PLS regression showed excellent results: the mean error of the prediction is less than 2% and the model quality is better than achieved with the PCA only. Furthermore the PCR showed similar good results.
Finally the SIMCA demonstrated the most complete prediction, since all substances at all pressures in all measurement directions have been correctly assigned with one single model.
In conclusion, MVA is a powerful method for analysis of THz molecular spectra. It can be used for qualitative and quantitative substance detection and identification, for example in medical or in security applications.
With a gas pre-processing system based on absorption and thermal desorption for example with porous carbon nano-sieves, the substance identification of little amounts should be possible. Due to the high adsorption selectivity VOCs can be concentrated by simultaneously filtering out N2, O2, CO2 and H2O. It is important to note that identification with MVA is possible even at gas pressures above the Doppler limit, where pressure broadening dominates. This enables the use of small waveguide absorption cells equipped with rather simple and light-weight vacuum pumps. In combination with upcoming compact THz transmitters and receivers in SiGe technology31 and a waveguide integration, the development of a versatile, miniaturized THz gas sensor seems feasible. However, for future real-world applications other issues for example gas collection and controlling external parameters such as the temperature has to be resolved.
Acknowledgements
P. F.-X. Neumaier acknowledges the support by the Helmholtz Research School on Security Technologies.References
- A. I. McIntosh, B. Yang, S. M. Goldup, M. Watkinson and R. S. Donnan, Chem. Soc. Rev., 2012, 4, 855 Search PubMed.
- C. F. Neese, I. R. Medvedev, G. M. Plummer, A. J. Frank, C. D. Ball and F. C. De Lucia, IEEE Sens. J., 2012, 12(8), 2565 CrossRef CAS.
- S. M. Cristescu, J. Mandon, F. J. M. Harren, P. Meriläinen and M. Högman, J. Breath Res., 2013, 7(017104), 1 Search PubMed.
- K. A. Bakeev, in Process analytical technology: spectroscopic tools and implementation strategies for the chemical and pharmaceutical industries, John Wiley & Sons, 2nd edn, 2010 Search PubMed.
- P. F. Taday, Philos. Trans. R. Soc., A, 2004, 362(1815), 351 CrossRef CAS PubMed.
- C. J. Strachan, P. F. Taday, D. A. Newnham, K. C. Gordon, J. A. Zeitler, M. Pepper and T. Rades, J. Pharm. Sci., 2005, 94(4), 837 CrossRef CAS PubMed.
- M. Tonouchi, Nat. Photonics, 2007, 1(2), 97 CrossRef CAS.
- T. W. Crowe, T. Globus, D. L. Woolard and J. L. Hesler, Philos. Trans. R. Soc., A, 2004, 362(1815), 365 CrossRef CAS PubMed.
- D. L. Woolard, E. R. Brown, A. C. Samuels, J. O. Jensen, T. Globus, B. Gelmont and M. Wolski, IEEE MTT-S Int. Microwave Symp. Dig., 2003, 2, 763 Search PubMed.
- W. Cao and Y. Duan, Clin. Chem., 2006, 52(5), 800 CAS.
- M. Phillips, J. Herrera, S. Krishnan, M. Zain, J. Greenberg and R. N. Cataneo, J. Chromatogr., 1999, 729(1–2), 75 CrossRef CAS.
- C. Wang and P. Sahay, Sensors, 2009, 9(10), 8230 CrossRef CAS PubMed.
- G. Peng, U. Tisch, O. Adams, M. Hakim, N. Shehada, Y. Y. Broza, S. Billan, R. Abdah-Bortnyak, A. Kuten and H. Haick, Nat. Nanotechnol., 2009, 4(10), 669 CrossRef CAS PubMed.
- G. Konvalina and H. Haick, Acc. Chem. Res., 2014, 47(1), 66 CrossRef CAS PubMed.
- H. Haick, Y. Broza, P. Mochalski, V. Ruzsanyi and A. Amann, Chem. Soc. Rev., 2014, 43, 1423 RSC.
- M. Hakim, Y. Broza, O. Barash, N. Peled, M. Phillips, A. Amann and H. Haick, Chem. Rev., 2012, 112(11) CrossRef CAS PubMed.
- T. H. Risby and S. F. Solga, Appl. Phys. B, 2006, 85(2–3), 421 CrossRef CAS PubMed.
- A. Berrington de González, M. Mahesh, K.-P. Kim, M. Bhargavan, R. Lewis, F. Mettler and C. Land, Arch. Intern. Med., 2009, 168(22), 2071 CrossRef PubMed.
- J. Hao and L. Wang, J. Air Waste Manage. Assoc., 2005, 55(9), 1298 CAS.
- L. S. Rothman, I. E. Gordon and A. Barbe, et al. , J. Quant. Spectrosc. Radiat. Transfer, 2009, 110(9–10), 533 CrossRef CAS PubMed.
- I. R. Medvedev, C. F. Neese, G. M. Plummer and F. C. De Lucia, Appl. Opt., 2011, 50(18), 3028 CrossRef CAS PubMed.
- W. Gordy and R. L. Cook, in Microwave Molecular Spectra, John Wiley & Sons, 1984 Search PubMed.
- A. D. Burnett, W. Fan, P. C. Upadhya, J. E. Cunningham, M. D. Hargreaves, T. Munshi, H. G. M. Edwards, E. H. Linfield and A. G. Davies, Analyst, 2009, 134(8), 1658 RSC.
- A. Gredilla, J. M. Amigo, S. Fdez-Ortiz de Vallejuelo, A. de Diego, R. Bro and J. M. Madariaga, Anal. Methods, 2012, 4, 676 RSC.
- R. Bro and A. K. Smilde, Anal. Methods, 2014, 6, 2812 RSC.
- W. Kessler, in Multivariate Datenanalyse für die Pharma-, Bio- und Prozessanalytik, John Wiley & Sons, 2007 Search PubMed.
- United States Environmental Protection Agency, Consolidated List of Chemicals Subject to the EPCRA, CERCLA and Section 112(r) of the Clean Air Act, Office of Solid Waste and Emerg. Response, 2012 Search PubMed.
- J. A. Kent and S. D. Barnicki, in Synthetic Organic Chemicals in Handbook of Industrial Chemistry and Biotechnology, Springer, US, 12th edn, 2013, ch. 10, p. 313 Search PubMed.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, in Production of Methanol: From Fossil Fuels and Bio-Sources to Chemical in Beyond Oil and Gas: The Methanol Economy, Wiley VCH, 2nd edn, 2009, ch. 12, p. 234 Search PubMed.
- Camo Software AS, in The Unscrambler Appendices: Method References, 2014 Search PubMed.
- K. Schmalz, Y. Mao, J. Borngraber, P. Neumaier and H.-W. Hübers, Electron. Lett., 2014, 50(12), 881 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |