
Palladium catalyzed Csp2–H activation for direct aryl hydroxylation: the unprecedented role of 1,4-dioxane as a source of hydroxyl radicals†
Kapileswar
Seth
,
Manesh
Nautiyal
,
Priyank
Purohit
,
Naisargee
Parikh
and
Asit K.
Chakraborti
*
Department of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research (NIPER), Sector 67, S. A. S. Nagar 160 062, Punjab, India. E-mail: akchakraborti@niper.ac.in; akchakraborti@rediffmail.com
First published on 4th November 2014
Abstract
A novel strategy for direct aryl hydroxylation via Pd-catalysed Csp2–H activation through an unprecedented hydroxyl radical transfer from 1,4-dioxane, used as a solvent, is reported with bio relevant and sterically hindered heterocycles and various acyclic functionalities as versatile directing groups.
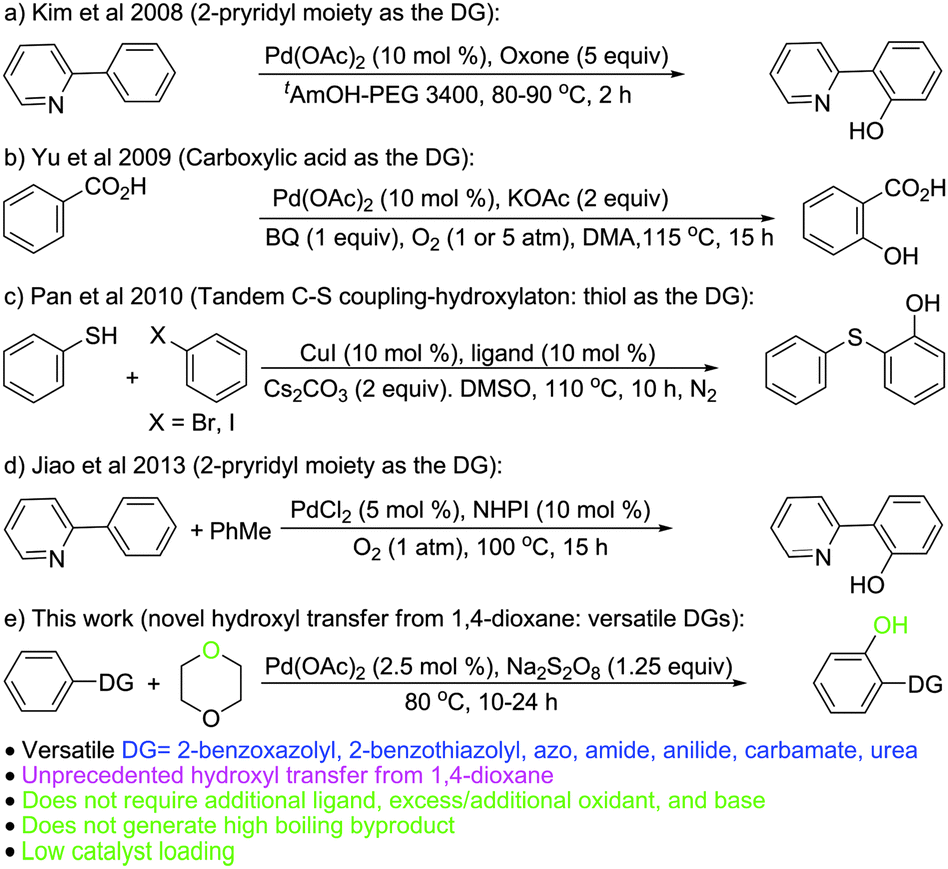
The wide occurrence of hydroxylated aromatics (phenols) in bioactive compounds1 makes them highly sought for synthetic targets. Aryl hydroxylation through C–H activation has emerged as a new synthetic route to phenols.2 However, this route represents indirect hydroxylation, as the reaction proceeds via the intermediate formation of the acyloxy arenes that are converted to hydroxyaromatics during the workup or on further hydrolytic transformation.3 The issue on direct aryl hydroxylation remains inadequately addressed (Scheme 1).4
 | ||
| Scheme 1 Different strategies for direct aryl hydroxylation. | ||
Fujiwara et al.4a revealed the feasibility of direct aryl hydroxylation but the low yield (∼5%) and turnover ratio (10–15 to Pd), poor selectivity, and harsh reaction conditions (15 atm O2, 15 atm CO, 180 °C, AcOH) make it less attractive. The strategy of Kim et al.4b involving the use of the 2-pyridyl moiety as the directing group (DG) for Csp2–H activation though affords good yields, requires excess oxidants (5 equiv.), is applicable to highly substituted DGs (2-pyridyl), and does not work with aryl moieties bearing o-substitution. The use of carboxylic acid as a DG by Yu et al.4c requires an excess of base, benzoquinone as an additional oxidant (in addition to O2 at 1–5 atm), and leads to decarboxylation (e.g. 1-naphthoic acid). The tandem C–S coupling-hydroxylation strategy by Pan et al.4d requires a special ligand, an excess of base, and aryl bromide/iodide as the reaction partner (additional reagent). The recent report on 2-pyridyl directed Csp2–H activation by Jiao et al.,4e though circumvents the limitations of the protocol by Kim et al.,4b requires N-hydroxyphthalimide (NHPI) and toluene as an additional reagent system (in addition to O2 at 1 atm) to form the benzyl radical that acts as the effective agent/species for hydroxyl radical generation and leads to the formation of high boiling by-products (benzaldehyde/benzoic acid).
Functionalised 2-(benzoxazol-2-yl)phenols have diverse biological activities.5 Thus, it would be attractive to use the 2-benzoxazolyl scaffold as the DG for Csp2–H activation for direct aryl hydroxylation to further potentiate their biological activity6 and as there is no report on direct aryl hydroxylation with the 2-benzoxazolyl moiety as the DG. 2-Phenylbenzoxazole (1a)7 was considered as a model substrate for oxidative hydroxylation via aryl Csp2–H activation under various conditions (Scheme 2).
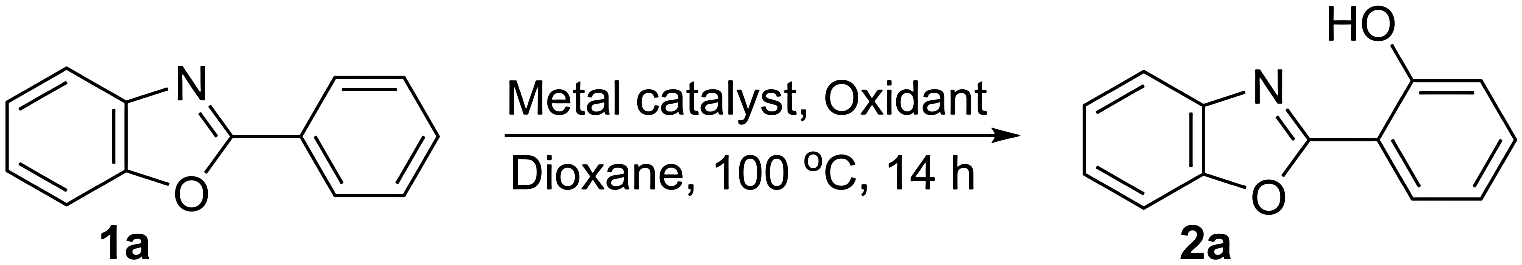 | ||
| Scheme 2 Direct hydroxylation of 1a to form 2a. | ||
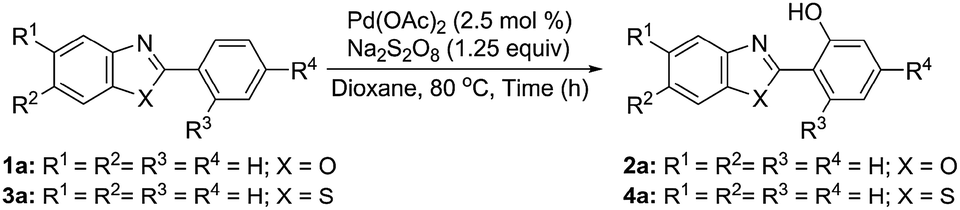
It appears that the limited reports on direct hydroxylation involve the generation of the oxygen/hydroxyl radical4b–d that adds on to the metal centre in its higher oxidation state in the cyclometallated intermediate and acts as the effective species for C–O bond formation. 1,4-Dioxane is reported to generate the hydroxyl radical upon pyrolysis.8 However, high temperature (1550–2100 K) is required to generate the hydroxyl radical by ring opening (homolytic C–O cleavage), isomerisation, and dissociation of the resulting linear species. We reasoned that the hydroxyl radical generation from 1,4-dioxane may be achieved under milder conditions in the presence of an oxidant that would abstract H from 1,4-dioxane and facilitate its dissociation via ring opening C–O cleavage involving the free radical mechanism. Thus, it was hypothesised that the use of 1,4-dioxane in combination with persulfate would serve the dual purpose of the reaction medium and the oxo/hydroxyl transfer agent without the requirement of any additional oxidant/reactant and base.
Treatment of 1a with different variations of the reaction parameters (ESI: Table S1A–E)† revealed the use of Pd(OAc)2 (2.5 mol%) and Na2S2O8 (1.25 equiv.) in 1,4-dioxane at 80 °C for 14 h to be the best operative reaction conditions affording 2a in 62% yield which could be further improved to 82% after reusing the unreacted 1a recovered after the first use. The lack of formation of 2a in any other solvent (ESI: Table S1D and E)† suggested 1,4-dioxane to be the hydroxyl transfer agent, as the comparable results using commercial grade as well as dry and degassed 1,4-dioxane under N2 or O2 eliminated the possibility of hydroxyl transfer from any dissolved oxygen/moisture. That the reaction forms the hydroxyarene directly was demonstrated by the fact that 2a was obtained in comparable yield (64%) avoiding any aqueous workup. However, the Pd(OAc)2-catalysed reaction of 1a with Na2S2O8 (1.5 equiv.) in HOAc–Ac2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) or Ac2O instead of 1,4-dioxane afforded O-acetylated 2a following a non-aqueous workup in 53 and 55% yields, respectively (ESI: Table S2A).† Thus, the persulfate-dioxane combination constitutes a novel reagent system for Pd-catalysed direct aryl hydroxylation.
:1) or Ac2O instead of 1,4-dioxane afforded O-acetylated 2a following a non-aqueous workup in 53 and 55% yields, respectively (ESI: Table S2A).† Thus, the persulfate-dioxane combination constitutes a novel reagent system for Pd-catalysed direct aryl hydroxylation.
Treatment of 1a under the reported conditions for acetoxylation/hydroxylation and minor variation (using 1,4-dioxane as solvent) did not produce a significant amount of 2a (ESI: Table S3A and B)† and established the distinctiveness of the present work.
The direct hydroxylation protocol is applicable to benzoxazoles bearing a substituent in the 2-aryl moiety as well as in the benzenoid ring of the benzoxazole scaffold (Table 1). The applicability of the direct hydroxylation towards various substituted 2-arylbenzothiazoles (Table 1),9 another bio relevant and sterically hindered DG, demonstrated the versatility with respect to the DG.10 The presence of Cl, Br, CF3, and NO2 in the 2-aryl moiety appears to be beneficial, as such substrates afforded the corresponding hydroxyarenes in better yields compared to that of the unsubstituted analogue 1a/3a. The effect of these substituents was more pronounced with the 2-benzothiazolyl moiety as the DG. The presence of an electron donating substituent such as the Me group decreased the yield.
|
|
|---|
| a The figure in the parentheses is the total yield based on recovery and reuse of the unreacted starting material after the first attempt. |
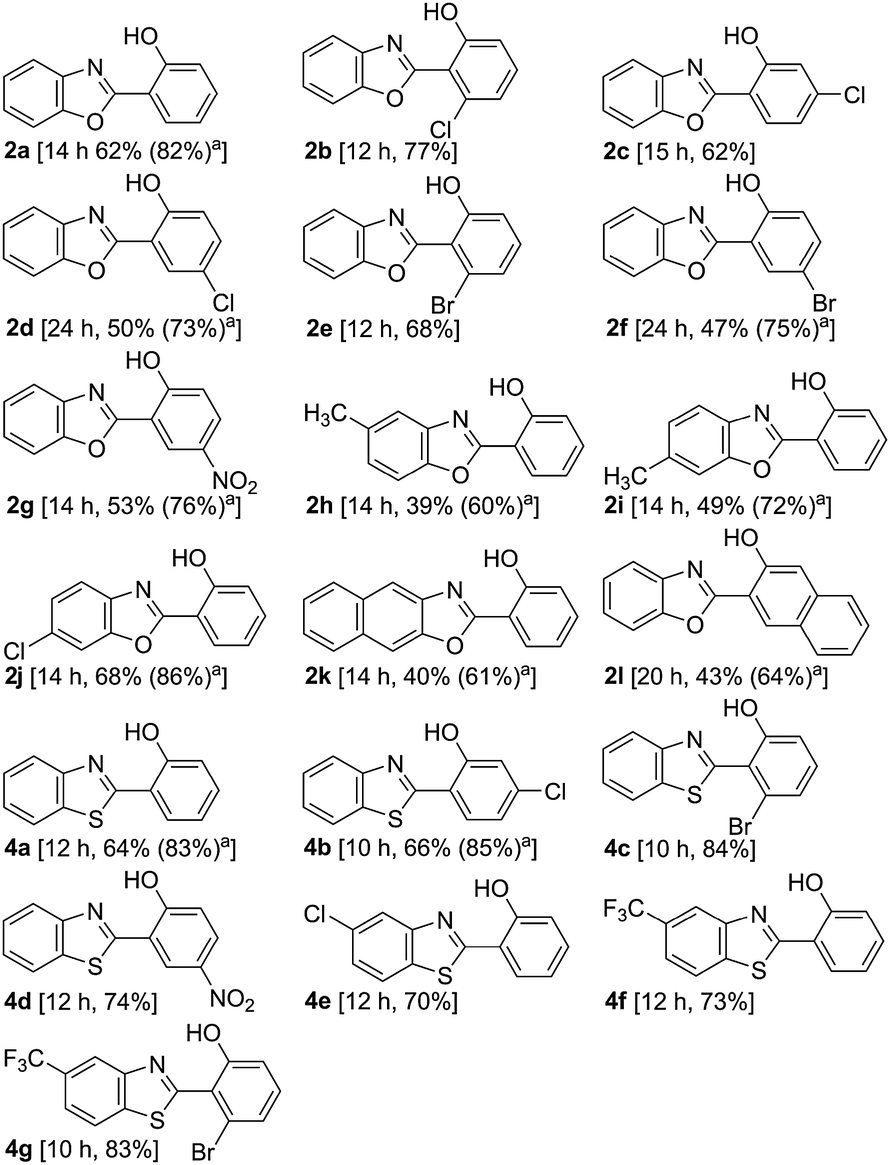
|
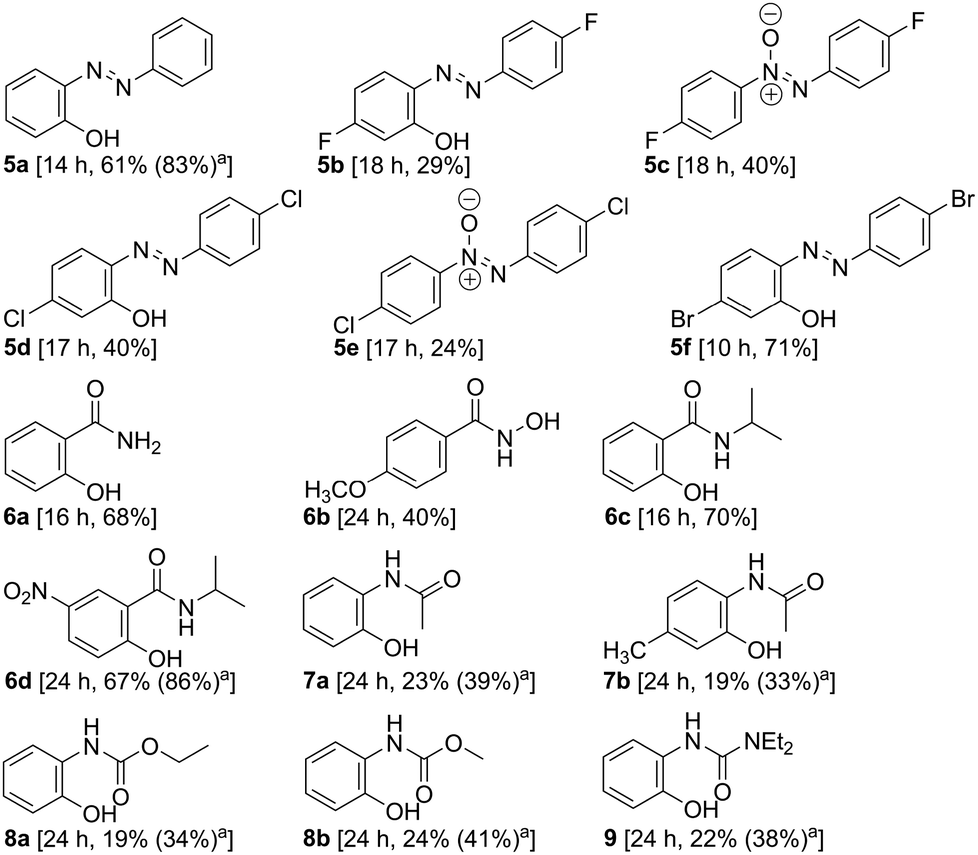
The results summarized in Table 2 demonstrate the further scope with acyclic DGs such as azo, amide, anilide, carbamate, and unsymmetrical urea. The F and Cl substituted azo benzenes produced the corresponding azoxy-benzene in 40 and 24% yields, respectively, along with the desired hydroxylated products. The chemoselective hydroxylation with substrates bearing halogen provides the scope of structural modification through Suzuki coupling11 to broaden the chemical space. Catalyst [Pd(OAc)2] was recovered and reused for five consecutive reactions affording comparable yields.†
|
|
|---|
| a The figure in the parentheses is the total yield based on recovery and reuse of the unreacted starting material after the first attempt. |
|
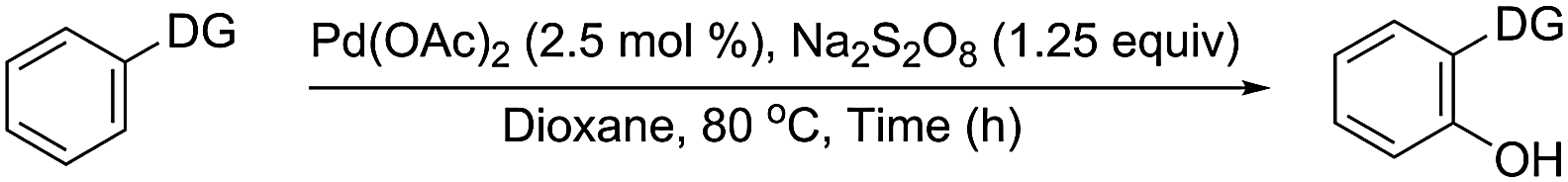
A plausible mechanism is depicted in Scheme 3. The Pd-catalyst forms complex I through coordination with the nitrogen centre of the DG and trigers aryl Csp2–H activation and ligand-assisted intra-molecular aryl proton abstraction in II to form cyclopalladated species III. Oxidative addition of the hydroxyl radical, formed by degradation of 1,4-dioxane with the persulfate anion, (Scheme 4) generates Pd(IV) species IV12 or PdIII–PdIII species V.13 The aryl C–O bond forming reductive elimination from IV/V leads to the hydroxyarene (phenol) and brings the Pd(II) compound back to the catalytic cycle to form I through ligand exchange (facilitated by intramolecular hydrogen bond formation between the newly inserted OH and the nitrogen atom of the DG).14
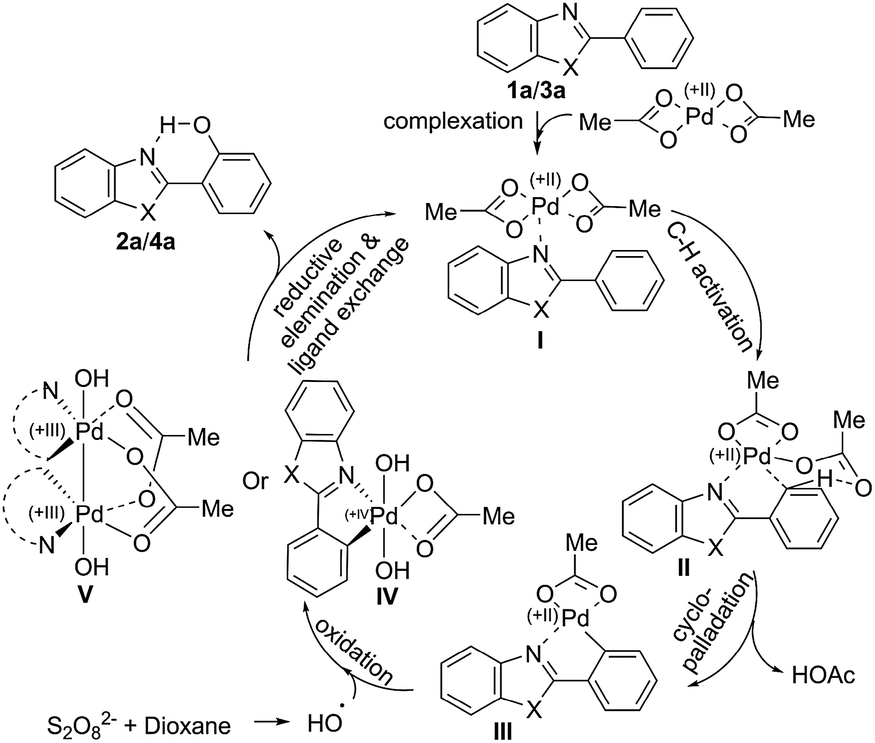 | ||
| Scheme 3 Plausible pathway for the direct aryl hydroxylation. | ||
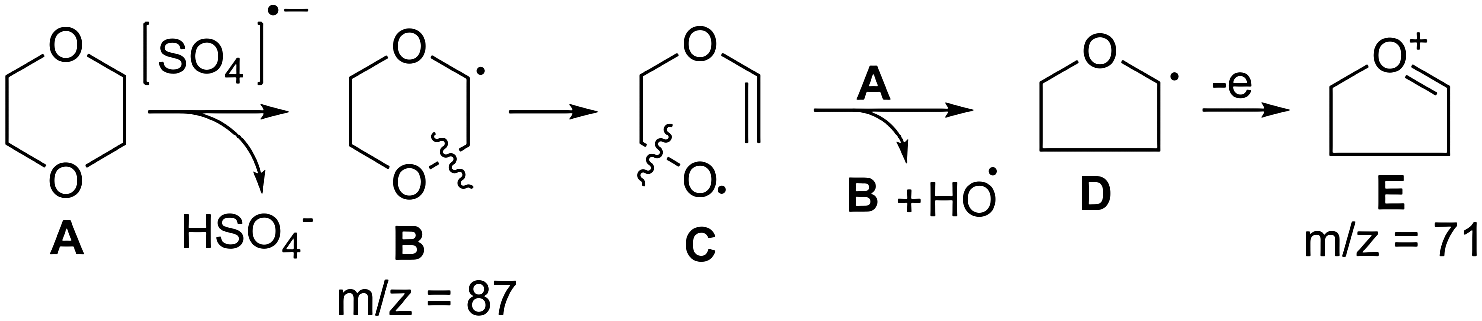 | ||
| Scheme 4 Generation of the OH radical from 1,4-dioxane and Na2S2O8. | ||
The generation of the hydroxyl radical from Na2S2O8 and 1,4-dioxane is presented in Scheme 4. The sulfate anion radical, generated from the persulfate anion,15 has the ability to abstract C–H hydrogen15a and reacts with 1,4-dioxane (A) to form the 1,4-dioxan-2yl radical (B). Homolytic ring C–O cleavage8 of B produces radical C which on further homolytic C–O cleavage is converted to the tetrahydrofuran-2-yl radical D. The liberated oxygen diradical is converted to the hydroxyl radical through H-abstraction from another molecule of 1,4-dioxane generating radical B to propagate the radical chain reaction. The relevant intermediates could be identified in the GC-MS spectra of aliquot samples (withdrawn at an interval of 2 h) of the reaction mixture of 1,4-dioxane and Na2S2O8 at 80 °C that showed ion peaks corresponding to B (m/z 87) (ESI: 10.2.3) and D/E (m/z 71) (ESI: 10.2.2) that compared well with the GC-MS and ms2 (of the ion at m/z 71) spectra of an authentic sample of THF.† Some of the daughter ions observed in the ms2 spectra of the ion at m/z 71 were also observed in the ms2 spectra of the ion at m/z 87 (e.g. 70.81 vs. 70.82; 42.93 vs. 42.87) suggesting that the ion at m/z 71 is derived from the ion at m/z 87 through loss of active oxygen species. The GC-MS spectra of aliquots of the reaction mixture during the treatment of 2-phenylbenzoxazole (1a) with Na2S2O8 and 1,4-dioxane at 80 °C revealed the presence of the ion peaks at m/z 87 and 71 corresponding to B and D/E, respectively (ESI: 10.5.3 and 10.5.2).†
The influence of ligand/counter anions on the catalytic potential of the Pd-compounds (ESI: Table S1B)† and the lack of improvement of yield using NH4OAc, KOAc and (KOAc + 18-C-6) as external base (ESI: Table S5A)† during the Pd-catalysed reactions implicate that the abstraction of the aryl proton during the C–H activation step occurs via an intramolecular process involving II. Thus the strong electron withdrawing CF3 group reduces the HB acceptor property of TFA in Pd(TFA)216 making it an inferior catalyst compared to Pd(OAc)2. The HB formation ability of the counter anion plays a key role in the formation of the reaction intermediate (transition state) in various organic reactions.17 The importance of the HB has also been realised in forming metal centred transition states.18
The involvement of radical species was corroborated by the fact that a drastic decrease in the product (2a) yield (17%) was observed in performing the Pd(OAc)2-catalysed reaction of 1a in the presence of a radical scavenger (TEMPO) (ESI: Scheme S5C).† The indispensable role of 1,4-dioxane is reflected in its ability to form radical species in the presence of persulfate15 or other oxidants.19
The present work provides a novel strategy for direct aryl hydroxylation via palladium-catalysed aryl Csp2–H activation with bioactive and sterically hindered heterocyclic moieties and other acyclic functionalities (azo, amide, anilide, carbamate, and urea) as versatile directing groups through unprecedented generation of hydroxyl radicals, as the effective aryl hydroxylation species, from 1,4-dioxane (used as a solvent).
Financial support from DST (SR/S1/OC-33/2008) and CSIR [senior research fellowship to KS], New Delhi, India is acknowledged.
Notes and references
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed.
- (a) Y. Rao, Synlett, 2013, 24, 2472 CrossRef CAS PubMed; (b) S. Enthaler and A. Company, Chem. Soc. Rev., 2011, 40, 4912 RSC; (c) D. A. Alonso, C. Nájera, I. M. Pastor and M. Yus, Chem. – Eur. J., 2010, 16, 5274 CrossRef CAS PubMed; (d) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed.
- (a) X. Li, Y.-H. Liu, W. Gu, J. Li, F.-J. Chen and B.-F. Shi, Org. Lett., 2014, 16, 3904 CrossRef CAS PubMed; (b) J. Gallardo-Donaire and R. Martin, J. Am. Chem. Soc., 2013, 135, 9350 CrossRef CAS PubMed; (c) W. Liu and L. Ackermann, Org. Lett., 2013, 15, 3484 CrossRef CAS PubMed; (d) X. Yang, G. Shan and Y. Rao, Org. Lett., 2013, 15, 2334 CrossRef CAS PubMed; (e) F. Yang and L. Ackermann, Org. Lett., 2013, 15, 718 CrossRef CAS PubMed; (f) P. Y. Choy and F. Y. Kwong, Org. Lett., 2013, 15, 270 CrossRef CAS PubMed; (g) G. Shan, X. Han, Y. Lin, S. Yu and Y. Rao, Org. Biomol. Chem., 2013, 11, 2318 RSC; (h) F. Mo, L. J. Trzepkowski and G. Dong, Angew. Chem., Int. Ed., 2012, 51, 13075 CrossRef CAS PubMed; (i) G. Shan, X. Yang, L. Ma and Y. Rao, Angew. Chem., Int. Ed., 2012, 51, 13070 CrossRef CAS PubMed; (j) V. S. Thirunavukkarasu and L. Ackermann, Org. Lett., 2012, 14, 6206 CrossRef CAS PubMed; (k) Y. Yang, Y. Lin and Y. Rao, Org. Lett., 2012, 14, 2874 CrossRef CAS PubMed; (l) M. R. Yadav, R. K. Rit and A. K. Sahoo, Chem. – Eur. J., 2012, 18, 5541 CrossRef CAS PubMed; (m) G.-W. Wang, T.-T. Yuan and X.-L. Wu, J. Org. Chem., 2008, 73, 4717 CrossRef CAS PubMed; (n) V. S. Thirunavukkarasu, J. Hubrich and L. Ackermann, Org. Lett., 2012, 14, 4210 CrossRef CAS PubMed; (o) Y. Leng, F. Yang, W. Zhu, Y. Wu and X. Li, Org. Biomol. Chem., 2011, 9, 5288 RSC.
- (a) T. Jintoku, K. Nishimura, K. Takai and Y. Fujiwara, Chem. Lett., 1990, 1687 CrossRef CAS; (b) S. H. Kim, H. S. Lee, S. H. Kim and J. N. Kim, Tetrahedron Lett., 2008, 49, 5863 CrossRef CAS PubMed; (c) Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 14654 CrossRef CAS PubMed; (d) R. Xu, J.-P. Wan, H. Mao and Y. Pan, J. Am. Chem. Soc., 2010, 132, 15531 CrossRef CAS PubMed; (e) Y. Yan, P. Feng, Q.-Z. Zheng, Y.-F. Liang, J.-F. Lu, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2013, 52, 5827 CrossRef CAS PubMed.
- (a) P. S. M. Sommer, R. C. Almeida, K. Schneider, W. Beil, R. D. Süssmuth and H.-P. Fiedler, J. Antibiot., 2008, 61, 683 CrossRef CAS PubMed; (b) S. K. Tipparaju, S. Joyasawal, M. Pieroni, M. Kaiser, R. Brun and A. P. Kozikowski, J. Med. Chem., 2008, 51, 7344 CAS.
- K. Seth, S. K. Garg, R. Kumar, P. Purohit, V. S. Meena, R. Goyal, U. C. Banerjee and A. K. Chakraborti, ACS Med. Chem. Lett., 2014, 5, 512 CrossRef CAS PubMed.
- (a) D. Kumar, S. Rudrawar and A. K. Chakraborti, Aust. J. Chem., 2008, 61, 881 CrossRef CAS; (b) R. Kumar, C. Selvam, G. Kaur and A. K. Chakraborti, Synlett, 2005, 1401 CAS.
- X. Yang, A. W. Jasper, B. R. Giri, J. H. Kiefer and R. S. Tranter, Phys. Chem. Chem. Phys., 2011, 13, 3686 RSC.
- N. Parikh, D. Kumar, S. Raha Roy and A. K. Chakraborti, J. Chem. Soc., Chem. Commun., 2011, 47, 1797 RSC.
- The protocol for indirect hydroxylation of 2-arylbenzothiazoles [ A. Banerjee, A. Bera, S. Guin, S. K. Rout and B. K. Patel, Tetrahedron, 2013, 69, 2175 CrossRef CAS PubMed ] is ineffective for 2-arylbenzoxazole†.
- K. Seth, P. Purohit and A. K. Chakraborti, Org. Lett., 2014, 16, 2334 CrossRef CAS PubMed.
- (a) J. M. Racowski, A. R. Dick and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 10974 CrossRef CAS PubMed; (b) K. Muñiz, Angew. Chem., Int. Ed., 2009, 48, 9412 CrossRef PubMed; (c) A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177 CrossRef CAS PubMed.
- D. C. Powers, M. A. L. Geibel, J. E. M. N. Klein and T. Ritter, J. Am. Chem. Soc., 2009, 131, 17050 CrossRef CAS PubMed.
- M. Santra, B. Roy and K. H. Ahn, Org. Lett., 2011, 13, 3422 CrossRef CAS PubMed.
- (a) N. Basickes, T. E. Hogan and A. Sen, J. Am. Chem. Soc., 1996, 118, 13111 CrossRef CAS; (b) D. D. Tanner and S. A. A. Osman, J. Org. Chem., 1987, 52, 4689 CrossRef CAS; (c) L. Dogliotti and E. Hayon, J. Phys. Chem., 1967, 71, 2511 CrossRef CAS.
- R. Chebolu, D. N. Kommi, D. Kumar, N. Bollineni and A. K. Chakraborti, J. Org. Chem., 2012, 77, 10158 CrossRef CAS PubMed and references therein.
- (a) A. K. Chakraborti, S. Raha Roy, D. Kumar and P. Chopra, Green Chem., 2008, 10, 1111 RSC; (b) A. K. Chakraborti and S. Raha Roy, J. Am. Chem. Soc., 2009, 131, 6902 CrossRef CAS PubMed; (c) S. Raha Roy and A. K. Chakraborti, Org. Lett., 2010, 12, 3866 CrossRef PubMed; (d) A. Sarkar, S. Raha Roy and A. K. Chakraborti, Chem. Commun., 2011, 47, 4538 RSC; (e) A. K. Chakraborti, L. Sharma and M. K. Nayak, J. Org. Chem., 2002, 67, 2541 CrossRef CAS PubMed; (f) M. K. Nayak and A. K. Chakraborti, Chem. Lett., 1998, 297 CrossRef CAS.
- K. Seth, S. Raha Roy, B. V. Pipaliya and A. K. Chakraborti, J. Chem. Soc., Chem. Commun., 2013, 49, 5886 RSC.
- Z.-Q. Liu, L. Zhao, X. Shang and Z. Cui, Org. Lett., 2012, 14, 3218 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data of all compounds and scanned spectra of new compounds. See DOI: 10.1039/c4cc06864e |
| This journal is © The Royal Society of Chemistry 2015 |