
[Me2C{SnCH(SiMe3)2}2]2. A μ-Me2C-bridged tetrastanna tetrahedrane†‡
Michael
Wagner
a,
Michael
Lutter
a,
Bernhard
Zobel
a,
Wolf
Hiller
a,
Marc H.
Prosenc
b and
Klaus
Jurkschat
*a
aLehrstuhl für Anorganische Chemie II, Technische Universität Dortmund, 44221 Dortmund, Germany. E-mail: klaus.jurkschat@tu-dortmund.de; Fax: +49 231 755 5048; Tel: +49 231 755 3800
bPhysikalische Chemie, TU Kaiserslautern, Germany
First published on 6th November 2014
Abstract
A spacer-bridged bis(organostannylene) was obtained. In the solid-state it adopts the structure of a doubly-capped tetrahedron. It reacts with elemental oxygen, O2, giving tin suboxides. Additionally, the first solid state structure of a spacer-bridged diorgano tin dihydride is reported.
Subvalent tin compounds like spacer-bridged bisstannylenes/distannenes and their transition metal complexes,1 metalloid clusters,2 Zintl ions3 and tin chalcogenide motifs containing tin–tin bonds4 have attracted much attention. While a variety of tetrahedron-shaped molecular compounds R4′E4 (E = C, Si, Ge)5–7 composed of group 14 elements have been synthesized, in the case of E = Sn, however, only the metallostannides M4Sn4 (M = Na, K, Rb, Cs) are known to adopt the structure of a tetrahedron.8 For compounds of the stoichiometry R8′′Sn4 (R′′ = t-Bu,9a R′′ = (CH3)3SiCH2,9b R′′ = 2,6-diethylphenyl,9c R′′ = 0.5C14H24Si29d,e) slightly twisted tetrastannacyclobutane rings were established by X-ray crystallography.
The reaction of 2,2-bis(dichloroorganostannyl)propane, Me2C(SnCl2R)2 [1, R = CH(SiMe3)2]10 with LiAlH4 provided the corresponding tetrahydride derivative Me2C(SnH2R)2, 2, as colourless crystalline material that exhibits good solubility in organic solvents such as benzene and diethyl ether (Scheme 1). Compound 2 is the only second example of a spacer-bridged bis(organotindihydride)11 and the first one the solid-state structure of which could be established.
 | ||
| Scheme 1 Synthesis of compounds 2, 3. | ||
The molecular structure of compound 2 is shown in Fig. 1. Selected interatomic distances and angles are given in the figure caption.
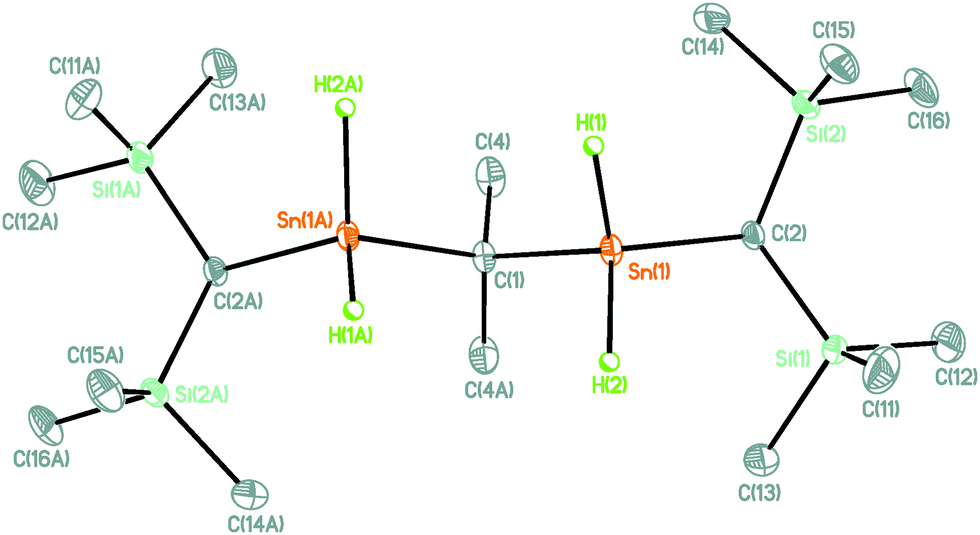 | ||
| Fig. 1 General view (SHELXTL) of 2 showing 30% probability displacement ellipsoids and the atom-numbering scheme. Generation of the second half of the molecule by symmetry operation C2 (symmetry code −x, y, −z + 1/2). Selected interatomic distances (Å): Sn(1)–C(1) 2.1637(18), Sn(1)–C(2) 2.1616(19), Sn(1)–H(1) 1.72(2), Sn(1)–H(2) 1.74(3). Selected interatomic angles (°): C(1)–Sn(1)–C(2) 114.40(9), C(1)–Sn(1)–H(1) 108.8(8), C(1)–Sn(1)–H(2) 108.1(11), C(2)–Sn(1)–H(1) 110.7(8), C(2)–Sn(1)–H(2) 111.6(12), H(1)–Sn(1)–H(2) 102.5(12), Sn(1)–C(1)–Sn(1A) 104.91(12). | ||
Compound 2 crystallizes in the monoclinic space group C2/c with four molecules in the unit cell. The twofold axis is located at C(1).
The Sn(1) atom shows a distorted tetrahedral environment with angles ranging between 102.5(12) (H(1)–Sn(1)–H(2)) and 114.40(9)° (C(1)–Sn(1)–C(2)). The Sn(1)–C(1)–Sn(2) angle of 104.91(12)° is slightly smaller than the corresponding angle in the organotin chloride 1 (106.31(18)°). The Sn(1)–H(1)/H(2) distances of 1.72(2)/1.74(3) Å are as expected for crystallographically determined ones.12 Its identity in solution was established by NMR spectroscopy. Thus, a 1H-coupled 119Sn NMR spectrum (ESI‡) displayed a triplet of pseudo-octet with 1J(119Sn–1H) coupling of 1713 Hz. A 1H NMR spectrum showed, in addition to the expected singlet resonances for the SiCH3, CH, and CCH3 protons, a singlet for the SnH protons at δ = 5.47 ppm [1J(1H–117/119Sn) 1632/1709 Hz, 3J(1H–117/119Sn) 12 Hz].
The reaction of compound 1 with two molar equivalents of (MesNacNacMg)2 (MesNacNac = [(MesNCMe)2CH]−, Mes = 2,4,6-Me3C6H2)13 gave a crude reaction mixture the 119Sn NMR spectrum of which showed exclusive formation of 3. From this reaction mixture air-sensitive blue single crystals of compound 3 were isolated (Scheme 1). Notably, attempts at obtaining compound 3 by the use of alternative reducing reagents such as sodium naphthalenide, potassium graphite, lithium cyclooctatetraenide, excess magnesium or dehydrogenation of compound 2 with pyridine failed. The molecular structure of compound 3 is shown in Fig. 2. Selected interatomic distances and angles are given in the figure caption. Compound 3 crystallizes in the monoclinic space group P21/c with four molecules in the unit cell.
 | ||
| Fig. 2 General view (SHELXTL) of 3 showing 30% probability displacement ellipsoids and the atom-numbering scheme. The hydrogen atoms and the disorder at C(30) are omitted for clarity. Selected interatomic distances (Å): Sn(1)–Sn(2) 3.2383(4), Sn(1)–Sn(3) 2.8287(4), Sn(1)–Sn(4) 2.8864(4), Sn(2)–Sn(3) 2.8457(3), Sn(2)–Sn(4) 2.8619(3), Sn(3)–Sn(4) 3.2263(3), Sn(2)–C(20) 2.192(3), Sn(1)–C(1) 2.221(3). Selected interatomic angles (°): C(1)–Sn(1)–C(10) 123.76(13), C(1)–Sn(1)–Sn(3) 92.23(10), C(1)–Sn(1)–Sn(4) 87.29(9), C(10)–Sn(1)–Sn(3) 121.06(9), C(10)–Sn(1)–Sn(4) 144.29(8), Sn(3)–Sn(1)–Sn(4) 68.729(9), Sn(1)–C(1)–Sn(2) 93.45(13). | ||
The structure can formally be derived from a tetraorgano tetrastanna tetrahedrane, which was modified under symmetry reduction by insertion of two dimethyl carbene moieties, Me2C, into two Sn–Sn bonds. As result of this, the Sn4 tetrahedron is strongly distorted and shows four short Sn–Sn distances ranging between 2.8287(4) (Sn(1)–Sn(3)) and 2.8864(4) (Sn(1)–Sn(4)) Å, and two long distances of 3.2263(3) (Sn(3)–Sn(4)) and 3.2383(4) (Sn(1)–Sn(2)) Å. The short distances are slightly longer than the ones observed for the α-Sn modification (2.81 Å)14 as well as for the 1,2,4,5-tetrastannacyclohexanes H2C(SnPh2SnPh2)2CH2 (2.783(1) Å)15 and Me2C(SnMe2SnMe2)2CMe2 (2.7753(8) Å),16 but shorter than those reported for the Zintl-salts M4Sn4 (M = Na, K, Cs, Rb) with 2.940(2) to 2.981(2) Å.8a The Sn(1), Sn(2), Sn(3), and Sn(4) atoms are each four-coordinated by two Sn and two carbon atoms and show strongly distorted tetrahedral environments with angles ranging between 68.576(9) (Sn(1)–Sn(4)–Sn(2)) and 147.01(9)° (C(20)–Sn(2)–Sn(3)). As result of the ring strain, the Sn(1)–C(1)–Sn(2) angle of 93.45(13)° is much smaller than the corresponding angle of 106.31(18)° reported for 1.10
A NBO analysis revealed for each Sn atom two covalent σ-type Sn–C and two covalent Sn–Sn bonds. However, no covalent bond was found for the long-distant Sn–Sn interaction. Second order perturbation analysis of the bonding situation revealed small contributions of Sn–C (bridge) bonds into the geminal bonded Sn–C antibonding orbitals. Also small contributions from Sn–Sn bond orbitals into the nonbridging Sn–C σ* orbitals are found. The NPA-charges for the Sn atoms are calculated to be 0.9–1.0. Charges on the bridging C-atoms are calculated to be −0.8 and for the end-on C-ligands −1.6 revealing a more carbene-like character of the bridging carbon ligands.
Upon dissolution of 3 in C6D6, color change from deep blue to red brown was observed that might be indicative of a structural change. The 119Sn NMR spectrum (Fig. S2, ESI‡) of this solution showed a resonance at δ = 16 ppm [(1J(119Sn–117Sn) = 3289 Hz, J(119Sn–117Sn) = 608 Hz)] the chemical shift of which is close to the calculated one for a symmetrized tetrahedron (D2d) of −44 ppm (ESI‡). However, the satellite-to-satellite-to-signal-to-satellite-to-satellite integral ratio of approx. 3.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6.6:79.6:6.9:3.8 does not fit with the tetrahedrane structure observed in the solid state. For such a structure, the above-mentioned integral ratio should be 7.7:3.8:77.0:3.8:7.7. Ruled out by the above-mentioned coupling pattern arguments can be the monomeric structure Me2C(SnR)2.
:6.6:79.6:6.9:3.8 does not fit with the tetrahedrane structure observed in the solid state. For such a structure, the above-mentioned integral ratio should be 7.7:3.8:77.0:3.8:7.7. Ruled out by the above-mentioned coupling pattern arguments can be the monomeric structure Me2C(SnR)2.
The J(119Sn–117Sn) coupling constant in 3 of 608 Hz fits well with the 2J(119Sn–117Sn) coupling constant of 599 Hz in compound 1.9 The 1J(119Sn–117Sn) coupling constant of 3289 Hz is similar to the coupling constant of 3330 Hz found in a spacer-bridged distannene reported by Wesemann and Henning.1h The 1H NMR spectrum of 3 in C6D6 shows three singlet resonances at δ = 0.31 (SiCH3), 0.50 [2J(1H–117/119Sn) 64 Hz, J(1H–117/119Sn) 10 Hz, CH], and 2.61 ppm [3J(1H–117/119Sn) 76 Hz, 4J(1H–117/119Sn) 21 Hz], respectively.17 For the latter signal, the existence of both the 3J and 4J couplings was unambiguously confirmed by a 117Sn decoupling experiment according to which the intensity of both pairs of satellites decreased by roughly 50% (ESI‡). A similar coupling pattern was observed for Me2C(SnMe2SnMe2)2CMe2 with 3J(1H–117/119Sn) 78 Hz and 4J(1H–117/119Sn) 8 Hz.18 The 13C NMR spectrum showed four resonances at δ = 4.3 [1J(13C–29Si) 51 Hz, SiCH3], 10.4 [1J(13C–29Si) 41 Hz, 1J(13C–117/119Sn) 171 Hz, 2J(13C–117/119Sn) 106 Hz, SiCH], 37.3 [2J(13C–117/119Sn) 92 Hz, CCH3], and 73.6 ppm [2J(13C–117/119Sn) 79 Hz, 1J(13C–117/119Sn) 101 Hz, CCH3].17 The coupling pattern and satellite-to-signal-to-satellite intensity distribution of the signal for the quaternary carbon atoms fits well with a simulated AX2X2′-type spectrum (A = C; X, X′ = Sn) (ESI‡).
The reaction of 3 in C6D6 with elemental oxygen (from air) was monitored by 119Sn NMR spectroscopy and revealed, via stepwise formation of organotin suboxides with Sn:O ratios of 4:2 (Sn4O2, 4a/4c), 4:3 (Sn4O3, 5) the adamantane-like diorganotin oxide [{R(O)Sn}2CMe2]2 (4b, R = (Me3Si)2CH)10 as final oxidation product (see ESI‡). The assignments are mainly based on number-of-signals- and coupling pattern-arguments. In addition, NMR spectra of crystals obtained along this route and which had not been subjected to X-ray diffraction showed to be a 50:50 mixture of compound 5 and 4b (see ESI‡).
Exposing blue single crystals of 3 that had been covered under oil, to air (Scheme 2) gave colorless crystals of compound 4 on top of the blue crystals of compound 3. X-ray diffraction analysis of the colorless crystals revealed these being monoclinic and not tetragonal like the previously reported adamantane-type diorganotin oxide 4b.10
 | ||
| Scheme 2 The reaction of 3 with elemental oxygen, O2, giving 4. | ||
A single crystal of better quality of compound 4 was also obtained from a toluene solution of compound 3 to which air had diffused into.
X-ray diffraction analysis of this crystal proved it being a co-crystallizate consisting of a 50:50 mixture of a diorganotin suboxide 4a, and the above-mentioned diorganotin oxide 4b being a result of the O(1) position that is only 50% occupied. The molecular structure of 4a is shown in Fig. 3 and that of 4b is given in the ESI.‡ The geometric data of the latter are close to the one published.10 The asymmetric unit of 4 is shown in the ESI.‡
 | ||
| Fig. 3 General view (SHELXTL) of 4a showing 30% probability displacement ellipsoids and the atom-numbering scheme. Generation of the second half of the molecule by symmetry operation C2 (symmetry code −x, y, −z + 1/2). The hydrogen atoms and methyl groups at the silicon atoms are omitted for clarity. Selected interatomic distances (Å): Sn(1′)–Sn(2′) 2.809(6), Sn(1′)–O(2) 1.971(9), Sn(1′)–C(3A) 2.178(8), Sn(1′)–C(10) 2.256(7), Sn(2′)–O(3) 1.956(3), Sn(2′)–C(3) 2.239(4), Sn(2′)–C(20) 2.275(4). Selected interatomic angles (°): C(10)–Sn(1′)–Sn(2′) 140.3(2), O(2)–Sn(1′)–Sn(2′) 92.2(2), Sn(2′)–C(3)–Sn(1B) 96.0(2), C(3A)–Sn(1′)–C(10) 111.2(3). | ||
Compound 4a shows a skeleton that resembles the structure of realgar, As4S4,4b in which the four arsenic atoms are replaced by four SnCH(SiMe3)2 moieties, and the four sulfur atoms by two Me2C units and two oxygen atoms. The Sn(1′) and Sn(2′) atoms are both tetracoordinated and exhibit strongly distorted tetrahedral environments with angles ranging between 92.2(2) (O(2)–Sn(1′)–Sn(2′)) and 140.3(2)° (C(10)–Sn(1′)–Sn(2′)). The Sn(1′)–Sn(2′) distance of 2.809(6) Å is slightly shorter as compared to the shortest of such bonds in compound 3. They are considerably shorter, however, than the Sn–Sn distances in the oxatristannacyclobutane derivative [O{Sn(C6H3Et2-2,6)2}3] (2.918(5)–3.263(5) Å).19 The Sn(1′)–Sn(1B) and Sn(1′)–Sn(2B) distances of 3.282(16) and 3.283(8) Å are similar to the longer distances in compound 3 as well. The Sn(1′)–O(2) (1.971(9) Å) and Sn(2′)–O(3) (1.956(3) Å) distances are as expected and compare well with the distances of 1.926(26) and 1.976(27) Å reported for the four-membered Sn3O compound mentioned above.19
The NPA-charges calculated for the Sn-atoms increase to 1.66–1.69 being in accord with an oxidation at the Sn-atoms. The charges calculated for the bridging carbon atoms decrease slightly to −0.9 and for the exocyclic SnC atoms by to −1.67 also revealing the increased ionic character of the Sn–ligand interaction upon oxidation.
In conclusion, a rare example of a spacer-bridged bis(organostannylene) (3) has been prepared by the reduction of an diorganotin dichloride with the Mg(I) compound (MesNacNacMg)2. The latter proved to be the reagent of choice for such reductions.20 The extremely air-sensitive compound 3 shows different structures in the solid state and in solution with the latter being not elucidated yet. Compound 3 reacts with elemental oxygen giving a mixture of the diorganotin suboxide 4a and the adamantane-shaped diorganotin oxide 4b. Further intermediates of the oxidation were observed by 119Sn NMR spectroscopy. Formally, the compounds 3, 4a, and 4b can be seen as products from the stepwise reactions of a tetraorgano tetrastanna tetrahedrane with dimethyl carbene, Me2C, and elemental oxygen, demonstrating some analogy to the reactivity of white phosphorus, P4. Compound 3 is isostructural with compounds obtained from the reaction of P4 with two molar equivalents of silylene, aluminylene or gallylene.21–23 This view implies that reactions of either yet unknown R4Sn4 or compound 3 with other carbene analogues or transition metal complexes might give novel organotin element clusters. Furthermore, the chemistry of other bicentric tin-containing Lewis acids used for anion complexation24 may be even further extended to produce subvalent compounds.
Notes and references
- (a) M. Bouška, L. Dostal, A. Růžička, L. Beneš and R. Jambor, Chem. – Eur. J., 2011, 17, 450 CrossRef PubMed; (b) M. Henn, M. Schürmann, B. Mahieu, P. Zanello, A. Cinquantini and K. Jurkschat, J. Organomet. Chem., 2006, 691, 1560 CrossRef CAS PubMed; (c) A. V. Zabula and F. E. Hahn, Eur. J. Inorg. Chem., 2008, 5165 CrossRef CAS; (d) M. Bouška, L. Dostal, F. de Proft, A. Růžička, A. Lyčka and R. Jambor, Chem. – Eur. J., 2011, 17, 455 CrossRef PubMed; (e) F. E. Hahn, A. V. Zabula, T. Pape, A. Hepp, R. Tonner, R. Haunschild and G. Frenking, Chem. – Eur. J., 2008, 14, 10716 CrossRef CAS PubMed; (f) A. V. Zabula and F. E. Hahn, Eur. J. Inorg. Chem., 2008, 5165 CrossRef CAS; (g) T. Kuwabara, J. D. Guo, S. Nagase and M. Saito, Angew. Chem., Int. Ed., 2014, 53, 434 CrossRef CAS PubMed; (h) J. Henning and L. Wesemann, Angew. Chem., Int. Ed., 2012, 51, 12869 CrossRef CAS PubMed; (i) S. Krupski, J. V. Dickschat, A. Hepp, T. Pape and F. E. Hahn, Organometallics, 2012, 31, 2078 CrossRef CAS; (j) J. Henning, K. Eichele, R. F. Finke and L. Wesemann, Organometallics, 2014, 33, 3904–3918 CrossRef CAS.
- (a) C. Schrenk and A. Schnepf, Main Group Met. Chem., 2013, 36, 161 CrossRef CAS; (b) A. Schnepf, Chem. Soc. Rev., 2007, 36, 745 RSC; (c) D. Nied and F. Breher, Chem. Soc. Rev., 2011, 40, 3455 RSC.
- (a) Zintl Ions, ed. T. Fässler, Struct. Bond., Springer-Verlag, Berlin Heidelberg, 2011, vol. 140 Search PubMed; (b) S. Scharfe, F. Kraus, S. Stegmaier, A. Schier and T. F. Fässler, Angew. Chem., Int. Ed., 2011, 50, 3630 CrossRef CAS PubMed.
- (a) Z. F. Fard, C. Müller, T. Harmening, R. Pöttgen and S. Dehnen, Angew. Chem., Int. Ed., 2009, 48, 4441 CrossRef CAS PubMed; (b) M. Weidenbruch, A. Stilter, H. Marsmann, K. Peters and H. G. von Schnering, Eur. J. Inorg. Chem., 1998, 1333 CrossRef CAS.
- G. Maier, S. Pfriem, U. Schäfer and R. Matusch, Angew. Chem., Int. Ed. Engl., 1978, 17, 520 CrossRef.
- N. Wiberg, C. M. M. Finger and K. Polborn, Angew. Chem., Int. Ed. Engl., 1996, 32, 1054 CrossRef.
- N. Wiberg, W. Hochmuth, H. Nöth, A. Appel and M. Schmidt-Amelunxen, Angew. Chem., Int. Ed. Engl., 1996, 35, 1333 CrossRef CAS.
- (a) F. Haarmann, D. Gruner, V. Bezugly, H. Rosner and Y. Grin, Z. Anorg. Allg. Chem., 2006, 632, 1423 CrossRef CAS; (b) M. Neumeier, F. Fendt, S. Gärtner, C. Koch, T. Gärtner, N. Korber and R. M. Gschwind, Angew. Chem., Int. Ed., 2013, 52, 4483 CrossRef CAS PubMed; (c) M. Waibel and T. F. Fässler, Z. Naturforsch., 2013, 68b, 732 CrossRef.
- (a) H. Puff, C. Bach, W. Schuh and R. Zimmer, J. Organomet. Chem., 1986, 312, 313 CAS; (b) V. K. Belsky, N. N. Zemlyansky, N. D. Kolosova and I. V. Borisova, J. Organomet. Chem., 1981, 215, 41 CrossRef; (c) C. J. Cardin, D. J. Cardin, M. A. Convery, M. M. Devereux and N. B. Kelly, J. Organomet. Chem., 1991, 414, C9 CrossRef CAS; (d) M. F. Lappert, W.-P. Leung, C. L. Raston and A. J. Thorne, J. Organomet. Chem., 1982, 233, C28 CrossRef CAS; (e) M. F. Lappert, W.-P. Leung, C. L. Raston, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1992, 775 RSC.
- (a) B. Zobel, M. Schürmann, K. Jurkschat, D. Dakternieks and A. Duthie, Organometallics, 1998, 17, 4096 CrossRef CAS; (b) B. Zobel, Dissertation, Universität Dortmund, 1998 Search PubMed.
- M. Herberhold, W. Milius, U. Steffl, K. Vitzithum, B. Wrackmeyer, R. H. Herber, M. Fontani and P. Zanello, Eur. J. Inorg. Chem., 1999, 145 CrossRef CAS.
- C. P. Sindlinger and L. Wesemann, Chem. Sci., 2014, 5, 2739 RSC.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem. – Eur. J., 2010, 16, 938 CrossRef CAS PubMed.
- R. Pöttgen, Z. Naturforsch., 2006, 61b, 677 Search PubMed.
- J. Meunier-Pieret, M. van Meerssche, M. Gielen and K. Jurkschat, J. Organomet. Chem., 1983, 252, 289 CrossRef.
- H. Preut and T. M. Mitchell, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 951 CrossRef.
- The 1H/13C NMR spectra contain impurities belonging to MesNacNac species (see ESI‡).
- B. Fabisch, Dissertation, Universität Dortmund, 1983 Search PubMed.
- C. J. Cardin, D. J. Cardin, M. A. Convery and M. M. Devereux, J. Organomet. Chem., 1991, 411, C3 CrossRef CAS.
- Similarly observed in: A. Sidiropoulos, C. Jones, A. Stasch, S. Klein and G. Frenking, Angew. Chem., Int. Ed., 2009, 48, 9701 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, M. Brym and M. Driess, Angew. Chem., Int. Ed., 2007, 46, 4511 CrossRef CAS PubMed.
- Y. Peng, H. Fan, H. Zhu, H. W. Roesky, J. Magull and C. H. Hughes, Angew. Chem., Int. Ed., 2004, 43, 3443 CrossRef CAS PubMed.
- G. Prabusankar, A. Doddi, C. Gemel, M. Winter and R. A. Fischer, Inorg. Chem., 2010, 49, 7976 CrossRef CAS PubMed.
- N. Chaniotakis, K. Jurkschat, D. Müller, K. Perdikaki and G. Reeske, Eur. J. Inorg. Chem., 2004, 2283 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Junzo Otera on the occasion of his retirement. |
| ‡ Electronic supplementary information (ESI) available: Experimental section and selected NMR spectra, 119Sn NMR-monitored oxidation study, crystallographic and computational details. CCDC 1010294–1010296. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc07417c |
| This journal is © The Royal Society of Chemistry 2015 |