
Silicon carbide coated with TiO2 with enhanced cobalt active phase dispersion for Fischer–Tropsch synthesis†
Yuefeng
Liu
*a,
Ileana
Florea
b,
Ovidiu
Ersen
b,
Cuong
Pham-Huu
*a and
Christian
Meny
b
aInstitut de Chimie et Procédés pour l'Energie, l'Environnement et la Santé (ICPEES), UMR 7515, CNRS-University of Strasbourg (UdS), 25, rue Becquerel, 67087 Strasbourg Cedex 02, France. E-mail: yuefeng.liu@unistra.fr; cuong.pham-huu@unistra.fr
bInstitut de Physique et Chimie des Matériaux de Strasbourg (IPCMS), UMR 7504, CNRS-University of Strasbourg (UdS), 23, rue du Loess, 67034 Strasbourg Cedex 02, France
First published on 3rd November 2014
Abstract
The introduction of a thin layer of TiO2 on β-SiC allows a significant improvement of the cobalt dispersion. This catalyst exhibits an excellent and stable catalytic activity for the Fischer–Tropsch synthesis (FTS) with high C5+ selectivity, which contributes to the development of a new active catalyst family in the gas-to-liquid process.
Fischer–Tropsch synthesis (FTS), which converts synthesis gas (a mixture of CO and H2) derived from coal, natural gas (also shale gas) or biomass into synthetic liquid fuels and chemicals, continues to attract more interest as a result of diminishing petroleum reserves and a quest for a clean fuel.1–3 The FTS reaction is strongly exothermic,4–7i.e. ΔH = −204 kJ molCO−1, and thus a large amount of heat was generated during the course of the reaction. In the case of insulator supports, the heat generated on the catalyst surface leads to the formation of local hot spots on the catalyst surface, which significantly affects the overall selectivity towards liquid hydrocarbon production.
Silicon carbide (β-SiC) has been recently reported to be an efficient support for fixed-bed FTS reaction due to its intrinsic medium thermal conductivity, which facilitates heat dissipation throughout the catalyst body, and thus avoids the formation of local hot spots, which are detrimental for the liquid hydrocarbon selectivity.8–11 The large porosity of the support also allows a rapid escape of the intermediate liquid hydrocarbons and water, and thus significantly contributes to the improvement of the liquid hydrocarbon selectivity as well.12 However, the metal–support interaction between the cobalt phase and the SiC surface is relatively weak, leading to the formation of medium to large cobalt particles, which are less efficient for the FTS reaction. In our previous attempts to improve SiC-based catalyst performance for the FTS reaction, the SiC surface was modified by introducing a dopant, which exhibits a higher metal–support interaction in order to achieve the better dispersion of the deposited cobalt phase.10 However, on such composite supports some large cobalt particles remain in contact with the SiC phase, which could lower the overall FTS performance of the catalyst.11,13 It is of interest to develop a new doping method to homogeneously decorate the SiC surface with a thin layer of TiO2 in order to efficiently disperse the cobalt particles, which, in turn, improves the FTS performance. It is expected that the future development of the FTS requires the introduction of active and stable catalysts. Herein, we report on the development of a highly active and stable FTS catalyst based on titania coated (TS-Cx where x is the TiO2 weight percent) high porosity industrial β-SiC containing well dispersed cobalt particles. The high dispersion of the cobalt particles on the thin TiO2 layer leads to a significant improvement of the catalyst activity for the FTS reaction compared to the undoped SiC and TiO2 doped SiC (TS-D) catalysts. To the best of our knowledge, the FTS activity of this SiC-based catalyst is the most active one among the different cobalt-based catalysts, either pure or doped with trace amounts of noble metal.
SiC mostly consists of a large amount of meso- and macropores (Fig. 1a). The transformation of the titanium precursor, deposited through an incipient wetness method on the SiC surface, into the TiO2 crystalline phase is performed by calcination of the sample in air at 600 °C for 5 h. The obtained TiO2 layer is well crystallized in a single anatase phase as confirmed by XRD and XPS analyses (Fig. S1 and S2, ESI†).14,15 The specific surface area (SBET) of the TiO2 coated SiC is in the range of 39–41 m2 g−1 (Table S1, ESI†), which is close to the pure SiC (40 m2 g−1). Such results could be attributed to the meso- and macropores of the support, which contribute to the homogeneous coating of the TiO2 layer on the SiC surface. The pore size distribution curves in the range of 2–150 nm derived from adsorption branches of the N2 isotherms using the BJH method are presented in Fig. 1b. TiO2 coated SiC and SiC supports display irregular and broad pore size distribution, which is different from those reported for SiO2 and Al2O3 supports.16,17 The TEM and energy filtered TEM (EFTEM) of the TiO2 coated SiC composite presented in Fig. 1c and d clearly show the presence of a thin and homogeneous TiO2 layer covering the surface of the SiC support. The similar trends of morphologies and elemental distribution (Fig. S3, ESI†) confirm the homogeneous coating of the support. The specific surface area of the catalyst, after depositing 10 wt% of cobalt, remains unchanged (ca. 40 m2 g−1) (Table 1), which indicates that the cobalt phase is relatively well dispersed on the support surface and no pore plugging is encountered.
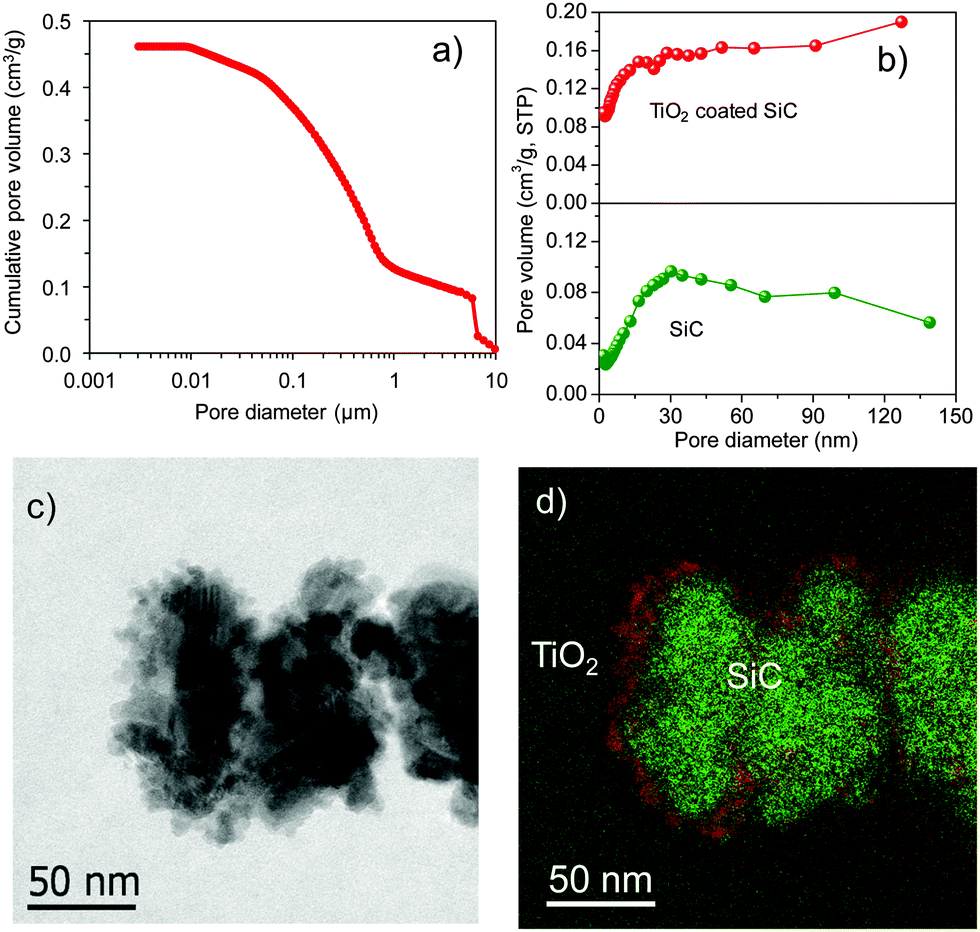 | ||
| Fig. 1 Characterization of the TiO2 coated SiC (TS-C10) support. (a) Pore size distributions measured by mercury intrusion of macroporous SiC sample, (b) pore size distribution using the BJH method of SiC and TiO2 coated SiC. (c) 2D TEM image of TiO2 coated SiC and (d) the corresponding EFTEM image of the classical 2D TEM map, which clearly shows the well distribution of TiO2 on the topmost surface of SiC (TiO2 (red), SiC (green)). | ||
| Sample | S BET (m2 g−1) | V total (cm3 g−1) | d 0Co (nm) | X CO (%) | Sel. (%) | CoTYc | α | |||
|---|---|---|---|---|---|---|---|---|---|---|
| CH4 | CO2 | C2–4 | C5+ | |||||||
| a All data were collected after 20 h time-on stream with stable catalytic performance. Reaction conditions: Co loading = 10 wt%, H2/CO = 2, 225 °C, 40 bar. b The particle size was calculated based on the 59Co NMR results of the cobalt atom fraction engaged in the different blocking temperature ranges. c Cobalt time yield (CoTY, 10−5 molCo gCo−1 s−1, molar CO conversion rate per gram of Co per hour). d Chain growth probability factor (α). e 10CS: 10 wt% of cobalt on SiC; 10CTS-D: 10 wt% of cobalt on SiC doped with TiO2 (with 17 wt% loading), gas velocity of 60 mL gcat−1 min−1. f SiC doped with a TiO2 supported catalyst, data from ref. 10. g SiC coated with a TiO2 supported catalyst (10 wt% of cobalt on SiC coated with 10 wt% TiO2). Gas velocity of 80 mL gcat−1 min−1. | ||||||||||
| 10CSe | 33 | 0.14 | 20 | 32.3 | 4.5 | 0 | 2.4 | 93.1 | 4.8 | 0.92 |
| 10CTS-De,f | 25 | 0.08 | 18 | 43.4 | 4.5 | 0 | 1.9 | 93.6 | 6.5 | 0.92 |
| 10CTS-C10g | 40 | 0.11 | 10 | 61.0 | 5.8 | 0.1 | 2.9 | 91.2 | 12.1 | 0.91 |
The TEM and the corresponding energy-dispersive X-ray (EDX) spectra of 10CoTS-C10 in different areas of the sample are presented in Fig. S4 (ESI†). The EDX spectra reveal that cobalt and titanium are localized in the same area, confirming the fact that cobalt is well dispersed on the surface of TiO2. Next, nil field spin-echo 59Co NMR is performed to understand the relationship between the structures of active sites and FTS activity. Fig. 2a and b show the 59Co NMR spectra recorded on cobalt supported on SiC (10CS) and TiO2 coated SiC (10CTS-C10) at 3 different temperatures. A significant contribution can be observed at 217 MHz in the NMR spectrum of the 10CS catalyst, showing the presence of a large number of Co atoms forming large face centered cubic (fcc) particles with a size larger than 60 nm.9,10,18,19 Such a contribution is not observed in Fig. 2b showing that the Co particles are significantly smaller in the presence of a TiO2 coated catalyst. From the temperature dependent NMR spectra, the fraction of cobalt atoms with different blocking temperature ranges (equivalent to different size ranges) as well as the relative percentage of cobalt nanoparticle (Co NP) sizes and the surface area are presented in Fig. 2c–e. On the other hand, on the 10CTS-C10 catalyst, 74% of cobalt atoms are engaged in the smallest cobalt particles (<10 nm, blocking temperature 2–4.2 K and 4.2–77 K), which is significantly higher than those of 10CS (28%) and 10CTS-D (37%) catalysts. It is expected that the high FTS performance (Table 1) obtained on this TiO2-coated catalyst is directly linked to the presence of small cobalt particles in strong interaction with the TiO2 coating layer.20,21 The average crystallite size of cobalt is about 20 nm for the 10CS catalyst, and about 10 nm for the 10CTS-C10 catalyst (Table 1), the latter being predicted to be the most active particle size for the FTS process (see discussion below).22 According to such results one can state that the introduction of the TiO2 phase on the SiC matrix significantly decreases the particle size of cobalt, probably by generating a higher chemical interaction with the metal salt precursor without modifying the reduction behavior of the sample. Such evidence is also found in the carbon-based supported cobalt catalyst.23
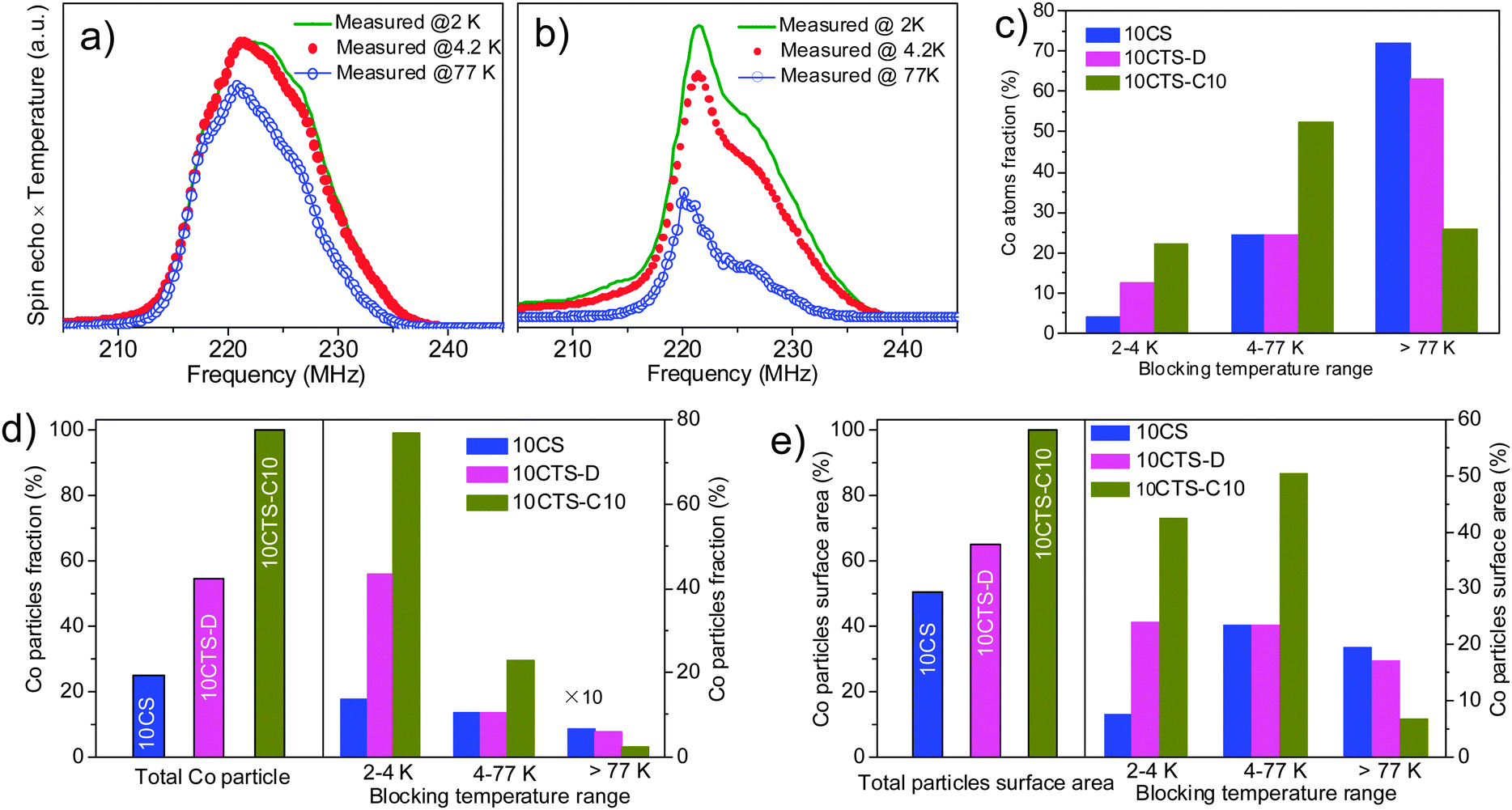 | ||
| Fig. 2 59Co NMR spectra of the (a) 10CS and (b) 10CTS-C10 catalysts after FTS test covered with solid wax. Recorded temperatures are 2, 4.2 and 77 K. (c) Co fraction atoms engaged in the different blocking temperature ranges (size ranges). (d) The total number of cobalt particles (left panel) and distribution of cobalt particles as a function of the blocking temperature (right panel) produced in the samples. (e) Total Co particle surface area (left panel) and the relative particle surface area as a function of the blocking temperature ranges (right panel). The data in (d) and (e) are normalized to the sample showing the largest available number of particles and surface area (assuming the same initial atomic cobalt content), respectively. | ||
The 10CS (cobalt on SiC), 10CTS-D (cobalt on SiC doped with TiO2) and 10CTS-C10 (10 wt% cobalt on SiC coated with 10 wt% TiO2) catalysts have been tested in FTS reaction, and the catalytic results are listed in Table 1. The doping or coating of SiC leads to a considerable improvement of the FTS performance compared to that of un-doped one (Table 1). The CO conversion increases from 32.3% to 61.0% after coating a layer of TiO2 (10 wt%) on the SiC support. The increase in the FTS catalytic activity is only accompanied by a slight decrease in selectivity for liquid hydrocarbons (C5+ selectivity of the 10CTS-C catalyst remains as high as 91%). The selectivity of CO2 for the tested catalysts is less than 0.2% (Table 1). It could be concluded that the water–gas shift reaction is not a side reaction over such cobalt-based catalysts. The carbon balance of the different experiments is >90 ± 4 wt%, which can be ascribed to the difference between the theoretical and experimental carbon balance with the error margin of the CO outlet flow rate and the liquid and solid hydrocarbon recovery. The relatively low carbon balance in the present work is due to the loss of the C6–C8 fractions (estimated to be about 5 wt%) during the heating of the trap according to our previous studies. It is thus expected that the C5+ selectivity will be even higher than the one reported if all the C6–C8 fractions are included in the calculation. The chain growth factor (α) determined on various tested catalysts (Table 1), which is calculated by using the Anderson–Schulz–Flory (ASF) model,3 is relatively high on the TiO2 coated SiC catalysts (0.91).
The different TiO2 mass loading (from 0 to 15 wt%) is also investigated, and results (Fig. S5 and S6, ESI†) show that the FTS rate increases significantly with an increase in TiO2 from 0 to 10wt%. However, the FTS rate goes down when the TiO2 mass loading is up to 15 wt%, which could be attributed to the stronger metal–support interaction between the TiO2 and the cobalt precursor as the thicker TiO2 layer is introduced (XRD results in Fig. S5, ESI†). It is also worth noting that the catalyst exhibits a relatively high stability and no deactivation was observed under the operating reaction conditions.
Moreover, one should expect that the FTS activity is mostly related to the amount of exposed cobalt surface area rather than to the distribution of cobalt atoms. It is known that cobalt surface active sites (atom surface area) are most probably activating the reaction by direct contact with the reactant molecules. As can be seen in Fig. 2e, the 10CTS-D catalyst, for example, contains 60% of cobalt atoms forming large particles (measured blocking temperature higher than 77 K with particle diameter ranging from 10 nm to 30 nm or even more) but it only represents less than 2% in terms of particle fraction, or only offers less than 20% of the total atom surface (active sites). It means that small particles (<10 nm) play the key role in FTS activity improvement. In the 10CTS-C10 catalyst, about 80% of cobalt atoms formed >90% of the fraction of the particles in the range of 3–4 nm and 4–10 nm, which contribute more than 90% to the particle surface area (3–4 nm and 4–10 nm offered 43% and 50% of atom surface area fraction). According to our NMR results, we speculate that the small particles (3–10 nm) have multiple active sites depending on the preparation and materials used as supports and are directly involved in the improvement of the FTS catalytic performance.24 These results are different from the results reported previously on cobalt supported on carbon nanofibers where low FTS activity is observed when the cobalt NP size is smaller than 8 nm.21,22,25 The peculiar interactions between the cobalt particles and the prismatic planes of the CNFs could be advanced to explain such a discrepancy.
We also ran the experiment using the cobalt loading up to 30 wt% under the realistic FTS reaction conditions (240 °C, 40 bar, and at a relatively high space velocity of 320 mL gcat−1 min−1). According to the results the thermal conductor TiO2–SiC-based catalyst exhibits a high and stable FTS performance for about 100 h on stream (Fig. 3). The specific rate is about 1.22 gC5+ gcatalyst−1 h−1 with a relatively high C5+ selectivity, i.e. 85.8% and a high chain-growth factor (Table S2 and Fig. S7, ESI†), which confirms the potential industrial interest of this catalyst in the FTS reaction.
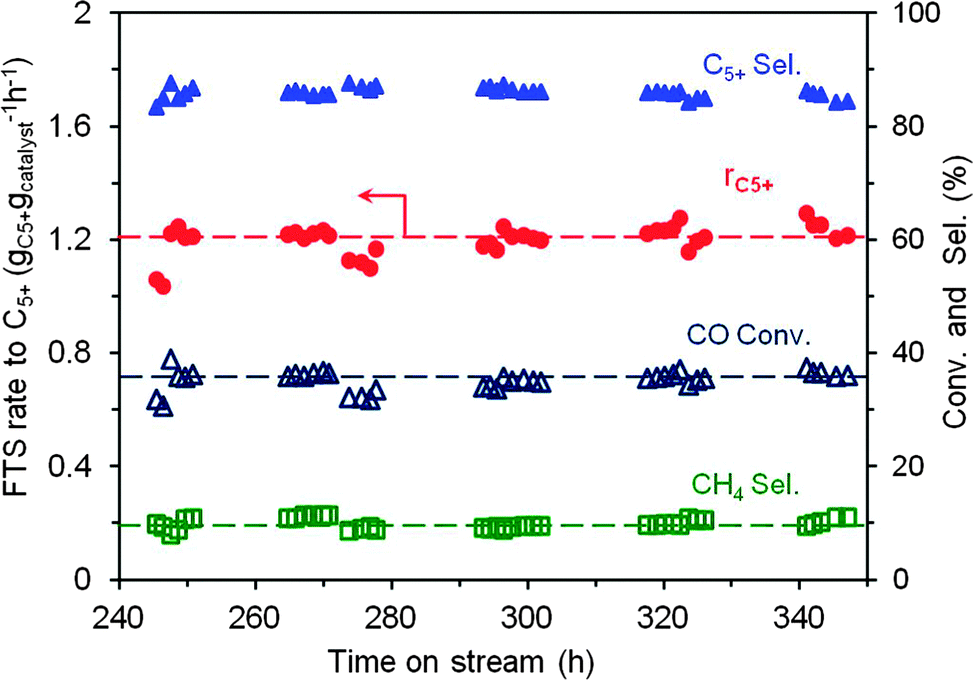 | ||
| Fig. 3 FTS rate and product selectivity as a function of time on stream on the 30CTS-C10 sample. The catalyst has already been tested in the FTS reaction under different conditions during 240 h on stream before performing this test. | ||
In conclusion, the introduction of a thin coated layer of TiO2 on the SiC surface significantly improves the metal–support interaction between the cobalt precursor and the support leading to a better dispersion of the cobalt active phase. The catalysts, either with low (10 wt%) or high (30 wt%) cobalt loading, exhibit a high and stable FTS activity and long chain hydrocarbon selectivity along with a long-term stability. The high dispersion of the deposited cobalt particles leading to the formation of small cobalt particles is advanced to explain the high FTS performance of the catalyst while the medium thermal conductivity of the support is expected to be at the origin of a high C5+ selectivity even at a relatively high reaction temperature. The meso- and macroporosity of the support is also responsible for the long-term stability of the catalyst by reducing the problem of pore plugging by liquid hydrocarbons produced during the reaction. The combination of the catalytic and the NMR results allows us to clearly evidence a direct relationship between the cobalt particle size and the FTS performance. It is expected that this catalyst could significantly promote the development of the FTS in the near future where the supply of high quality liquid fuels becomes the core process in several sectors.
The authors would like to thank SICAT Co. (http://www.sicatcatalyst.com) for offering SiC materials. Y. F. Liu thanks the grant from China Scholarship Council (CSC). F. Vigneron, T. Romero and P. Bernhardt are gratefully acknowledged for experimental assistance and SEM/XPS analysis.
Notes and references
- J. R. Rostrup-Nielsen, Science, 2005, 308, 1421–1422 CrossRef CAS PubMed.
- G. M. Whitesides and G. W. Crabtree, Science, 2007, 315, 796–798 CrossRef CAS PubMed.
- H. M. T. Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Science, 2012, 335, 835–838 CrossRef PubMed.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS PubMed.
- E. Iglesia, Appl. Catal., A, 1997, 161, 59–78 CrossRef CAS.
- E. de Smit and B. M. Weckhuysen, Chem. Soc. Rev., 2008, 37, 2758–2781 RSC.
- Q. Zhang, J. Kang and Y. Wang, ChemCatChem, 2010, 2, 1030–1058 CrossRef CAS.
- A. R. de la Osa, A. De Lucas, J. Diaz-Maroto, A. Romero, J. L. Valverde and P. Sanchez, Catal. Today, 2012, 187, 173–182 CrossRef CAS PubMed.
- B. de Tymowski, Y. F. Liu, C. Meny, C. Lefevre, D. Begin, P. Nguyen, C. Pham, D. Edouard, F. Luck and C. Pham-Huu, Appl. Catal., A, 2012, 419, 31–40 CrossRef PubMed.
- Y. Liu, B. de Tymowski, F. Vigneron, I. Florea, O. Ersen, C. Meny, P. Nguyen, C. Pham, F. Luck and C. Pham-Huu, ACS Catal., 2013, 3, 393–404 CrossRef CAS.
- Y. Liu, O. Ersen, C. Meny, F. Luck and C. Pham-Huu, ChemSusChem, 2014, 7, 1218–1239 CrossRef CAS PubMed.
- J. Kang, K. Cheng, L. Zhang, Q. Zhang, J. Ding, W. Hua, Y. Lou, Q. Zhai and Y. Wang, Angew. Chem., Int. Ed., 2011, 50, 5200–5203 CrossRef CAS PubMed.
- I. Florea, Y. F. Liu, O. Ersen, C. Meny and C. Pham-Huu, ChemCatChem, 2013, 5, 2610–2620 CrossRef CAS.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- Y. Wang, H. J. Sun, S. J. Tan, H. Feng, Z. W. Cheng, J. Zhao, A. D. Zhao, B. Wang, Y. Luo, J. L. Yang and J. G. Hou, Nat. Commun., 2013, 4, 2214, DOI:10.1038/ncomms3214.
- J. P. Hong, W. Chu, P. A. Chernavskii and A. Y. Khodakov, Appl. Catal., A, 2010, 382, 28–35 CrossRef CAS PubMed.
- O. Borg, S. Erib, E. A. Blekkan, S. Storsaeter, H. Wigum, E. Rytter and A. Holmen, J. Catal., 2007, 248, 89–100 CrossRef CAS PubMed.
- C. Meny and P. Panissod, in Nuclear Magnetic Resonance in Ferromagnetic Multilayers and Nanocomposites: Investigations of their Structural and Magnetic Properties, ed. G. A. Webb, Modern Magnetic Resonance, Springer, Heidelberg, Germany, 2006, pp. 1493–1498 Search PubMed.
- P. Panissod and C. Meny, Appl. Magn. Reson., 2000, 19, 447–460 CrossRef CAS.
- W. Chu, L. N. Wang, P. A. Chernavskii and A. Y. Khodakov, Angew. Chem., Int. Ed., 2008, 47, 5052–5055 CrossRef CAS PubMed.
- J. P. den Breejen, P. B. Radstake, G. L. Bezemer, J. H. Bitter, V. Froseth, A. Holmen and K. P. de Jong, J. Am. Chem. Soc., 2009, 131, 7197–7203 CrossRef CAS PubMed.
- G. L. Bezemer, J. H. Bitter, H. P. C. E. Kuipers, H. Oosterbeek, J. E. Holewijn, X. D. Xu, F. Kapteijn, A. J. van Dillen and K. P. de Jong, J. Am. Chem. Soc., 2006, 128, 3956–3964 CrossRef CAS PubMed.
- Y. Liu, J. Luo, M. Girleanu, O. Ersen, C. Pham-Huu and C. Meny, J. Catal., 2014, 318, 179–192 CrossRef CAS PubMed.
- Z. J. Wang, S. Skiles, F. Yang, Z. Yan and D. W. Goodman, Catal. Today, 2012, 181, 75–81 CrossRef CAS PubMed.
- G. Prieto, A. Martinez, P. Concepcion and R. Moreno-Tost, J. Catal., 2009, 266, 129–144 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization techniques such as XRD, XPS, SEM-EDX, TEM-EDX and FTS results (Fig. S1–S7 and Tables S1 and S2). See DOI: 10.1039/c4cc07469f |
| This journal is © The Royal Society of Chemistry 2015 |