
Ammonium iodide-induced sulfonylation of alkenes with DMSO and water toward the synthesis of vinyl methyl sulfones†
Xiaofang
Gao
,
Xiaojun
Pan
,
Jian
Gao
,
Huawen
Huang
,
Gaoqing
Yuan
* and
Yingwei
Li
*
School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou, 510640, P. R. China. E-mail: gqyuan@scut.edu.cn; liyw@scut.edu.cn
First published on 4th November 2014
Abstract
A novel ammonium iodide-induced sulfonylation of alkenes with DMSO and water toward the synthesis of vinyl methyl sulfones is described. The process proceeded smoothly under metal-free conditions with high stereoselectivity and good functional group tolerance. The reaction mechanism was revealed to proceed through a domino reaction of oxidation and elimination after the radical addition to alkenes.
Vinyl sulfones are important compounds in organic and medicinal chemistry due to their versatile reactivities and potential biological activities.1 In the last few decades, the synthesis of vinyl sulfones has attracted more and more attention from chemists.2–6 Traditional pathways for the preparation of vinyl sulfones typically include the direct oxidation of vinyl sulfides and Heck reaction of aryl halides with vinyl sulfones.2,3 However, these methods suffer from poor selectivity or expensive starting materials. In recent years, some efficient metal-catalyzed approaches have been investigated such as cross-coupling reactions of olefins and their derivatives (vinyl halides, stannanes, and arylboronic acids) with sulfone reagents including sulfinate salts, sulfonyl chlorides, or sulfonyl hydrazides (Scheme 1a).4 Recently, some researchers have achieved this transformation in the absence of transition-metal catalysts.5 Despite remarkable improvement and efficiency of such reactions, the discovery of sulfone sources from inexpensive and readily available reagents is highly desired. Very recently, Loh and co-workers published an alternative Cu-catalyzed methyl sulfonylation of alkynes using DMSO and O2 as a methyl sulfone source (–SO2Me) (Scheme 1b).6 Herein, we disclose a novel and efficient ammonium iodide-induced sulfonylation of alkenes in which DMSO and H2O are used as the starting materials to provide the source of –SO2Me (Scheme 1c). To the best of our knowledge, such a route for the synthesis of vinyl methyl sulfones has not been reported under metal free conditions.
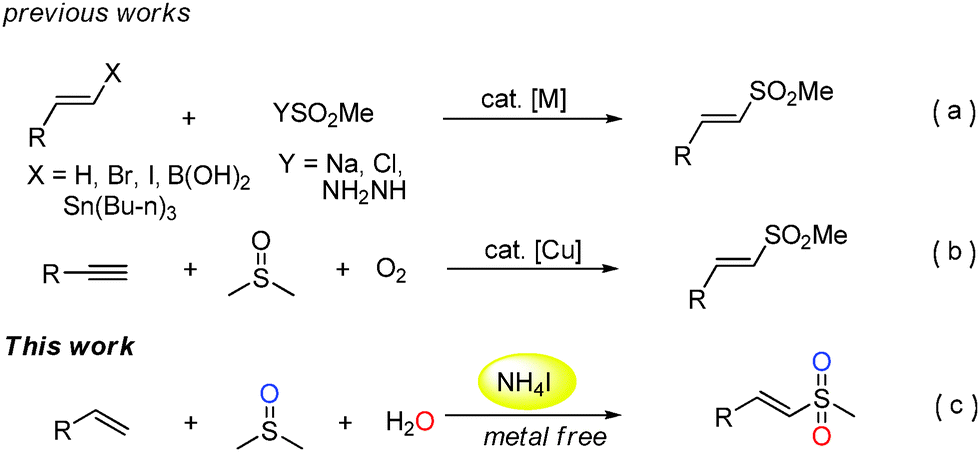 | ||
| Scheme 1 Different routes for the synthesis of vinyl methyl sulfones. | ||
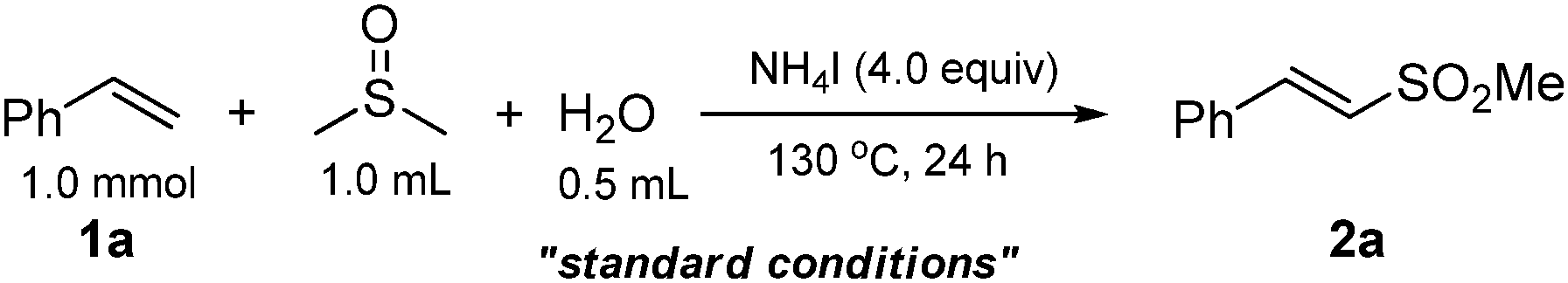
Using styrene (1a) and DMSO as the substrates, optimal conditions were first examined for the sulfonylation. After screening a variety of reaction parameters, the sulfonylation of 1a gave 2a in an excellent yield with sole stereoselectivity under the standard conditions (Table 1, entry 1: 4.0 equiv. NH4I, 0.5 mL H2O, 1.0 mL DMSO, at 130 °C for 24 h; optimization details, see ESI†). The effects of the other factors on the reaction are shown in entries 2–13. Neither the organic tetrabutylammonium iodide (n-Bu4NI) nor the inorganic KI did afford the desired product (entries 2 and 3), suggesting that the iodide anion is not the real form for NH4I participated in this conversion. No desired transformation was observed employing a similar ammonium salt NH4Br (entry 4), ruling out the function of NH4+ in this reaction. When using 2.0 equiv. of I2 instead of 4.0 equiv. of NH4I, 2a was not obtained with only benzoic acid as the product, whereas 15% yield of 2a was determined with 0.5 equiv. of I2 (entries 5 and 6). In addition, when the reaction was performed with aqueous ammonia, the yield of the product was not improved (entry 7). However, in the presence of catalytic amounts of I2, no desired product was obtained only recovering the unreacted styrene (entry 8). These results clearly indicated that I2 was indeed the actual form to promote this conversion and its concentration had a great impact on the selectivity of reaction. It was also observed that both the amount of NH4I added and reaction temperature were important parameters for a high yield (entries 10–12). In the absence of water, the yield of the target product dramatically decreased (entry 13).
|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yielda/% |
| a Determined by 1H NMR (internal standard: 1,3,5-trimethyl benzene). b Run for 32 h. c 1.0 equiv. of radical trapper was used. | ||
| 1 | None | 98 |
| 2 | Bu4NI instead of NH4I | 0 |
| 3 | KI instead of NH4I | 0 |
| 4 | NH4Br instead of NH4I | 0 |
| 5 | 2.0 equiv. of I2 instead of NH4I | 0 |
| 6 | 0.5 equiv. of I2 instead of NH4I | 15 |
| 7 | 0.5 equiv. of I2 + 0.5 mL NH3·H2O instead of NH4I | 17 |
| 8 | 10 mol% I2 instead of NH4I | 0 |
| 9 | HI instead of NH4I | 8 |
| 10 | 1.0 equiv. of NH4I | 0 |
| 11 | 100 °C instead of 130 °C | 0 |
| 12 | 120 °C instead of 130 °C | 39 |
| 13 | Without adding H2O | 12 |
| 14b | Under N2 atmosphere | 91 |
| 15c | Adding TEMPO | 0 |
| 16c | Adding BHT | 0 |
With the optimal conditions in hand, we next evaluated the substrates scope and limitations of our protocol (Tables 2 and 3). In general, the reactions of alkenes bearing electron-withdrawing groups gave slightly higher yields of product than those containing electron-donating moieties, as well as much faster reaction rates (2a–2n). All of the reactions proceeded with excellent regioselectivity, and various functional groups such as methyl, methoxyl, fluoro, chloro, bromo, ester, cyanide, nitro, and trifluoromethyl groups at the meta or para positions of the aromatic ring were reasonably well tolerated under the standard conditions. The reaction of disubstituted alkenes was also efficient (2o, 2q). Polyaryl substrates such as 2-naphthalene and 9-phenanthrene retarded the reaction even after an extended reaction time, albeit in moderate yields (2s–2t), possibly due to the steric interference. In addition, heteroaromatic olefins proceeded smoothly to afford the target products in good yields (2r, 2u). Gratifyingly, interminal alkenes were also productive, exclusively forming (E)-sulfonyl product (2v) either from the cis- or trans-β-methylstyrene. However, allylbenzene failed to give the corresponding product (2w), and a similar result was also observed with alkyl alkenes as substrates. In contrast conjugated diene afforded the target product in a moderate yield (2x), indicating that conjugative effect played an important role in stabilizing the intermediate.7
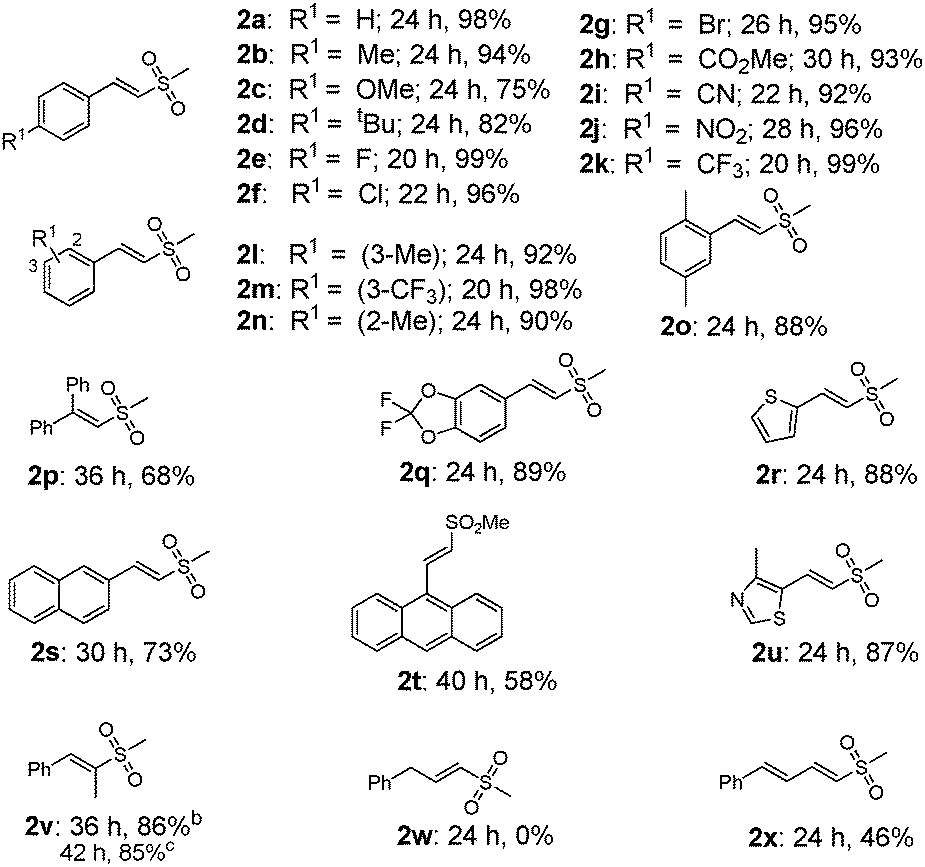
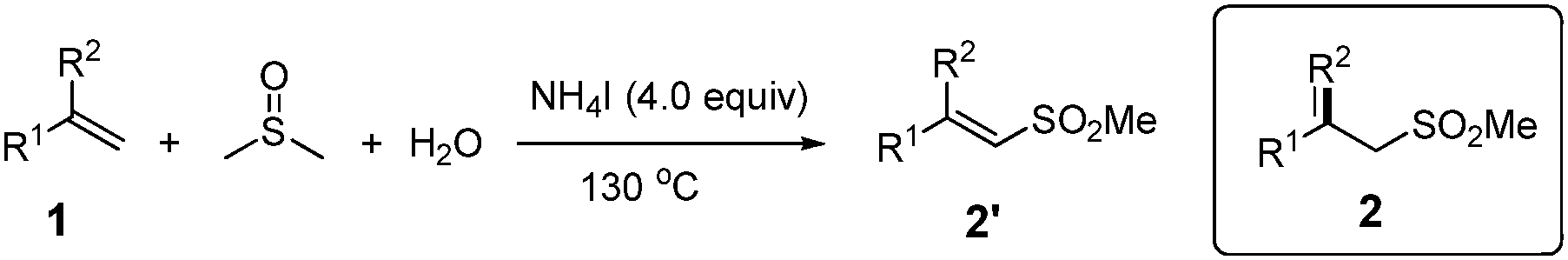
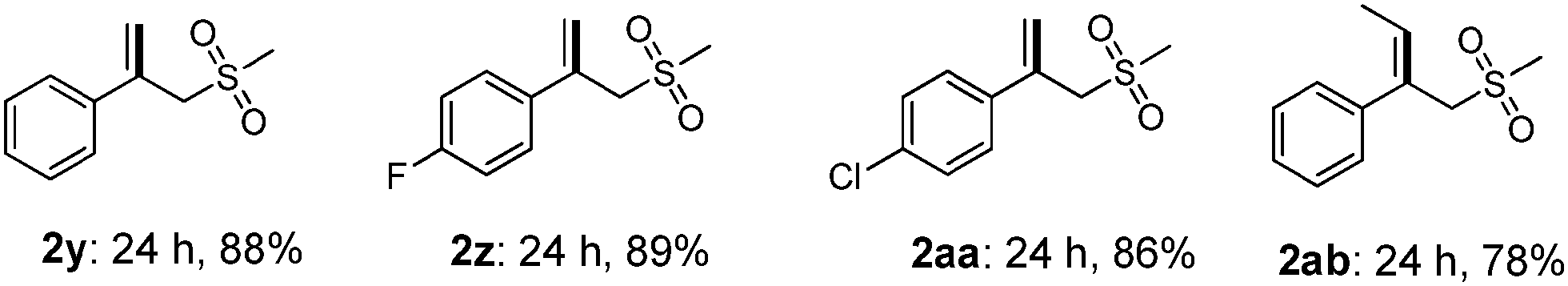
Interestingly, when α-methylstyrene (1y) was applied as the substrate, an isomerization product (2y) was obtained instead of the desired vinyl sulfone (Table 3). Other similar olefins were tested under the standard conditions, producing the isomerized products in good yields (2z–2ab) as well. These results suggested that a different elimination protocol existed for 1,1-disubstituted olefins. This isomerization result is consistent with the previous studies by Jiang4b and Li.5c
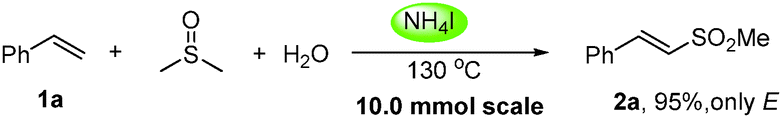
Moreover, the reaction is scalable and practical as an excellent yield (95%) of 2a was obtained when the reaction was performed on a 10.0 mmol scale (eqn (1)).8
 | (1) |
 | (2) |
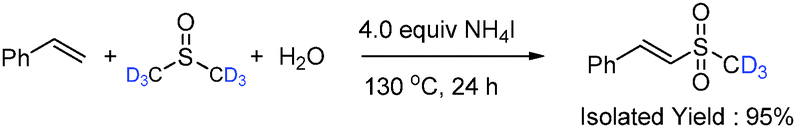 | (3) |
In order to investigate the reaction mechanism, a series of control experiments were carried out. Under the protection of N2, the target product 2a could be obtained in an excellent yield (Table 1, entry 14), indicating that the oxygen atom of the sulfone group in 2a does not come from O2. The isotopic labeling experiment with H218O was conducted (eqn (2)).8 These experimental results make sure that one oxygen atom of the sulfone group of 2a is from H2O and the other results from DMSO. The deuterium labeling experiment with DMSO-d6 (eqn (3)) confirms that methylthiyl (MeS) originates from DMSO. Furthermore, a reaction profile was obtained in the sulfonylation of styrene (1a) under the standard conditions (Fig. 1). Within a reaction time of 4 h, 2-(methylthio)-1-phenylethanol (3a) was firstly detected in 20% yield, and then 2-(methylsulfonyl)-1-phenylethanol (4a) and (E)-(2-(methylsulfonyl)vinyl)benzene (2a) were produced. During the reaction process, 3a and 4a remained in a low concentration in the reaction mixture and disappeared at the end of the reaction. Simultaneously, the concentration of 2a increased with the reaction time, thus implying that 3a and 4a were the intermediates in this conversion. Indeed, discrete 3a and 4a both could be completely converted to 2a under the otherwise identical conditions.8 In addition, when the reaction was performed in the presence of a stoichiometric amount of TEMPO or BHT under the standard conditions, 2a was not obtained (Table 1, entries 15 and 16). These results suggested that the present reaction presumably proceeded through a radical pathway.
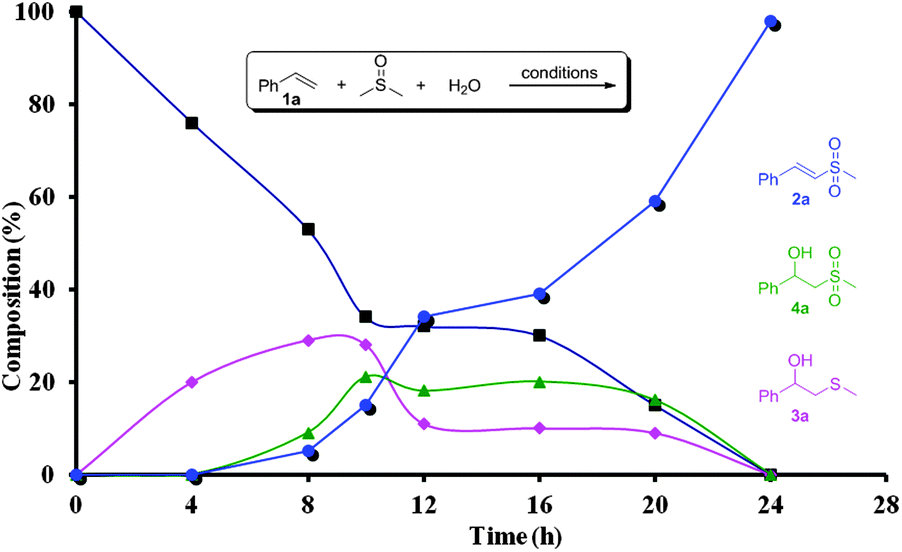 | ||
| Fig. 1 Reaction profile in the NH4I-induced sulfonylation of 1a. | ||
Based on the above results, a plausible reaction mechanism is proposed in Scheme 2. First, the radical initiator I2 and precursor MeSH are generated through a series of reactions, as shown in eqn (4)–(6).9–11 Then, I2 is decomposed to form an iodine radical (eqn (7)),12 which attacks on MeSH to give a methylthiyl radical MeS˙ (eqn (8)).13 Subsequently, the radical MeS˙ is added to the alkene to generate a radical intermediate I.14 At the same time, the MeS˙ abstracts hydrogen from water to afford a hydroxyl radical (OH˙). The rapid coupling reaction between the radical intermediate I and OH˙ produces intermediate 3a, which is further oxidized by DMSO15 and H2O2 resulting from self-coupling of the radical OH˙,16 to form intermediate 4a. Finally, with the assistance of the newly formed sulfone group, 2a is generated via acid-catalysed dehydration of 4a.
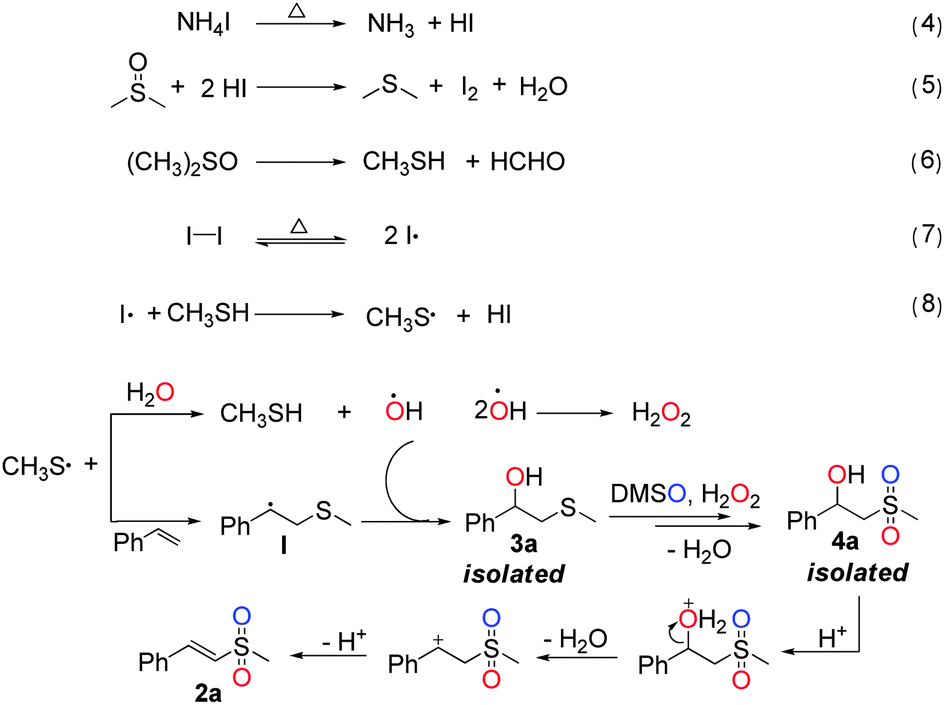 | ||
| Scheme 2 Proposed reaction mechanism. | ||
In conclusion, we have developed a novel and efficient protocol of ammonium iodide-induced sulfonylation of olefins with DMSO and water, achieving excellent stereoselectivity and high functional group tolerance. In addition, this procedure provides a new valuable method of generating a methylthiyl radical (MeS˙) from DMSO. Further studies on the details of the reaction mechanism and applications in organic synthesis are currently underway in our laboratory.
We thank the National Natural Science Foundation of China (21172079, 21322606, and 21436005), and Guangdong Natural Science Foundation (10351064101000000) for financial support.
Notes and references
- For reviews, see: (a) B. A. Frankel, M. Bentley, R. G. Kruger and D. McCafferty, J. Am. Chem. Soc., 2004, 126, 3404 CrossRef CAS PubMed; (b) M. V. Reddy, N. M. Iqbal, K. A. Robell, A. D. Kang and E. P. Reddy, J. Med. Chem., 2008, 51, 86 CrossRef CAS PubMed; (c) J.-N. Desrosiers and A. B. Charette, Angew. Chem., Int. Ed., 2007, 46, 5955 CrossRef CAS PubMed.
- (a) M. Tiecco, L. Testaferri, M. Tingoli, D. Chianelli and M. Montanucci, J. Org. Chem., 1983, 48, 4795 CrossRef CAS; (b) P. Caramella, E. Albini, T. Bandiera, A. C. Coda, P. Grünanger and F. M. Albini, Tetrahedron, 1983, 39, 689 CrossRef CAS.
- A. Battace, T. Zair, H. Doucet and M. Santelli, Synthesis, 2006, 3495 CAS.
- (a) N. Taniguchi, Synlett, 2011, 1308 CrossRef CAS PubMed; (b) X. Li, Y. Xu, W. Wu, C. Jiang, C. Qi and H. Jiang, Chem. – Eur. J., 2014, 20, 1 CrossRef CAS PubMed; (c) M. Bian, F. Xu and C. Ma, Synthesis, 2007, 2951 CAS; (d) S. S. Labadie, J. Org. Chem., 1989, 54, 2496 CrossRef CAS; (e) F. Huang and R. A. Batey, Tetrahedron, 2007, 63, 7667 CrossRef CAS PubMed; (f) A. Kar, I. A. Sayyed, W. F. Lo, H. M. Kaiser, M. Beller and M. K. Tse, Org. Lett., 2007, 9, 3405 CrossRef CAS PubMed.
- (a) S. Tang, Y. Wu, W. Liao, R. Bai, C. Liu and A. Lei, Chem. Commun., 2014, 50, 4496 RSC; (b) Y. Xu, X. Tang, W. Hu, W. Wu and H. Jiang, Green Chem., 2014, 16, 3720 RSC; (c) X. Li, X. Xu and C. Zhou, Chem. Commun., 2012, 48, 12240 RSC; (d) S. Liang, R. Zhang, G. Wang, S. Chen and X. Yu, Eur. J. Org. Chem., 2013, 7050 CrossRef CAS; (e) P. Katrun, S. Chiampanichayakul, K. Korworapan, M. Pohmakotr, V. Reutrakul, T. Jaipetch and C. Kuhakarn, Eur. J. Org. Chem., 2010, 5633 CrossRef CAS.
- Y. Jiang and T.-P. Loh, Chem. Sci., 2014, 5, 4939 RSC.
- B. Giese, Angew. Chem., Int. Ed., 1983, 22, 753 CrossRef.
- See the ESI† for details.
- A. Smith and R. P. Calvert, J. Am. Chem. Soc., 1914, 36, 1363 CrossRef CAS.
- H. Gilman and J. Eisch, J. Am. Chem. Soc., 1955, 77, 3862 CrossRef CAS.
- V. J. Traynelis and W. L. Hergenrother, J. Org. Chem., 1964, 29, 221 CrossRef CAS.
- J. Gromada and K. Matyjaszewski, Macromolecules, 2001, 34, 7664 CrossRef CAS.
- A. Fava, G. Reichenbach and U. Peron, J. Am. Chem. Soc., 1967, 89, 6696 CrossRef CAS.
- (a) C. Walling and W. Helmreich, J. Am. Chem. Soc., 1959, 81, 1144 CrossRef CAS; (b) L. Lunazzi, G. Placucci and L. Grossi, J. Chem. Soc., Perkin Trans. 2, 1981, 703 RSC.
- S. Searles Jr and H. R. Hays, J. Org. Chem., 1958, 23, 2028 CrossRef.
- (a) E. Brillas and I. Sirés, Chem. Rev., 2009, 109, 6570 CrossRef CAS PubMed; (b) J. Kiwi, K. Kalyanasundaram and M. Graetzel, Struct. Bonding, 1982, 49, 39 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization of products, and NMR spectral charts. See DOI: 10.1039/c4cc07606k |
| This journal is © The Royal Society of Chemistry 2015 |