
Label-free, isothermal and ultrasensitive electrochemical detection of DNA and DNA 3′-phosphatase using a cascade enzymatic cleavage strategy†
Shufeng
Liu
*,
Tao
Liu
and
Li
Wang
Key Laboratory of Sensor Analysis of Tumor Marker, Ministry of Education, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, China. E-mail: sliu@qust.edu.cn
First published on 31st October 2014
Abstract
Label-free and ultrasensitive electrochemical assays of target DNA and T4 polynucleotide kinase phosphatase (PNKP) were developed, which took full advantage of three enzymes to realize the signal readout and amplification. It can ultimately achieve the low detection limits of 10 fM and 1 mU mL−1 for target DNA and PNKP, respectively.
With the rapid development of biosensor technology, more and more biomarkers for nucleic acids and proteins, have been thoroughly disclosed to be correlated with the occurrence and development of some diseases such as cancers.1 In order to satisfy the requirement of profiling trace amounts of biomarkers, and then to serve for the clinical diagnosis and medical treatment of diseases, amplified detection toward various target analytes is always highly pursued.2 Up to now, different signal amplification strategies, especially cascade or autocatalytic amplification strategies, have been developed for the detection of nucleic acid or nucleic acid related bio-analytes.3 They are often involved in the individual or combined use of various nucleases including endonuclease, exonuclease and polymerase, etc. to achieve recycling of the target itself and/or the concomitant generation of large amounts of target-associated nucleic acid sequences. This indicates indirect amplification towards target number and thus contributes to the striking improvement of the detection sensitivity towards target analytes.4 It should be noted that the signal amplification strategies are more preferentially combined with the fluorescence technique. By comparison, electrochemical methods can provide significant advantages, such as its inherent signal stability, low cost, high sensitivity and ease of calibration. Also, the instrumentation can be easily integrated with some miniaturized devices or lab-on-a-chip platforms.5 Although great advances have been made for the fabrication of amplified electrochemical biosensors by combining some signal amplification strategies,6 they are often hindered by complex probe design or detection procedures, and even unsatisfactory detection performance. Sometimes, the developed method is not easily extended for the detection towards more analytes beyond nucleic acids. Therefore, upgrading the detection sensitivity by combining a proper signal amplification strategy to make it easier-to-use and further extending the detection analytes beyond nucleic acids remain urgent challenges in the development of ultrasensitive electrochemical biosensors. On the other hand, the development of label-free electrochemical assays would substantially simplify the procedure and allow targets to be profiled with high confidence, particularly for diagnostic purposes at point-of-care.7
Herein, a one-pot cascade enzymatic cleavage strategy was firstly developed for the label-free and ultrasensitive electrochemical detection of nucleic acids (Scheme 1). A hairpin DNA probe (HP1) was designed and immobilized onto a gold electrode surface, which could largely inhibit the diffusion property of [Fe(CN)6]3−/4− towards the electrode surface. In the presence of the target DNA, it can hybridize with the loop region of HP1 and make the HP1 stem open. Then, an engaging primer anneals with the open stem and allows polymerization induced by Bst polymerase, which displaces the target DNA and synthesizes a DNA duplex according to the HP1 probe. The displaced target DNA is free to bind to another HP1 on the electrode, initiating a new cycle. With each reaction cycle, the target DNA is regenerated and a duplex DNA probe is formed. This constitutes the recycling I stage in Scheme 1. The duplex DNA probe produced from the recycling I stage contains a newly generated nicking endonuclease recognition site. In the case of no target, the HP1 remains inactive, and the recognition sequence remains as a single strand, which is an unsuitable substrate for the nicking endonuclease. After nicking of the 5′-end of duplex DNA, the duplex DNA probe will be removed from the electrode, resulting in the enhancement of the diffusion ability of [Fe(CN)6]3−/4− towards the electrode surface.7a,b Moreover, the nicking-generated 5′-phosphorylated blunted termini of the dissociated duplex DNA probe can be recognized by lambda exonuclease, which catalytically digests the 5′-phosphorylated strands of the DNA duplexes and releases the corresponding complementary strands.8 Importantly, the released complementary strands can directly hybridize with the HP1 on the electrode surface to form complementary DNA duplexes, which can be further recognized and cleaved by nicking endonuclease. The dissociated duplexes can again be digested by lambda exonuclease to recycle the complementary strands. This constitutes the recycling II stage as indicated in Scheme 1. By following the mechanism, the target DNA can lead to two independent recycling processes, which results in a cascade amplification format for the catalytic removal of the HP1 from the electrode surface, and ultimately largely promotes the diffusion property of [Fe(CN)6]3−/4− towards the electrode surface.
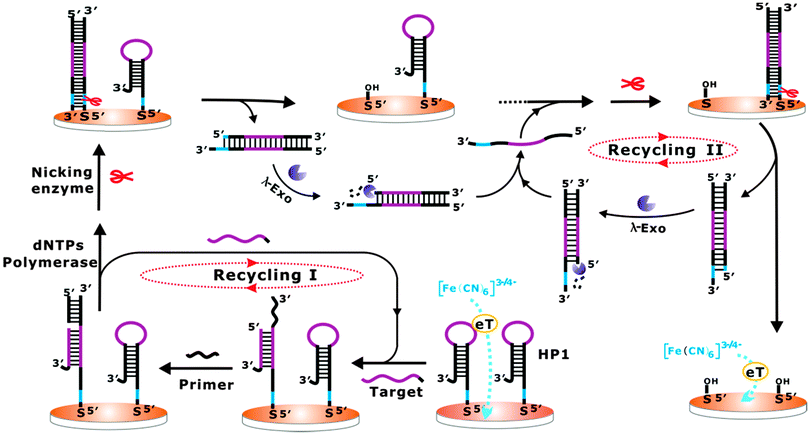 | ||
| Scheme 1 Schematic illustration of the fabricated cascade enzymatic amplification system for target DNA detection. | ||
The feasibility of the proposed cascade enzymatic cleavage strategy for label-free electrochemical DNA detection was verified by the differential pulse voltammograms (DPV) experiments shown in Fig. 1A. A distinct reduction peak current could be observed at the bare gold electrode (curve a in Fig. 1A). After the assembly of HP1 and MCH on the electrode surface, this reduction peak current was evidently inhibited (curve b in Fig. 1A). In the presence of 0.1 nM target DNA, after the cascade enzymatic cleavage process, the peak current was largely restored (curve d in Fig. 1A). However, in the absence of target DNA, an almost unchanged reduction peak compared with the HP1 and MCH assembled electrode could only be observed (curve c). Electrochemical impedance spectra (EIS) were also recorded (Fig. 1B), which were well in accordance with the DPV results, and strongly verified the feasibility of the fabricated cascade enzymatic cleavage system for label-free electrochemical detection of target DNA.
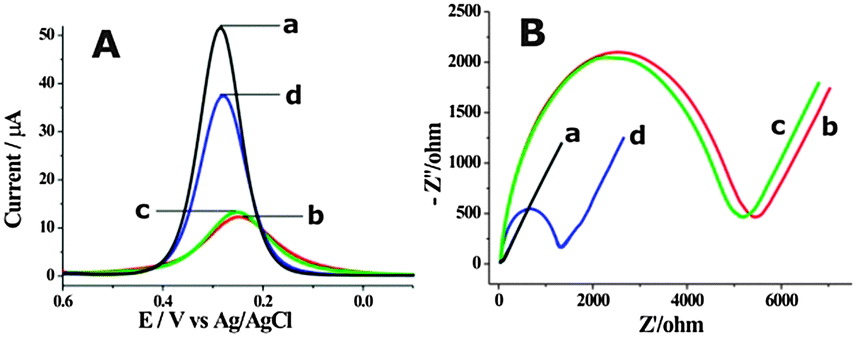 | ||
| Fig. 1 The DPV (A) and EIS (B) responses of the fabricated cascade enzymatic cleavage sensing system toward 0.1 nM target DNA (curve d) and blank solution (curve c) in 5 mM [Fe(CN)6]3−/4− and 1 M KNO3. Curves a and b are for bare gold electrode and HP1 assembled electrode, respectively. | ||
In order to achieve the best sensing performance, the corresponding experimental conditions were optimized. The probe DNA concentration of 1 μM could achieve the best electrochemical response towards target DNA detection (Fig. S1, ESI†). The optimized amounts of nicking endonuclease and lambda exonuclease were chosen as 10 and 7.5 U, respectively (Fig. S2, ESI†). The optimized response time in the current three-enzyme system and reaction temperature were determined as 60 min and 37 °C, respectively (Fig. S3, ESI†).
Furthermore, the detection performance of the fabricated label-free electrochemical biosensing system was investigated using target DNA with different concentrations. As shown in Fig. 2A, the DPV peak current increased with the concentration of target DNA from 0 to 0.1 nM, indicating that the removal of HP1 from the electrode surface was highly dependent on the concentration of target DNA. Fig. 2B shows the DPV peak current of [Fe(CN)6]3−/4− as a function of the concentration of target DNA. A good linear correlation of the DPV peak currents with the target DNA concentrations ranged from 10 fM to 500 fM could be obtained. The detection limit was found to be as low as 10 fM, which was superior or comparable to those of previously reported electrochemical methods (Table S2, ESI†). The specificity of the developed cascade enzymatic cleavage strategy was further investigated by using four kinds of DNA sequences, including complementary target DNA (TD), single-base mismatched DNA (1MT), two-base mismatched DNA (2MT), and non-complementary DNA (NC) (Fig. S4, ESI†). It demonstrated a good performance to discriminate between a perfect complementary target and the base mismatched targets, and held great potential for single nucleotide polymorphism analysis.
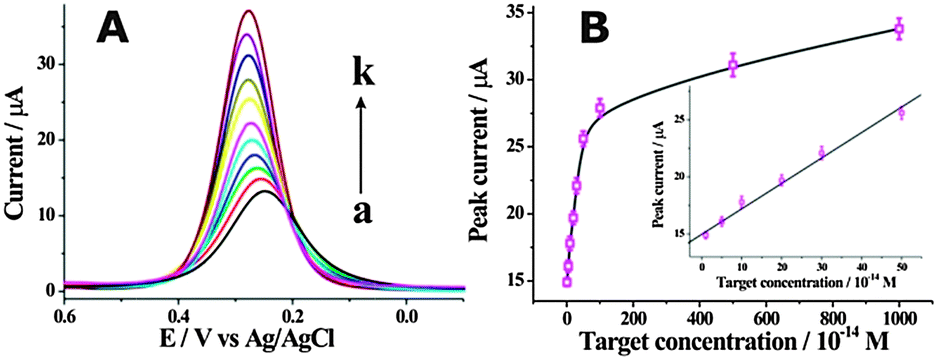 | ||
| Fig. 2 (A) DPV responses corresponding to the analysis of different concentrations of target DNA. The concentrations of target DNA for the curves (a) to (k) are: (a) 0 M, (b) 10 fM, (c) 50 fM, (d) 100 fM, (e) 200 fM, (f) 300 fM, (g) 500 fM, (h) 1 pM, (i) 5 pM, (j) 10 pM, (k) 0.1 nM. (B) Calibration curve corresponding to the DPV peak currents for variable concentrations of target DNA. Inset shows the linear relationship between the DPV peak current and the target DNA concentration. | ||
To further prove the signal amplification effect of the developed cascade enzymatic cleavage strategy, the detection of target DNA was also conducted in the case of no lambda exonuclease, as schematically demonstrated in Fig. S5A (ESI†). The electrochemical response also increased with the concentration of target DNA, ranging from 0 to 10 nM (Fig. S5B, ESI†). It demonstrated a linear relationship of DPV peak current with target DNA concentration, ranging from 1 pM to 100 pM. A detection limit of only 1 pM for target DNA could be obtained (Fig. S5C, ESI†). Also, the cascade enzymatic cleavage strategy by using three enzymes could realize a more rapid electrochemical response before reaching equilibrium compared with that in the absence of lambda exonuclease, indicating again that the cascade amplification process for target DNA by using three enzymes indeed occurred (Fig. S3A, ESI†). To further evaluate the practical utility of the proposed DNA biosensor, we further challenged the detection towards target DNA spiked in a relatively complex biological matrix (2% fetal bovine serum). Comparable responses were obtained for the detection of target DNA in both buffer and diluted serum (Fig. S6, ESI†), indicating the potential for the application in a relatively complex biological sample.
The developed cascade enzymatic cleavage strategy could be further adapted for electrochemical detection towards a wide spectrum of target molecules. Herein, as a proof-of-concept, DNA 3′-phosphatase was chosen as the model analyte of interest, which plays important roles in the repair of strand breaks induced by DNA damaging agents. DNA blocking lesions, for example DNA terminated with a 3′-phosphate resulting from exposure to both external genotoxic agents and endogenous sources of stress, would prevent the polymerization and ligation events necessary to re-establish the continuity of a DNA strand, which has been proven to be mutagenic and detrimental to survival.9 Thus, the removal of the abnormal termini is a prerequisite for DNA damage repair. T4 polynucleotide kinase phosphatase (PNKP), as a typical DNA 3′-phosphatase, can effectively restore the 3′-phosphate termini to hydroxyl groups by virtue of its 3′-phosphodiesterase activity.10 The principle design of our approach for PNKP is illustrated in Scheme 2. As far as we know, this is the first time that DNA 3′-phosphatase has been electrochemically detected using a cascade enzymatic cleavage strategy. The assembled hairpin DNA probe (HP2) with a 3′-phosphoryl end on the electrode surface can resist the DNA polymerase elongation reaction, which is only initiated at the 3′-hydroxyl end of the primer. In the presence of PNKP, the HP2 can be dephosphorylated into a 3′-hydroxyl group, which then can serve as the preferred substrate for Bst polymerase. Following the polymerase elongation reaction, a long double-helix hairpin product can be produced, which contains a newly generated nicking endonuclease recognition site. After its recognition and cleavage by nicking endonuclease, the duplex hairpin DNA can be removed from the electrode, resulting in the enhancement of the diffusion of [Fe(CN)6]3−/4− towards the electrode surface. The further digestion of the dissociated duplex hairpin DNA by lambda exonuclease leads to the release of the corresponding complementary strands, which can then hybridize with another HP2 to form the partially complementary DNA duplexes. It then triggers a new cleavage and digestion recycle operated by nicking endonuclease and lambda exonuclease. Following this principle, the PNKP activity could be achieved for amplified detection.
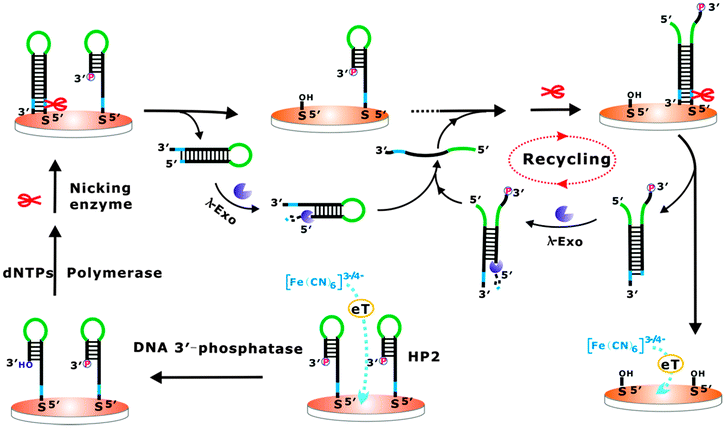 | ||
| Scheme 2 The schematic illustration of the fabricated cascade enzymatic cleavage sensing system for the detection of DNA 3′-phosphatase. | ||
The biosensor fabrication and feasibility for PNKP activity detection was verified by CV, DPV and EIS methods (Fig. S7, ESI†). These characterizations provide comparable and consistent evidence towards PNKP activity detection. We also synthesized a HP3 probe with a 3′-hydroxyl termini as a substitute of HP2. In the absence of PNKP, a remarkable electrochemical response after the incubation of three enzymes could be still observed, which was comparable with that obtained for HP2 in the presence of 1 U mL−1 PNKP (Fig. S8A, ESI†). This further strongly verified that the electrochemical response was indeed initiated by the dephosphorylation reaction of PNKP. The amplification effect towards PNKP activity detection using three enzymes was also compared with those obtained in a series of control experiments (Fig. S8B, ESI†). We also conducted time optimization of the dephosphorylation reaction (Fig. S9, ESI†). The optimized dephosphorylation reaction time was determined as 60 min.
To further confirm the ability of the described cascade enzymatic cleavage strategy to sensitively detect PNKP activity, a series of different concentrations of PNKP ranged from 0 to 2 U mL−1 were measured (Fig. 3A). The electrochemical intensity was observed to increase with the concentration of PNKP. A calibration curve of the DPV peak currents with the PNKP concentration was plotted and a good linear relationship between DPV peak current and PNKP concentration ranging from 1 mU mL−1 to 20 mU mL−1 could be obtained (Fig. 3B). The directly measured detection limit could be obtained as low as 1 mU mL−1, which was much lower than previously reported fluorescence methods.10b,c The PNKP activity detection was also conducted in the absence of lambda exonuclease as illustrated in Fig. S10A (ESI†). A low detection limit of about 50 mU mL−1 toward PNKP activity could be obtained (Fig. S10B, ESI†), indicating again the robust amplification ability of the developed cascade enzymatic cleavage sensing strategy. The selectivity of the described method for PNKP activity detection was also compared with other interfering proteins (Fig. S11, ESI†). A remarkable electrochemical response was only observed in the presence of PNKP, providing an excellent selectivity for the DNA 3′-phosphatase assay.
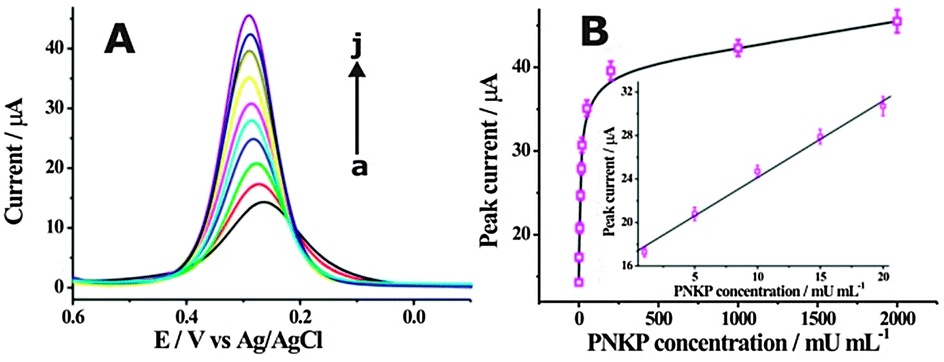 | ||
| Fig. 3 (A) DPV responses obtained at different concentrations of PNKP. The measured PNKP concentrations (from a to j) were 0, 1, 5, 10, 15, 20, 50, 200, 1000, 2000 mU mL−1, respectively. (B) The calibration curve of DPV peak current with PNKP concentration. The inset shows the linear relationship of the DPV peak current with PNKP concentration. | ||
In conclusion, the label-free and ultrasensitive detection of DNA and DNA 3′-phosphatase was developed for the first time using a cascade enzymatic cleavage strategy, in which the three enzymes were finely tuned for the electrochemical signal readout and amplification. It also exhibits the distinct advantages of simplicity in probe design, biosensor fabrication and detection process. Furthermore, it should be fairly easy to extend for the detection of a wide spectrum of analytes. Therefore, it opens a promising avenue to develop label-free and ultrasensitive electrochemical biosensors.
This work was funded by the National Natural Science Foundation of China (No. 21475072 and 21005043), and the Basic Research Program of Qingdao (No. 13-1-4-214-jch).
Notes and references
- (a) A. Sassolas, B. D. Leca-Bouvier and L. J. Blum, Chem. Rev., 2008, 108, 109 CrossRef CAS PubMed; (b) S. L. Wood, M. A. Knowles, D. Thompson, P. J. Selby and R. E. Banks, Nat. Rev. Urol., 2013, 10, 206 CrossRef CAS PubMed; (c) C. L. Sawyers, Nature, 2008, 452, 548 CrossRef CAS PubMed; (d) A. P. F. Turner, Chem. Soc. Rev., 2013, 42, 3184 RSC.
- (a) S. Song, Y. Qin, Y. He, Q. Huang, C. Fan and H. Y. Chen, Chem. Soc. Rev., 2010, 39, 4234 RSC; (b) A. A. Lubin and K. W. Plaxco, Acc. Chem. Res., 2010, 43, 496 CrossRef CAS PubMed.
- (a) F. Wang, C. H. Lu and I. Willner, Chem. Rev., 2014, 114, 2881 CrossRef CAS PubMed; (b) L. Yan, J. Zhou, Y. Zheng, A. S. Gamson, B. T. Roembke, S. Nakayama and H. O. Sintim, Mol. BioSyst., 2014, 10, 970 RSC; (c) J. Lei and H. Ju, Chem. Soc. Rev., 2012, 41, 2122 RSC.
- (a) X. Zuo, F. Xia, Y. Xiao and K. W. Plaxco, J. Am. Chem. Soc., 2010, 132, 1816 CrossRef CAS PubMed; (b) R. Duan, X. Zuo, S. Wang, X. Quan, D. Chen, Z. Chen, L. Jiang, C. Fan and F. Xia, J. Am. Chem. Soc., 2013, 135, 4604 CrossRef CAS PubMed; (c) W. Zhou, X. Gong, Y. Xiang, R. Yuan and Y. Chai, Anal. Chem., 2014, 86, 953 CrossRef CAS PubMed; (d) H. Zhou, S. J. Xie, S. B. Zhang, G. L. Shen, R. Q. Yu and Z. S. Wu, Chem. Commun., 2013, 49, 2448 RSC; (e) F. Wang, L. Freage, R. Orbach and I. Willner, Anal. Chem., 2013, 85, 8196 CrossRef CAS PubMed; (f) S. Bi, L. Li and Y. Cui, Chem. Commun., 2012, 48, 1018 RSC; (g) S. Liu, C. Zhang, J. Ming, C. Wang, T. Liu and F. Li, Chem. Commun., 2013, 49, 7947 RSC; (h) F. Wang, J. Elbaz, C. Teller and I. Willner, Angew. Chem., Int. Ed., 2011, 50, 295 CrossRef CAS PubMed; (i) F. Gao, J. Lei and H. Ju, Chem. Commun., 2013, 49, 4006 RSC.
- (a) N. J. Ronkainen, H. B. Halsall and W. R. Heineman, Chem. Soc. Rev., 2010, 39, 1747 RSC; (b) A. A. Lubin, B. V. S. Hunt, R. J. White and K. W. Plaxco, Anal. Chem., 2009, 81, 2150 CrossRef CAS PubMed; (c) F. Xuan, X. Luo and I. M. Hsing, Anal. Chem., 2012, 84, 5216 CrossRef CAS PubMed.
- (a) S. Liu, C. Wang, C. Zhang, Y. Wang and B. Tang, Anal. Chem., 2013, 85, 2282 CrossRef CAS PubMed; (b) H. Ji, F. Yan, J. Lei and H. Ju, Anal. Chem., 2012, 84, 7166 CrossRef CAS PubMed; (c) H. Li, Z. Sun, W. Zhong, N. Hao, D. Xu and H. Y. Chen, Anal. Chem., 2010, 82, 5477 CrossRef CAS PubMed; (d) J. Zhuang, D. Tang, W. Lai, G. Chen and H. Yang, Anal. Chem., 2014, 86, 8400 CrossRef CAS PubMed.
- (a) H. Xu, L. Wang, H. Ye, L. Yu, X. Zhu, Z. Lin, G. Wu, X. Li, X. Liu and G. Chen, Chem. Commun., 2012, 48, 6390 RSC; (b) Y. Cao, S. Zhu, J. Yu, X. Zhu, Y. Yin and G. Li, Anal. Chem., 2012, 84, 4314 CrossRef CAS PubMed; (c) Y. Ren, H. Deng, W. Shen and Z. Gao, Anal. Chem., 2013, 85, 4784 CrossRef CAS PubMed; (d) L. Kashefi-Kheyrabadi, M. A. Mehrgardi, E. Wiechec, A. P. F. Turner and A. Tiwar, Anal. Chem., 2014, 86, 4956 CrossRef CAS PubMed.
- (a) J. W. Little, J. Biol. Chem., 1967, 242, 679 CAS; (b) K. Hsieh, Y. Xiao and H. T. Soh, Langmuir, 2010, 26, 10392 CrossRef CAS PubMed.
- Y. J. Song, K. G. Qu, C. Zhao, J. S. Ren and X. G. Qu, Adv. Mater., 2010, 22, 2206 CrossRef CAS PubMed.
- (a) S. W. Yang, A. B. Burgin Jr, B. N. Huizenga, C. A. Robertson, K. C. Yao and H. A. Nash, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 11534 CrossRef CAS; (b) L. Lin, Y. Liu, X. Zhao and J. H. Li, Anal. Chem., 2011, 83, 8396 CrossRef CAS PubMed; (c) W. Zhu, Z. Zhao, Z. Li, J. Jiang, G. Shen and R. Yu, J. Mater. Chem. B, 2013, 1, 361 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional information. See DOI: 10.1039/c4cc08140d |
| This journal is © The Royal Society of Chemistry 2015 |