
The accurate calculation of the band gap of liquid water by means of GW corrections applied to plane-wave density functional theory molecular dynamics simulations†
Changming
Fang
*a,
Wun-Fan
Li
a,
Rik S.
Koster
a,
Jiří
Klimeš
b,
Alfons
van Blaaderen
a and
Marijn A.
van Huis
a
aDebye Institute for Nanomaterials Science and Center for Extreme Matter and Emergent Phenomena, Utrecht University, Princetonplein 5, 3584 CC Utrecht, The Netherlands. E-mail: C.Fang@uu.nl
bUniversity of Vienna, Faculty of Physics and Center for Computational Materials Science, Sensengasse 8/12, A-1090 Vienna, Austria
First published on 31st October 2014
Abstract
Knowledge about the intrinsic electronic properties of water is imperative for understanding the behaviour of aqueous solutions that are used throughout biology, chemistry, physics, and industry. The calculation of the electronic band gap of liquids is challenging, because the most accurate ab initio approaches can be applied only to small numbers of atoms, while large numbers of atoms are required for having configurations that are representative of a liquid. Here we show that a high-accuracy value for the electronic band gap of water can be obtained by combining beyond-DFT methods and statistical time-averaging. Liquid water is simulated at 300 K using a plane-wave density functional theory molecular dynamics (PW-DFT-MD) simulation and a van der Waals density functional (optB88-vdW). After applying a self-consistent GW correction the band gap of liquid water at 300 K is calculated as 7.3 eV, in good agreement with recent experimental observations in the literature (6.9 eV). For simulations of phase transformations and chemical reactions in water or aqueous solutions whereby an accurate description of the electronic structure is required, we suggest to use these advanced GW corrections in combination with the statistical analysis of quantum mechanical MD simulations.
I. Introduction
Because of their importance in the natural sciences, the structural and physical properties of different phases of water have been the subject of intensive investigations by scientists and engineers for over a hundred years.1–5 A lot of effort has been made to understand the structural and transport properties of water. At present the structures of gaseous and solid H2O (ice) phases are well established, based on the early work by Fowler and Bernal who proposed that (1) each H2O molecule can be regarded as an ‘element’ and (2) in building the structures of ice phases, each H2O element is in tetragonal coordination with hydrogen bonds (O⋯H) of about 1.9 Å.5 The variations of O⋯H ordering in the tetragonal coordination create dozens of ice phases, which at present are still under discussion.5–12 Obviously, there is no long-range ordering in liquid water. However, at short-range the structure of water is topologically the same as those of ice, but with flexible bond-lengths and angles of H2O tetragons.3,5,6,12–14To successfully perform simulations of chemical reactions in aqueous solutions, it is imperative that not only the atomic structure, but also the electronic structure is well described, whereby the electronic band gap can serve as the most important criterion. Many studies have already been performed on the electronic band gap of liquid water, but in the cases where accurate computational methods were used, these were applied to too few water molecules to be representative of liquid water. And in the cases where large numbers of water molecules were actually considered, methods such as cluster calculations were used which are known to be less accurate in predicting electronic properties.
Here we aim for high accuracy by applying GW corrections to plane-wave density functional theory (PW-DFT) molecular dynamics (MD) simulations using a van der Waals density functional and a statistical analysis by the time-averaging of relevant physical parameters ‘measured’ during the MD simulations. We will now first discuss the experimental studies whereby the electronic structures of gas, water, and ice phases were investigated.2,15–25 Shibaguchi and co-workers investigated the electronic structure of ice by means of a combination of X-ray photoemission spectroscopy (XPS), Ultraviolet photoemission spectroscopy (UPS), and optical absorption spectroscopy, and obtained a band gap of 8.75 eV.17 Painter and co-workers measured the reflectance of liquid water between 1050 and 3000 Å16 and found that the imaginary index of refraction starts at around 7.1 eV. From the measured real and imaginary parts of the index of refraction and dielectric constant, the authors suggested an exciton transition at 8.3 eV, an interband transition at 9.6 eV, and an electronic band gap of 9.0 eV.16 Other measurements of photoemission spectra provide a range of values for the band gap of water; 6.9 eV,21,22 7.0,23 8.7 eV,24 8.9 eV25 and 9.0 eV.16 As reviewed by Winter and co-workers, such a variance in experimental data for the insulating ice phases and water is caused by many factors, such as electric charging, work functions, surface sensitivity (electron escape depths are typically ∼10–20 Å), vapour–solid or vapour–liquid mixing, impurities, etc.2,19
Many theoretical and simulation studies have also been performed to study the structural and electronic properties of water phases. To study the structure and dynamics simple empirical potentials are widely used.26–30 Moreover, ab initio Hartree–Fock (HF) and post-HF methods were employed to study water molecules and clusters, as well as ice phases.11,12,21,22,34–36 Finally, first-principles density functional theory (DFT) has also been widely used to study the structural and electronic properties of ice phases and liquid water.31–37
In theoretical studies of ice and liquid water, there are several issues to be noted. First, the standard DFT functionals, such as the local density approximation (LDA) and different GGA functionals, which are local or semi-local, do not describe accurately van der Waals interactions. This is an important issue since van der Waals interactions were shown to play an important role in water and ice phases, as well as other solids.39–50 For example, Santra and co-workers have recently investigated the ice phases at non-zero pressure using different van der Waals DFT exchange–correlation functionals and observed significant improvements of phase transition pressures of the ice phases compared to that of the PBE (after the names of authors, Perdew, Burke and Ernzerhof) functional.41,42 The van der Waals density functional was also found to describe water adsorption on metals well.43,44,49 Second, most of the calculations performed so far have focused on the description of the ground states of ice phases and water. However, experimental measurements of electronic properties, such as band gaps, are related to excitations of electrons. To describe the excitations another scheme needs to be used, such as the GW approximation (whereby the self-energy is obtained from Green's function G and the electronic screening effect W) which describes well the eigenstates of one-electron quasi-particle (QP) levels which relate to electronic excitations.31,51–56 This method, but in a non-self-consistent G0W0 fashion, was used by Hermann and co-workers to obtain the band gap of Ice-XI.31,32 Moreover, most first-principles molecular dynamics simulations employed super-cells of 100 to 200 atoms, which can represent only a small number of molecules, and most simulations provide information about only one or a few configurations. This is not sufficient to obtain statistically meaningful results.56,57 Consideration of a large number of configurations with sufficient number of molecules to probe all relevant structures is needed. This approach was applied to analyse statistically the dominant defects in hydrogenated amorphous silicon nitrides covering different stoichiometries.51,57 The electronic structures were successfully simulated, including the energy gaps for the amorphous semiconductors with different types of defects.56,57
In the present manuscript, we report our results of a first-principles study on the H2O molecule, its dimer and Ice-XI, a low-temperature and low-pressure H2O phase with well-established H ordering8,10,11,26–32 as well as molecular dynamics simulations of water at 300 K, using the van der Waals density functional developed by Dion39 and implemented to the code VASP by Klimeš and co-workers.40 We also employ the self-consistent GW0 approach52,54 for Ice-XI. The almost linear relationship that was obtained between the QP (quasi-particle) energies and K–S (Kohn–Sham) eigenvalues is used to obtain the electronic structure including the band gap of water. Furthermore, we also analyse the charge distributions for H2O molecules, H2O dimers, and charge transfer between H2O molecules in water using the Bader charge approach58 which has been successfully applied to many molecules59 and crystalline solids.58,60 Distributions of charges at O and H were obtained for the simulated water, as well as for the isolated H2O monomer, its dimer and Ice-XI. The obtained electronic structures and the energy gaps for the ice phase and liquid water are in good agreement with available experimental data. This is not only useful for having a better understanding of the intrinsic properties of water, but also suggests that the same approach can be successfully applied to aqueous solutions that are widely used in various scientific disciplines and in industrial processes.
The paper is arranged as follows. In Section II, we introduce the (super)cells which were employed for the structural optimization of an isolated H2O molecule and its dimer, as well as of water with the experimental density at room temperature. The details of the computational methods are also described. The optimized structures of the isolated H2O molecule, its dimer and the lattice parameters of Ice-XI are presented in Section III.A. The structural and chemical bonding details and electronic properties of first-principles molecular dynamics simulated water are presented in Section III.B. The electronic properties of Ice-XI obtained using the DFT van der Waals density functional are discussed together with the GW0 corrections in Section III.C. In Section III.D, we discuss the electronic structure of water using the GW corrections. The corrected curves of the density of states (DOS) are compared with the experimental measurements (mainly photoemission spectra). Finally a brief summary is presented in Section IV.
II. Details of computational techniques
For all the structural optimizations and total energy calculations of the isolated molecules and molecular dynamics simulations, we employed the Vienna code VASP (Vienna Ab initio Simulation Package)57,58 which is based on the density functional theory (DFT) within the Projector-Augmented Wave (PAW) method.59,60 Dion's van der Waals density functional which includes nonlocal contributions was employed for the correlation energy term.39,40,44 The generalized gradient approximation61 (GGA) within the optB88 functional was employed for the exchange term. The electronic wave functions were sampled on dense grids (20 × 10 × 10 or 396 irreducible k-points) in the Brillouin zone (BZ) of the crystals, using the Monkhorst and Pack method62 for the total energy and electronic structure calculations of liquids and solids. Structural optimizations were performed by the relaxation of both lattice parameters and coordinates of atoms for the crystals, while only atomic coordinates in a fixed cube were relaxed for the molecules.It is noteworthy that water and the ice phase contain strong local bonds, O 2p–H 1s, and therefore, the calculated valence electron total energy is expected to depend on the cut-off energies. We make a systematic investigation on the total energy calculations using different cut-off energies for the wave functions (Ecut) and for the augmentation functions (Eaug), which is about 30% higher than the corresponding Ecut for a H2O molecule. The calculated results (see Fig. 1) show strong variations in the low energy range (Ecut/Eaug = 100/200–250/400 eV) for the calculated bond-length, the H–O–H angle and the total valence electron energy. When Ecut/Eaug is at least 300/450 eV, there is little dependence of the O–H bond-length (within 0.001 nm) and the H–O–H angle (within 0.1°) on the energy set, but still a weak dependence of the calculated total energy on the energy set. The calculated total valence electron energy becomes stable when the cut-off energy is over 650 eV (within 1 meV per atom). Therefore, to ensure cut-off independent results in all total energy calculations the cut-off energy of the wave functions was set to be 750 eV, and the cut-off energy of the augmentation functions was set to be 1000 eV. The low sensitivity of the H–O–H angle and the O–H bond-length on the cut-off energy also allows us to use lower cut-off energies for the molecular-dynamics simulations. We used a cut-off energy of 320 eV, and only the Γ point in the Brillouin zone (BZ) in all molecular-dynamics simulations for the sake of finding a reasonable balance between computational feasibility and the accuracy of the results since the cell is large.
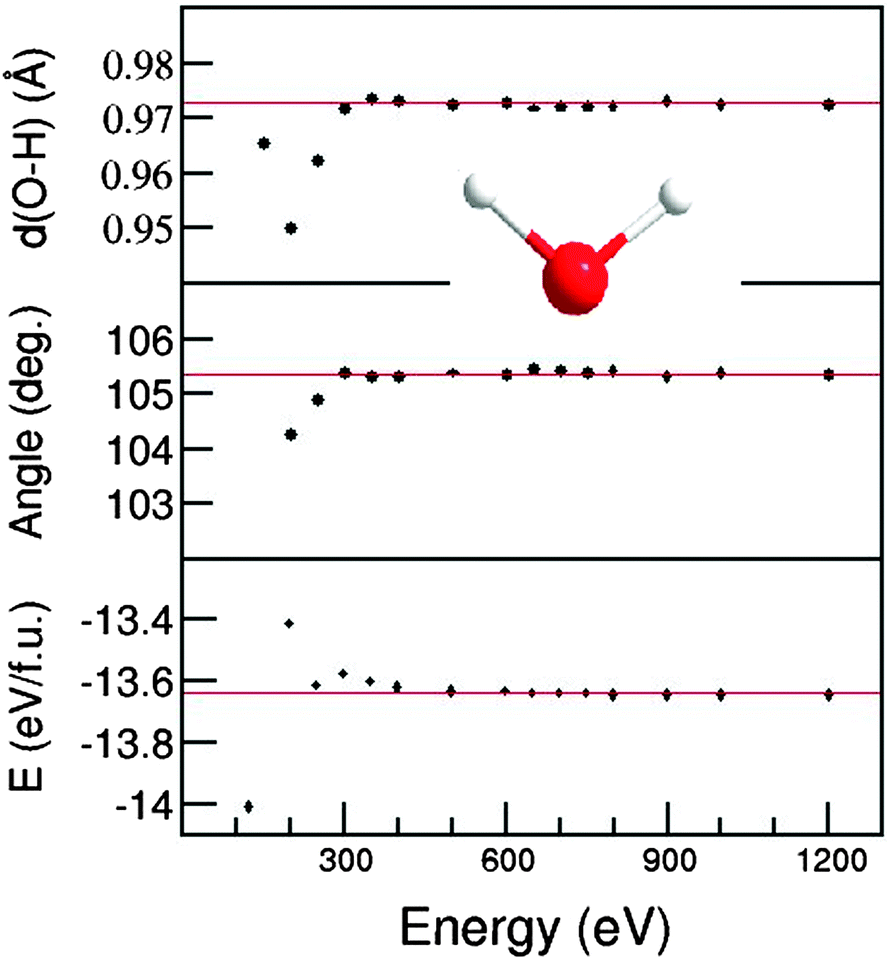 | ||
| Fig. 1 Convergence of the O–H bond-length (top), the H–O–H angle (middle) and the calculated total energy (bottom) as a function of the cut-off energy for an isolated H2O molecule (the schematic structure being the inset) using the van der Waals density functional. The red solid lines indicate the converged values. | ||
III. Calculated results and discussion
III.A. Structures of the H2O molecule, the H2O dimer, and the Ice-XI phase
We first discuss the properties of a water monomer, a dimer, and Ice-XI. Tables 1–4 list the calculated results for an isolated H2O molecule and its dimer (Fig. 2b), as well as the Ice-XI phase with ordered hydrogen arrangements11,12,26 (see Fig. 2a). The calculated results are compared with recent theoretical studies.11,33,34,38,47,63,67| Methods | d(O–H)(Å) | Angle H–O–H (°) | |
|---|---|---|---|
| This work | optB88-vdW | 0.973 | 105.40 |
| Xu and Goddard35,36 | HF | 0.941 | 106.2 |
| PBEPBE | 0.971 | 104.0 | |
| X3LYP | 0.961 | 105.1 | |
| Casassa, et al.11,12 | HF-CRY-c | 0.940 | 106.2 |
| B3LYP-CRY-c | 0.961 | 105.1 | |
| PW91-CRY-c | 0.968 | 104.3 | |
| Silvestralli + Parrinello34 | GGA-BLYP | 0.972 | 104.4 |
| Jurečka38 | HF-CCSDT | 0.959 | 104.2 |
| Exp.15 | 0.957 | 104.5 | |
| Exp.16 | 0.958 | 104.45 |
| Methods | d(O–H)(Å) range | d(O–O)(Å) | E form (eV/f.u.) | |
|---|---|---|---|---|
| a The O–H distance with hydrogen bonding with another O. | ||||
| This work | optB88-vdW | 0.97/0.98 | 2.866 | −0.236 |
| Xu and Goddard35,36 | HF | 0.941/0.945 | 3.048 | −0.16 |
| PBEPBE | 0.970/0.981 | 2.899 | −0.28 | |
| X3LYP | 0.957/0.968 | 2.908 | −0.24 | |
| Casassa, et al.11,12 | HF-CRY-c | 0.944a | 3.042 | −0.12 |
| B3LYP-CRY-c | 0.969 | 2.925 | −0.21 | |
| PW91-CRY-c | 0.979 | 2.880 | −0.24 | |
| Jurečka38 | HF-CCSDT | −0.218 | ||
| Exp.12,17 | 2.976 | −0.24 | ||
| Method | Lattice pars. (Å), V (Å3/f.u.) | Atomic coordinates |
|---|---|---|
| optB88-vdW (this work) | a = 4.343 (−3.5%) | O1 4a(0,0.6502,0.0586) |
| b = 7.609 (−2.5%) | O2 4a(0,0.3190,0.9358) | |
| c = 7.035 (−4.0%) | H1 4a(0,0.6571,0.2017) | |
| V = 29.06 (−9.6%) | H2 4a(0,0.5226,0.0197) | |
| H3 8b(0.6870,0.7526,0.9811) | ||
| HF-CRY11 | a = 4.57 (+1.5%) | |
| b = 8.40 (+7.7%) | ||
| c = 7.69 (+4.9) | ||
| V = 36.85 (14.6%) | ||
| B3LYP11 | a = 4.42 (−1.8%) | |
| b = 7.85 (0.7%) | ||
| c = 7.23 (−1.3) | ||
| V = 31.36 (−2.5%) | ||
| Neutron diffraction65 | a = 4.5019 | O1 4a(0,0.6648,0.0631) |
| b = 7.7978 | O2 4a(0,0.3255,0.9360) | |
| c = 7.3280 | H1 4a(0,0.6636,0.1963) | |
| V = 32.16 | H2 4a(0,0.5363,0.0183) | |
| H3 8b(0.6766,0.7748,0.9817) |
| Structure | O | H |
|---|---|---|
| Monomer | −1.28 | 0.64 |
| Dimer | −1.18 to −1.19 | 0.55 to 0.61 |
| Ice_XI | −1.30, −1.31 | 0.64 to 0.67 |
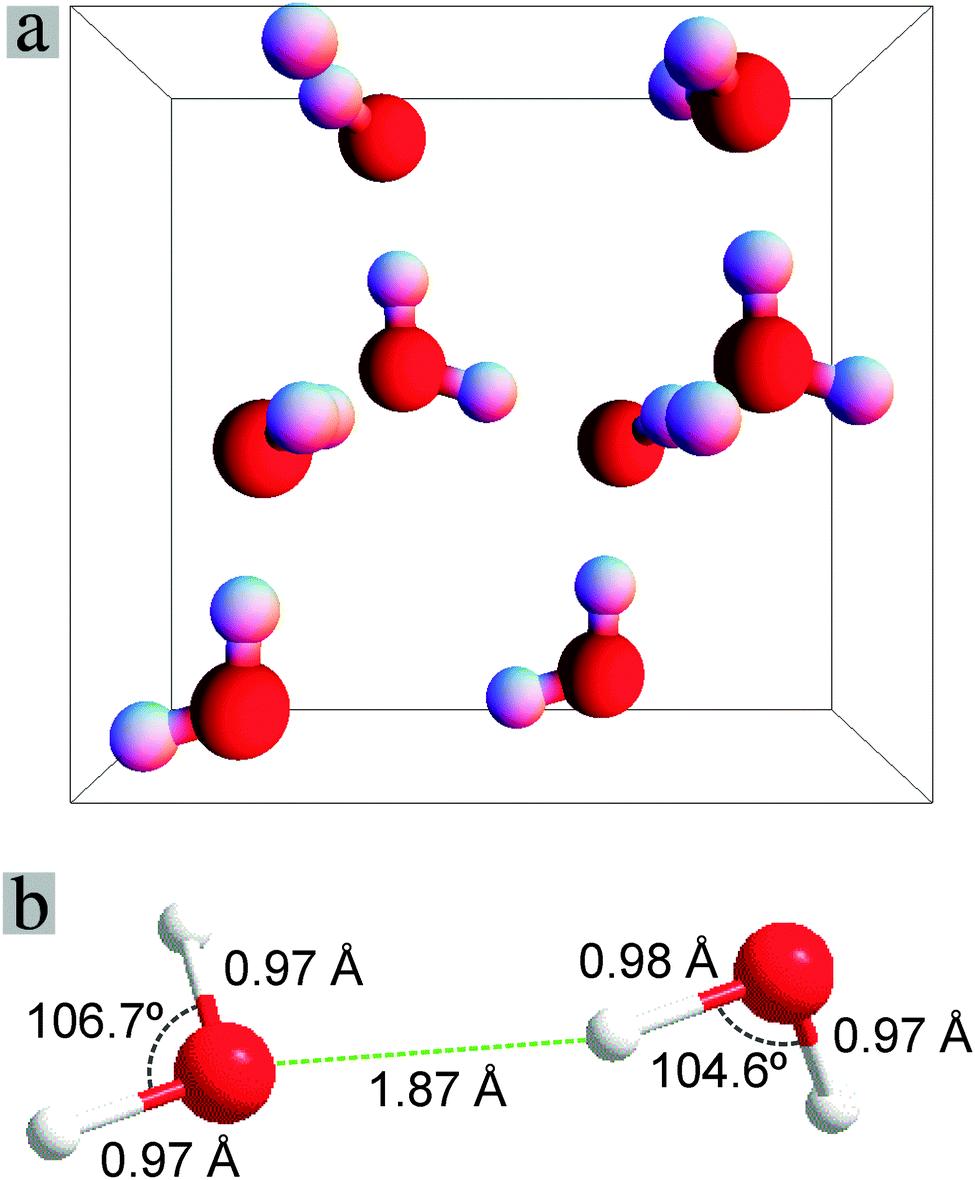 | ||
| Fig. 2 (a) Schematic structure of ice Ice-XI with the projection along the [001] direction. (b) Structure of a H2O dimer, the unit for the angles is degree, for the O–H and O⋯H bonds (bond lengths in Å). | ||
The O–H bond-length in the H2O monomer is 0.973 Å, close to the experimental values within 0.02 Å.64 There are many theoretical studies using various functionals, including the ‘hybrid’ B3LYP method which adopted an all-electron 6311G(p,d) basis set,11,12 X3LYP which contains van der Waals interactions,35,35 and the ab initio Hartree–Fock method within coupled-cluster singles, doubles, triples (HF-CCSDT) wave function.38 Here we compare our results with a few of them (Table 1). The obtained O–H bond is close to those obtained by the ab initio HF methods and by first-principles DFT approximations (Table 1). The presently calculated H–O–H angle is just about 1.0° larger than the measured one, and in good agreement with former theoretical calculations, as shown in Table 1.
Table 2 and Fig. 2b also show the calculated results for the H2O dimer. The calculated bonds and angles are in line with the former calculations. The calculated O–H bond for the H participating in the hydrogen bond (0.98 Å) is slightly longer than the other 3 short O–H bonds (0.97 Å). This difference (0.01 Å) was also obtained by other DFT and HF calculations.11,33,34,38 The calculated binding energy is close to the experimental value, with slight overestimation, as observed previously.12,27
The ground state of H2O is the Ice-XI phase.9–12,26–30,67–69 The crystal structure of Ice-XI is orthorhombic with space group Cmc21 (nr. 36). The structure at 5 K was determined by Leadbetter and co-workers using neutron diffraction (Table 3)69 and investigated theoretically by several groups using different approaches.11,26,27,32,41,42
We performed full structural optimization of this ice phase using the van der Waals density functional optB88-vdW. The results are shown in Table 3. It is clear that our calculated lattice parameters are 2.5–4.0% smaller than the corresponding experimental values. That difference is similar to the PW91 results obtained by Casassa and co-workers.11,12 We also note that HF calculations overestimate the volume while better agreement with the experiment can be obtained with much more computationally expensive correlated methods, such as the Random Phase Approximation.47
The Bader charge analysis58 shows that there is charge transfer of 0.64 electrons per hydrogen atom from H to O. In the H2O dimer, the charges are almost the same for the two O ions (−1.18 electrons), which is about 0.10 electrons less than that of an isolated molecule, in agreement with former calculations.58 There are also some differences between the charges on the H atoms: the H of the long O–H bond has a charge of +0.55 electrons, which is smaller than the charges (about 0.60 electrons) on the other three H atoms. For the ice phase, the charge on the two oxygen ions is about −1.30 electrons and the H ions are charged in the range of 0.64–0.67 electrons, as shown in Table 4.
III.B. Charges in water at 300 K
The thermal history of water simulated at 300 K is shown in Fig. 3 which also includes the variation of the cohesive energy for the water system on time at 300 K. It is clearly shown that the energy lowers down with time and reaches the equilibrium after about 3.5 ps (or 7000 iterations). | ||
| Fig. 3 The thermal history of temperature (a) and total valence electron energy (b) of water simulated at a temperature of 300 K. | ||
First we briefly discuss the structure of the simulated water. We monitored the processes of the molecular dynamics simulations. We observed no sign of charged ions/clusters, neither a single proton (H+) or (H3O)+, (H5O2)+, nor (OH)− or (H3O2)− or (H3O2)− clusters. This is understandable considering the high formation energies of such ions or charged clusters.2,6,22 In addition, the Born–Oppenheimer approximation was used in our molecular dynamics simulations, so that proton tunnelling is neglected. However, we observed a 2(H2O) cluster with a very short intermolecular (O–H) bond of 1.4 Å (Fig. S1, ESI†), which exists for about 50 fs. Fig. 4a shows a typical configuration of water obtained from the first-principles molecular dynamics simulations at 300 K. We analysed several configurations obtained for each of about 0.5 ps time-gap. The O–H bonds distributions in H2O molecules together with the Bader charge distributions are shown in Fig. S2 (ESI†). Apparently there is a strong similarity among the distributions of the intra-molecule bonds for the different configurations (snapshots at different times) with significant deviations from a regular Gaussian-type distribution. This is also true for the Bader charge distributions (Fig. S2, ESI†).
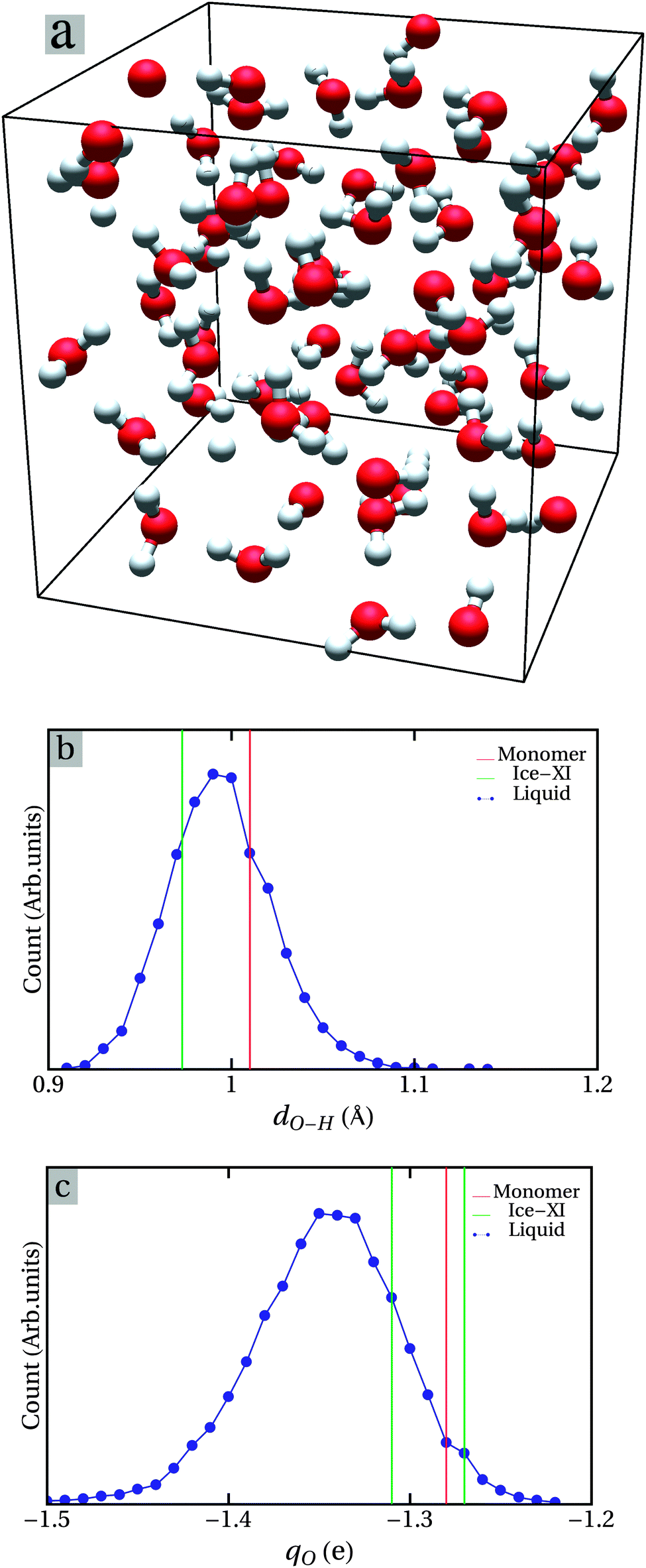 | ||
| Fig. 4 Snapshot of one typical water structure during the simulations at 300 K (a), the distributions of O–H bond lengths (b) and the Bader charge of O ions (c) time-averaged over a period of 2 ps during the simulation of water at 300 K (about 100 samples). | ||
As discussed previously, compared with the size of a drop of water (∼μm) the small number of water molecules in the cell (64H2O molecules, 192 atoms, size ∼nm) can on the one hand be regarded as being local. The fluctuations of bonding in time, on the other hand, reflect the stochastic nature of liquid water. Therefore, time-averaging can and should be used to determine the physical properties of water. Here we simulate these properties by time-averaging over 2 ps.
The results are shown in Fig. 4 and Fig. S2 (ESI†). It is clear that the time-averaged distributions show more or less regular Gaussian-type distributions, in comparison to the large scattering observed between individual snapshot configurations (Fig. S2, ESI†). The O–H bond-lengths in water range from 0.92 to 1.08 Å with the width of a half-peak height of 0.1 Å, centred at 0.99 Å, which is between that of the isolated H2O monomer (0.97 Å) and that of the Ice-XI phase (1.01 Å). This corresponds to the model of Bernal and co-workers.3–5 As shown in Fig. 4c, Bader's charge at O sites of water is between −1.22 e (electrons) and −1.48 e with a broad peak at about −1.34 e. The averaged value (about 1.34 e) of the charges at O ions is slightly larger than those of an isolated H2O monomer and Ice-XI, although the averaged O–H bond-lengths of water are close to those of the monomer and ice. This can be understood from the Brown bond theory66,67 where the valence (Vi) of the central atom to the ith neighbour is defined as
| Vi = exp[−(Ri − R0)/A] |
III.C. Electronic structure of Ice-XI and water at 300 K
To have an overview of the electronic properties of different phases of water, the eigenvalues of an isolated H2O molecule (5a), its dimer (5b), the total density of states for Ice-XI (5c), and one snapshot of liquid water (5d) are displayed in Fig. 5.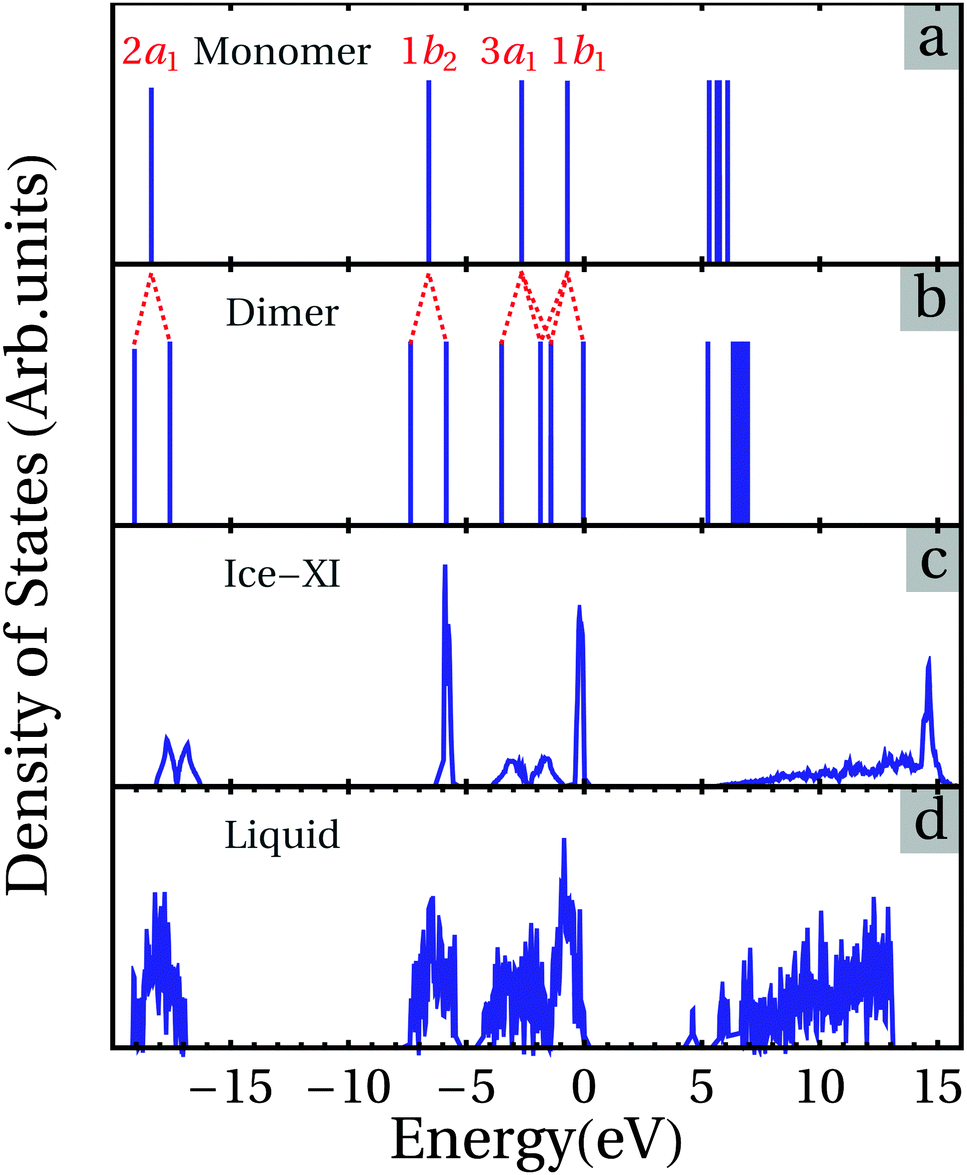 | ||
| Fig. 5 The DFT-van der Waals density functional calculations of eigen-energies of an isolated H2O, and its dimer; the tDOS of the ice phase and a snapshot of water simulated at 300 K with notable localization of electronic states. | ||
Fig. 6 shows the calculated partial and total density of states (pDOS and tDOS) (6a) and the corresponding dispersion curves along the high symmetry lines of the Brillouin Zone (BZ) for the orthorhombic Ice-XI phase using Dion's DFT-v/d Waals density functional.39,40 The eigenvalues of the isolated H2O molecule originate from the interactions between O 2s, O 2p, and H 1s states.15–19,33,73 There are early assignments for the four occupied eigenstates, 2a1 (O 2s with some H 1s character), 1b2 and 3a1 (O 2p hybridized with H 1s), 1b1 (non-bonding O 2p), and the lowest unoccupied 4a1 (O 2s 2p and H 1s) and 2b2 (O 2p and H 1s) states. 2a1, 1b2 and 3a1 belong to bonding states while 4a1 and 2b2 have anti-bonding characters.
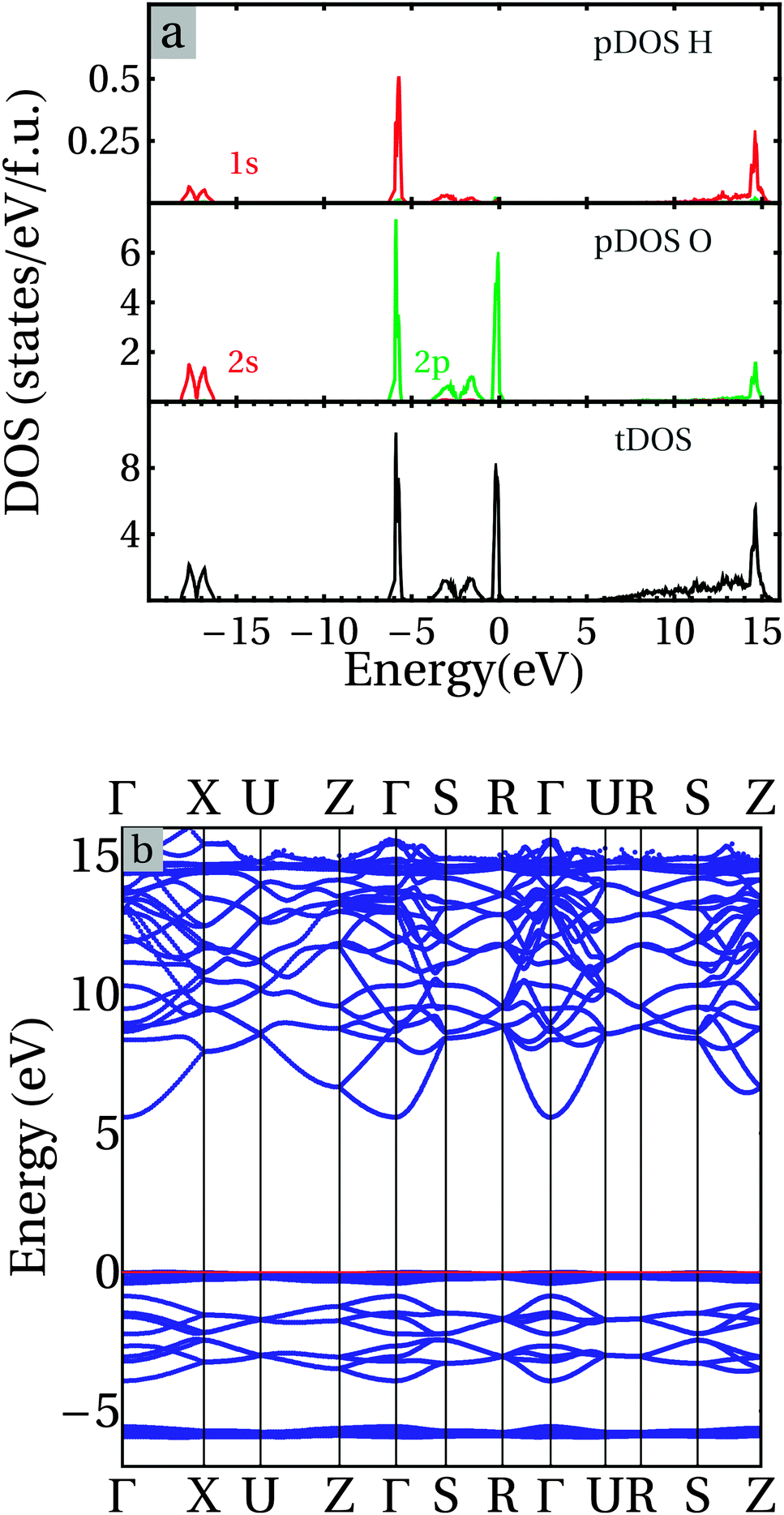 | ||
| Fig. 6 (a) Partial densities of states (pDOS), (b) dispersion curves along the high symmetry lines in the Brillouin zone (BZ) for the ground state of the orthorhombic Ice-XI phase. The Fermi level is set to be the top of the valence band. | ||
For the H2O dimer, the distortion of the molecules and the interactions between the molecules (Fig. 2) cause splitting (about 1.5 eV) of the 1b2 states, and further hybridizations between 3a1 and 1b1 states that induce the lowering of new 3a1 and shifting up of new 1b1, together with the creation of two new hybridized states (bonding 3a1/1b1 and anti-bonding (3a1/1b1)* states). The lowest unoccupied states 4a1 are also lowered in energy due to further hybridization. Clearly the energy gap between the occupied and unoccupied states is strongly reduced in comparison to that of the H2O molecule.
Fig. 5 also includes the tDOS for Ice-XI for the sake of comparison. Overall the results for the Ice-XI phase agree well with the early symmetry study by Parravicini and Resca who used the Bloch sums formed by H2O molecule orbitals as an expansion set for cubic ice,68,72 and the recent DFT-GGA calculations by Prendergast and co-workers for the Ice-Ih phase,34 considering the strong similarities between the different H2O ordering of these phases.3–6,10–12 Here we briefly discuss the electronic structure of Ice-XI, with the partial DOS and dispersion curves along the high symmetry lines in Fig. 6. There are five separated bands. We use here the labels used in the early assignments15,16,73 to designate the bands. Two of them, band 1b2 at −6 eV, and band 1b1 at the top of the valence band (set to 0 eV), show an atomic-orbital behavior with very small band widths. The lowest one (2a1) belongs to the O 2s states hybridized with H 1s orbitals with two peaks at about −17.7 eV and −16.8 eV, respectively. The width of the O 2s bands is about 2 eV, with the Fermi level being set at the top of the valence band. This indicates strong interactions between the O 2s and H 1s states. However, they do not contribute to the chemical bonding since both bonding and antibonding states are fully occupied.
There is a gap of about 10.4 eV between the O 2s band (2a1) and the O 2p (1b2) states with a significant hybridization with H 1s states. The very narrow band indicates a strongly localized character of the O 2p–H 1s bonding corresponding to direct bonding. The next band with two peaks at about −3.2 eV (bonding 3a1) and −1.6 eV (anti-bonding 3a1*) is dominated by O 2p states mixed with some H 1s character. The top valence band belongs to the non-bonding O 2p states (1b1). This band is very narrow and shows a strong localized character in nature. The lower part of the conduction band (from about 5.6 to about +14.5 eV) is dominated by O 2p/3s states mixed with some H 1s character. These states in the lower conduction band show significant dispersion over 5 eV. There is a peak at about 14.5 eV dominated by O 2p–H 1s states (anti-bonding, 4a1).
The van der Waals density functional produced an energy gap of 5.55 eV. The top of the valence band is at (1/4,0,0) in the Brillouin Zone while the bottom of the conduction band is at Γ(0,0,0). The direct energy gap at Γ is 5.58 eV, just slightly larger than the indirect gap. The calculated energy gap is smaller than the experimental value of 8.75 eV,17 which is not surprising since DFT functionals are known to underestimate the energy gaps of insulators and semiconductors.52,72
We return to Fig. 5 for the electronic structure of a snapshot of water at 300 K. Though similar to that of the Ice-XI phase, the tDOS curves of water at 300 K are more dispersed: there are localized states with tailed states near the band gap. As mentioned before, the present cell containing 64H2O molecules can be regarded as a representative of a small fraction of a real system. Therefore, to have a better understanding of an amorphous or a liquid system simulated with a limited number of atoms, statistics are required based on a number of samples, rather than based on one or a few samples, to produce physically meaningful results.32,54,72 Water is an insulator with strong local distortions, though topologically similar to that of ice phases.1–3 Correspondingly the DOS curve is composed of very localized states (Fig. 5). As shown in Fig. 4b there are broad O–H bonds and the distributions of charges at O (Fig. 4c) and at H (Fig. S2, ESI†) in water. It is also notable that although there is a short hydrogen bond in the 2(H2O) clustering (Fig. S1, ESI†), this clustering contributes nothing to the top of the valence band and the bottom of the conduction band, as shown in Fig. S5 (ESI†).
That is understandable since the electronic structure of water is determined mainly by the short O–H bonds in the water molecule. In fact our analysis showed a change of the Fermi level in a range of about 0.9 eV for water in 2 ps (Fig. S3, ESI†). Meanwhile, as shown in Fig. S3 (ESI†), the lowest unoccupied eigen-energies vary, albeit only slightly (0.1 eV). The major change of the DOS comes from the top of valence states due to the wide local bonding. Therefore, the integrated tDOS can be considered as a representative of the electronic properties of water as a whole (Fig. 5). Clearly the integrated tDOS have broader bandwidths for each band. There are tailed states at the edges of the valence band and conduction bands. This results in a band gap of about 3.9 eV for the present calculations, largely due to the tailed states at the top of the valence band. The overall electronic characteristics are shown in Table 5.
| Author year | Method | (3a1/3a1*)b-1b1 | (1b2/1b2*)b-1b1 | (2a1–2a1*)b-1b1 | ΔEgap | |
|---|---|---|---|---|---|---|
| a There is only one peak for each of the 2a1, 1a2 and 3a1 states for one H2O molecule (Fig. 5). b The states are overlapped for liquid water. | ||||||
| H2O mono. | This work | optB88-vdW | 1.94a | 5.88a | 17.64a | 6.02 |
| Couto 2005 | GGA_MPW1 | 2.133/ | 6.033 | 12.433 | ||
| Banna 1986 | Exp. | 2.2071 | 6.1871 | 20.0271 | ||
| H2O dimer | This work | optB88-vdW | 3.47/1.82/1.38 | 7.33/5.82 | 19.04/17.54 | 5.29 |
| Couto 2005 | GGA_MPW1 | 3.7/2.1/1.433 | 7.5/6.133 | 23.0/21.333 | 11.3333 | |
| HF | 3.5/2.0/1.333 | 7.3/6.133 | 24.6/23.233 | 13.9733 | ||
| Ice-XI | This work | optB88-vdW | (2.99/1.47) | 5.60 | 17.06 | 5.55 |
| GW0 | (3.36/1.65) | 6.29 | 18.20 | 9.17 | ||
| Banna 1986 | Exp. | 1.971 | 5.371 | 18.771 | 8.7571 | |
| Nordlund 2008 | Exp. | (2.40/1.80)19 | 6.0419 | |||
| Liquid | This work | optB88-vdW | 1.9 | 5.7 | 17.3 | ∼3.9 |
| S_GW0 | 2.1 | 6.4 | 19.5 | ∼7.3 | ||
| Shibaguchi 1977 | Exp. | 2.3517 | 6.1817 | 19.4017 | ||
| Banna 1986 | Exp. | 2.271 | 6.471 | 20.071 | ||
| Winter 2004 | Exp | 2.3419 | 6.3819 | 19.7419 | ||
| Nordlund 2008 | Exp | 2.5020 | 6.3720 | |||
| Coe 1997, 2001 | Exp. | 6.921,22 | ||||
III.D. GW0 corrections and comparison with experimental observations
It is clear from Table 5 that there are many differences between the calculated electronic properties of the ice phase and water and the experimental photoemission spectra (PES) measurements. This is due to the fact that the DFT approaches describe well the ground states of electronic properties of materials, while most experimental methods, e.g. PES, are sensitive to the excitation of electrons. The GW method, a beyond-DFT approach, is very suitable to describe the excitation processes of sp electrons (a quasi-particle (QP) approach) and has been successfully applied to many systems.49,50,52,53,70 For example, Hermann and co-workers performed a non-self-consistent G0W0 correction for ice.31,32 The requirements for computational capability limit us to the Ice-XI phase. Fig. 7 shows the results of partially self-consistent GW0 corrections for Ice-XI.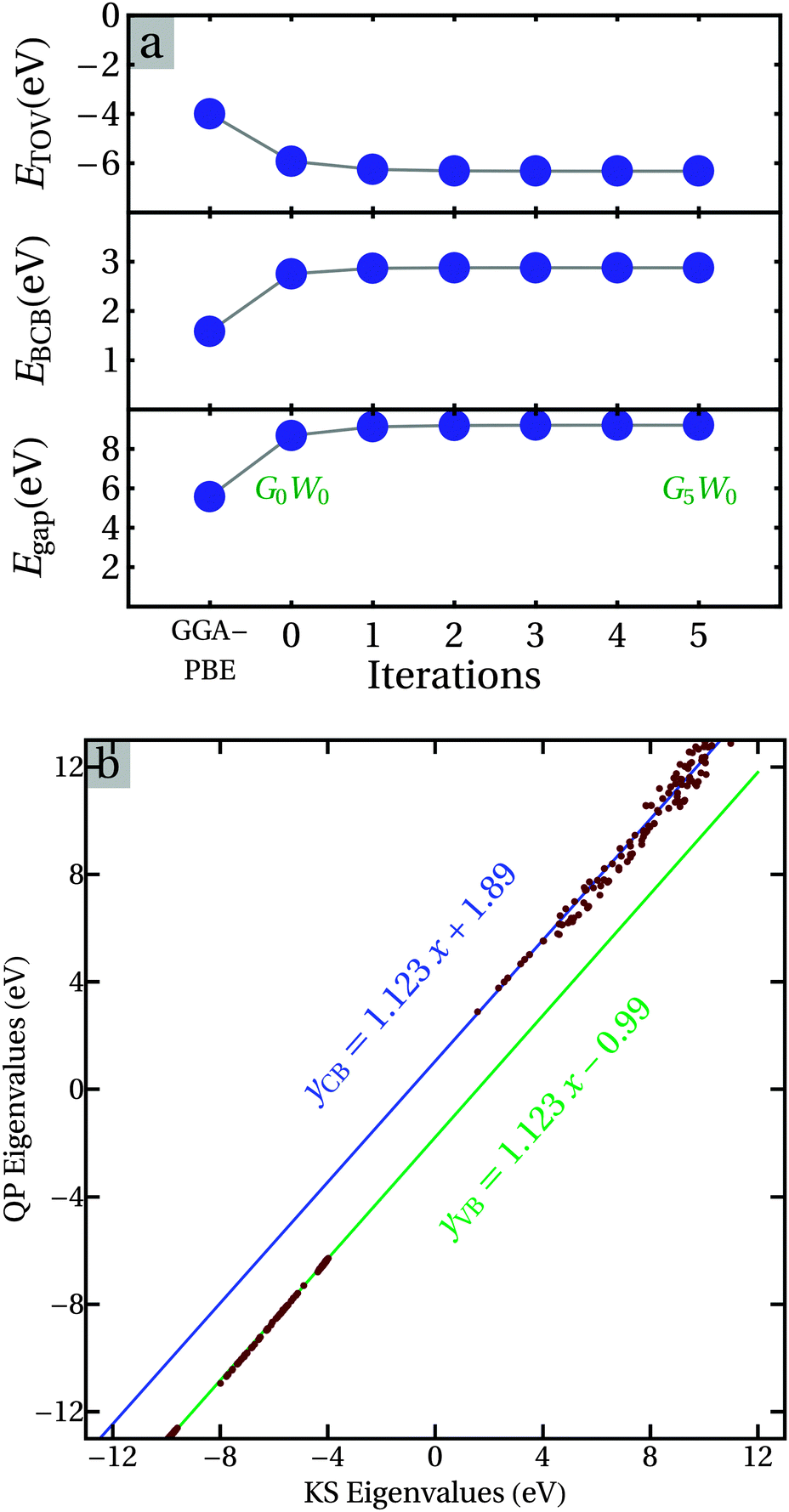 | ||
| Fig. 7 Convergence of the partially self-consist GW0 calculations (a), and relationships between the DFT-GGA K–S eigenvalues and GW0 QP energies (b). The green and blues lines represent the fitting functions. | ||
The calculations show that the direct GW0 correction has already improved the K–S energy gap (DFT-PBE) significantly. About 3 iterations are required to reach convergence (Fig. 7a). Fig. 7b shows that there is almost linear relationship between the QP energies and the Kohn–Sham (KS) eigenvalues for the sp electrons in Ice-XI. The first itineration (G0W0) provided an energy gap of about 8.66 eV which is strongly enhanced as compared to that (5.55 eV) from the van der Waals density functional theory. This G0W0 value is also close to that (about 8.8 eV) obtained by Hermann and co-workers.31,32 Furthermore, the QP energy gap from the GW0 correction is about 9.2 eV, considerably larger than that of the KS energy gap, as shown in Table 5. This obtained QP band gap is close to the experimental value, 8.75 eV.71
Considering the topological similarity between the regular structure of the ice phase and water, it is reasonable to use the obtained linear relationships between the QP energies and KS eigenvalues of Ice-XI as a correction to the electronic structure of water. Fig. 8 shows the obtained tDOS curves for simulated water in comparison with the PES spectra.19,20,75Table 5 lists the obtained electronic characteristics of the eigen-energies of water and the ice phase. It is clear that the obtained vdW-DFT tDOS curves have the peak mismatch with the experimental observations.
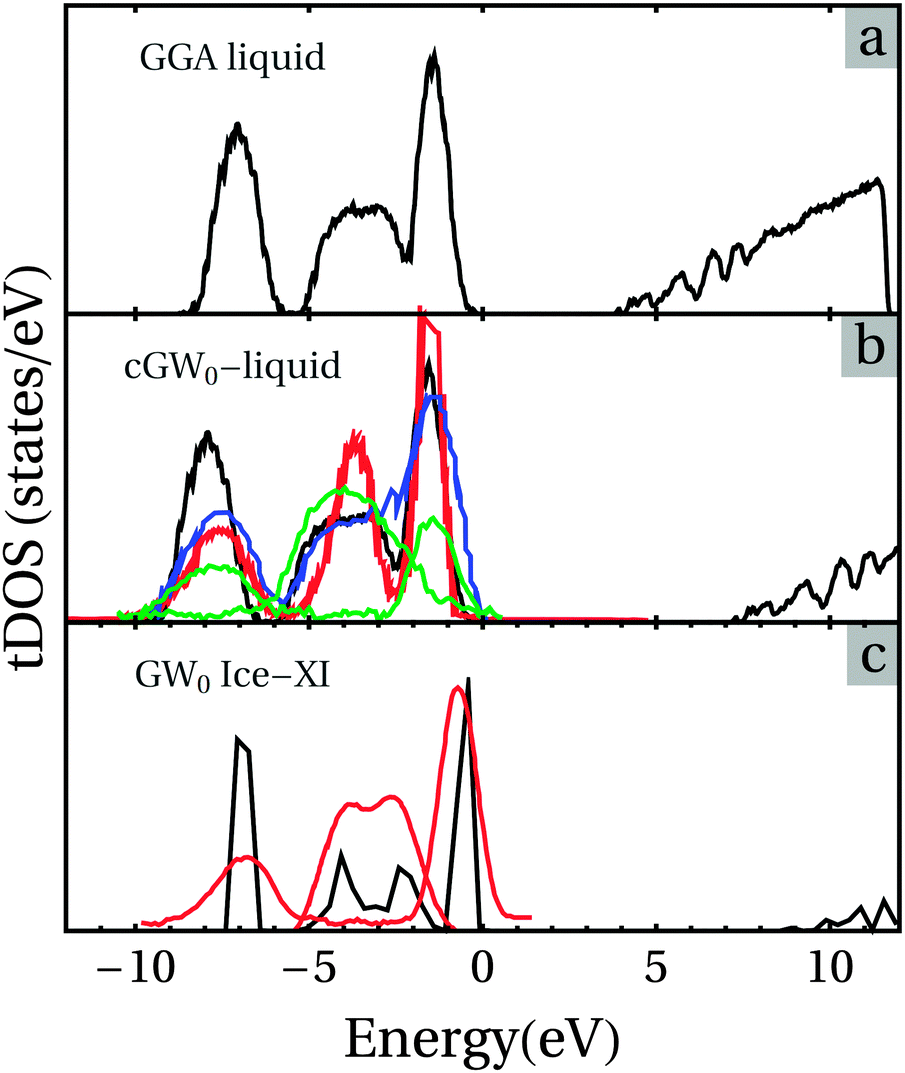 | ||
| Fig. 8 The time-averaged tDOS curves obtained by the optB88-vdW calculations (a) and its QP tDOS (b) of water with the GW-correction using the relationships between the DFT KS-eigenvalues and the GW0 QP energies (see Fig. 6b and the corresponding text). The GW0 tDOS curve for the Ice-XI phase is shown in figure (c) for the sake of comparison. The Fermi level for all the curves is set to be the top of the valence bands. The colored curves in (b) and (c) are experimental measurements: in (c) the red curve for ice,20 in (b) for water from ref. 75 (red line), from ref. 19 (blue line) and from ref. 20 (green lines). The black line represents the calculated results using the GW0 corrections. | ||
This can be understood from the fact that standard DFT provides the electronic properties of a solid at its ground state while the PES measurements are related to excitons of electrons, as mentioned before. There is excellent agreement between valence bands of the QP tDOS and the PES observations (also in Fig. 8). The peaks of the hybridized O 2s and H 1a states (2a1) match well with the PES measurements (not shown in Fig. 8). Naturally, the heights/intensities of the peaks are dependent on the cross-sections of the photons with the characteristics of electrons, but the positions of the peaks can be easily recognized and are listed in Table 5. The obtained energy gap for the integrated tDOS of water at 300 K is about 7.3 eV for the QP, which is much higher than that of the KS gap (about 3.9 eV). The QP gap of 7.3 eV is in between the experimental values ranging from about 6.9 to 9.0 eV. The energy gap found in the present work is slightly larger than the predicted values (6.01 to 6.55 eV) obtained from a combination of configuration interaction (CI) and time-dependent DFT (TD-DFT) calculations.76,77 In particular, the QP gap is in very good agreement with the recent experimental measurements of the band gap of 6.9 eV.21,22
IV. Conclusions
Using the first-principles van der Waals density functional, we performed structural optimizations for the isolated H2O monomer, its dimer, and the crystalline ice phase. The obtained results agree well with the available experimental values and former theoretical calculations. First-principles molecular-dynamics simulations for water at 300 K showed the high stability of the structural element H2O, whereby configurations of the H2O elements retain tetragonal coordination with distortions. The Bader charge analysis showed that water is strongly ionic with a charge transfer of 0.64e/H to O.To describe the electronic structure and in particular the electronic band gap of liquid water with high accuracy, we have employed the GW correction based on the relationship between the Kohn–Sham (KS) eigenvalues and the quasi-particle (QP) eigenvalues of the ice phase. Moreover, we have taken about one hundred snapshot configurations of liquid water with 192 atoms in the simulation cell, and performed statistical time averaging to obtain a more reliable estimate of the physical properties. Overall, the self-consistent GW0 corrections produce electronic structures and electronic band gaps of both Ice-XI and water, which are in excellent agreement with experimental observations. The present knowledge is helpful in understanding many processes and reactions in water and aqueous solutions, such as sol–gel silica processes.74,78 We suggest this approach as an accurate method to investigate the electronic properties of aqueous solutions and chemical reactions therein.
Acknowledgements
We thank Dr Bing Liu (UU, NL) for useful discussions. MvH acknowledges a VIDI grant from the Dutch Science Foundation NWO. JK was supported by the Austrian Science Fund (FWF) within the SFB ViCoM (Grant F 41).References
- V. Bartels-Rausch, J. H. E. Bergeron, R. Cartwright, J. L. Escribano, H. Finney, P. J. Grothe, J. Gutijėrrez, W. F. Haapala, J. B. C. Kuhs, S. D. Pettersson, C. I. Price, D. J. Sainz-Diaz, G. Stokes, E. S. Strazzulla, H. Thomson, H. Trinks and N. Uras-Aytemiz, Rev. Mod. Phys., 2012, 84, 885 CrossRef.
- B. Winter and M. Fauber, Chem. Rev., 2006, 106, 1176–1211 CrossRef CAS PubMed.
- J. D. Bernal, Proc. R. Soc. London, Ser. A, 1958, 247, 421 CrossRef CAS.
- J. D. Bernal, Proc. Phys. Soc. A, 1949, 62, 537 CrossRef.
- R. H. Fowler and J. D. Bernal, Trans. Faraday Soc., 1933, 29, 1049–1056 RSC.
- J. D. Bernal and R. H. Fowler, J. Chem. Phys., 1933, 1, 515 CrossRef CAS PubMed.
- B. Q. Christner, C. E. Morris, C. M. Foreman, R. M. Cai and D. C. Sands, Science, 2008, 319(5867), 1214 CrossRef CAS PubMed.
- H. Iglev, M. Schmeisser, K. Simeonidis, A. Thaller and A. Laubereau, Nature, 2006, 439(7073), 183–186 CrossRef CAS PubMed.
- B. J. Murray, D. A. Knopf and A. K. Bertram, Nature, 2006, 434(7030), 202–205 CrossRef PubMed.
- Y. Fang, B. Xiao, J. M. Tao, J. W. Sun and J. P. Perdew, Phys. Rev. B, 2013, 87, 214101 CrossRef.
- S. Casassa, M. Calatayud, K. Doll, C. Minot and C. Pisani, Chem. Phys. Lett., 2005, 409, 110 CrossRef CAS PubMed.
- C. Vega, J. L. F. Abascal and P. G. Debenedetti, Phys. Chem. Chem. Phys., 2011, 13, 19660 RSC.
- J. L. Finney, J. Phys.: Conf. Ser., 2007, 57, 40–52 CrossRef CAS.
- J. R. Errington and P. G. Debenedetti, Nature, 2001, 409, 318–321 CrossRef CAS PubMed.
- R. E. Verrall and W. A. Senior, J. Chem. Phys., 1968, 50, 2746 CrossRef PubMed.
- L. R. Painter, R. N. Hamm, E. T. Arakawa and R. D. Birkhoff, Phys. Rev. Lett., 1968, 21, 282–284 CrossRef CAS.
- T. Shibaguchi, H. Onuki and R. Onanka, J. Phys. Soc. Jpn., 1977, 42, 152–158 CrossRef.
- N. Timneanu, C. Caleman, J. Hajdu and D. van der Spoel, Chem. Phys., 2004, 299, 277 CrossRef CAS PubMed.
- B. Winter, R. Weber, W. Widdra, M. Dittmar, M. Fauber and I. V. Hertel, J. Phys. Chem. A, 2004, 108, 2625 CrossRef CAS.
- D. Nordlund, M. Odelius, H. Bluhm, H. Ogasawara and L. G. W. Pettersson, Chem. Phys. Lett., 2008, 460, 86 CrossRef CAS PubMed.
- J. V. Coe, A. D. Earhart, M. C. Cohen, G. J. Hoffman, H. W. Sarkas and K. H. Bowen, J. Chem. Phys., 1997, 107, 6023 CrossRef CAS PubMed.
- J. V. Coe, Int. Rev. Phys. Chem., 2001, 20, 33–58 CrossRef CAS.
- D. Grand, A. Bernas and E. Amouyal, Chem. Phys., 1979, 44, 73 CrossRef CAS.
- A. Bernas, C. Ferradini and J.-P. Jay-Gerin, Chem. Phys., 1997, 222, 151 CrossRef CAS.
- T. Goulet, A. Bernas, C. Ferradini and J.-P. Jay-Gerin, Chem. Phys. Lett., 1990, 170, 492 CrossRef CAS.
- G. T. Barkema and J. de Boer, J. Chem. Phys., 1993, 99, 2059 CrossRef CAS PubMed.
- G. T. Barkema and M. W. J. Newman, Phys. Rev. E, 1998, 57, 1155 CrossRef CAS.
- H. Itoh, K. Kawamura, T. Hondoh and S. Mae, J. Chem. Phys., 1998, 109, 4894 CrossRef CAS PubMed.
- N. Guisoni and V. B. Henriques, J. Chem. Phys., 2001, 115, 5238 CrossRef CAS PubMed.
- R. Ramirez, N. Neuerburg and C. P. Herro, J. Chem. Phys., 2012, 137, 134503 CrossRef CAS PubMed.
- A. Hermann and P. Schwerdtfeger, Phys. Rev. Lett., 2011, 106, 187403 CrossRef.
- A. Hermann, N. W. Ashcroft and R. Hoffmann, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 745 CrossRef CAS PubMed.
- P. Cabral do Couto, S. G. Estácio and B. J. Costa Cabral, J. Chem. Phys., 2005, 123, 054510 CrossRef CAS PubMed.
- P. L. Silvestrelli and M. Parrinello, J. Chem. Phys., 1999, 111, 3572 CrossRef CAS PubMed.
- X. Xu and W. A. Goddard III, J. Chem. Phys., 2004, 121, 4068 CrossRef CAS PubMed.
- X. Xu and A. Goddard III, J. Phys. Chem. A, 2004, 108, 2305 CrossRef CAS.
- D. Nordlund, H. Ogasawar, K. J. Andersson, M. Tatarkhanov, M. Salmerón, L. G. M. Petterson and A. Nilson, Phys. Chem. Chem. Phys., 2006, 8, 1985–1993 RSC.
- P. Jurečka, J. Šponer, J. Černy and P. Hobza, Phys. Chem. Chem. Phys., 2006, 8, 1985–1993 RSC.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- J. Klimeš, D. R. Bowler and A. Michelides, J. Phys.: Condens. Matter, 2010, 22, 022201 CrossRef PubMed.
- B. Santra, J. Klimeš, D. Alfe, A. Tkatchenko, B. Slater, A. Michaelides, R. Car and M. Scheffler, Phys. Rev. Lett., 2011, 107, 185701 CrossRef.
- B. Santra, J. Klimeš, D. Alfe, A. Tkatchenko, B. Slater, A. Michaelides, R. Car and M. Scheffler, J. Chem. Phys., 2013, 139, 154702 CrossRef PubMed.
- J. Carrasco, J. Klimeš and A. Michaelides, J. Chem. Phys., 2013, 138, 024708 CrossRef PubMed.
- G. Román-Peréz and J. M. Soler, Phys. Rev. Lett., 2009, 103, 096102 CrossRef.
- G. Graziano, J. Klimeš, F. Fernandez-Alonso and A. Michaelides, J. Phys.: Condens. Matter, 2012, 24, 424216 CrossRef PubMed.
- J. Klimeš and A. Michaelides, J. Chem. Phys., 2012, 137, 120901 CrossRef PubMed.
- M. Macher, J. Klimeš, C. Franchini and G. Kresse, J. Chem. Phys., 2014, 140, 084502 CrossRef CAS PubMed.
- R. G. Parr, Annu. Rev. Phys. Chem., 1983, 34, 631 CrossRef CAS.
- V. G. Ruiz, W. Liu, E. Zojer, M. Scheffler and A. Tkatchenko, Phys. Rev. Lett., 2012, 108, 146103 CrossRef.
- P. Rinke, A. Qteish, J. Neugebauer, C. Feysoldt and M. Schffler, New J. Phys., 2006, 7, 126 CrossRef.
- G. Kresse, M. Marsman, L. E. Hintzsche and E. Flage-Larsen, Phys. Rev. B, 2012, 85, 045205 CrossRef.
- L. E. Hintzsche, C. M. Fang, T. Watts, M. Marsman, G. Jordan, M. W. P. E. Lamers, A. W. Weeber and G. Kresse, Phys. Rev. B, 2012, 86, 235204 CrossRef.
- E. Flage-Larsen, O. M. Løvvik, C. M. Fang and G. Kresse, Phys. Rev. B, 2013, 88, 165310 CrossRef.
- R. F. W. Bader, T. T. Nguyen and Y. Tal, Rep. Prog. Phys., 1981, 44, 893 CrossRef.
- C. M. Fang, M. A. Van Huis, J. Jansen and H. W. Zandbergen, Phys. Rev. B, 2011, 84, 094102 CrossRef.
- C. M. Fang, M. H. F. Sluiter, M. A. van Huis and H. W. Zandbergen, Phys. Rev. B, 2012, 86, 134114 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B, 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B, 1994, 49, 14251 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B, 1994, 50, 17953–17979 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1999, 54, 11169 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188 CrossRef.
- J. C. G. Pereira, C. R. A. Catlow, G. D. Price and R. M. Almeida, J. Sol-Gel Sci. Technol., 1997, 8, 55 CAS.
- L. A. Curtiss and J. A. Pople, J. Mol. Spectrosc., 1975, 55, 1 CrossRef CAS.
- A. J. Leadbetter, R. C. Ward, J. W. Clark, P. A. Tucker, T. Matsuo and H. Suga, J. Chem. Phys., 1985, 82, 424 CrossRef CAS PubMed.
- I. D. Brown, The Chemical Bond in Inorganic Chemistry. IUCr Monographs in Crystallography 12, Oxford Science Publications, Oxford University Press, Oxford, U.K., 2002 Search PubMed.
- I. D. Brown, Chem. Rev., 2009, 109, 6858–6919 CrossRef CAS PubMed.
- G. P. Parravicini and L. Resca, Phys. Rev. B, 1973, 8, 3009 CrossRef.
- W. A. Goddard III and W. J. Hunt, Chem. Phys. Lett., 1974, 24, 464 CrossRef.
- L. E. Hintzsche, C. M. Fang, M. Marsman, G. Jordan, M. W. P. E. Lamers, A. W. Weeber and G. Kresse, Phys. Rev. B, 2013, 88, 155204 CrossRef.
- M. S. Banna, B. H. McQuaide, R. Malutzki and V. Schmidt, J. Chem. Phys., 1986, 84, 4739 CrossRef CAS PubMed.
- M. E. Tuckerman, D. Marx and M. Parrinello, Nature, 2002, 417, 925 CrossRef CAS PubMed.
- A. Hassanali, M. K. Prakash, H. Eshet and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20410 CrossRef CAS PubMed.
- T. T. Trinh, A. P. J. Jansen, R. A. Van Santen and E. J. Meijer, J. Phys. Chem. C, 2009, 113, 2647 CAS.
- K. C. Chen, T. Tsuchiya and J. D. Mackenzie, J. Non-Cryst. Solids, 1986, 81, 227 CrossRef CAS.
- G. Brancat, N. Rega and V. Barone, Phys. Rev. Lett., 2008, 100, 107401 CrossRef.
- P. C. do Couto and B. J. Costa Cabral, J. Chem. Phys., 2007, 126, 014509 CrossRef PubMed.
- F. F. Xia, D. W. Zeng, H. B. Yi and C. H. Fang, J. Phys. Chem. A, 2013, 117, 8468 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures are available. See DOI: 10.1039/c4cp04202f |
| This journal is © the Owner Societies 2015 |