
Decarboxylation of oleic acid over Pt catalysts supported on small-pore zeolites and hydrotalcite
M.
Ahmadi
a,
A.
Nambo
a,
J. B.
Jasinski
b,
P.
Ratnasamy†
b and
M. A.
Carreon
*c
aDepartment of Chemical Engineering, University of Louisville, Louisville, KY 40292, USA
bConn Center for Renewable Energy Research, University of Louisville, Louisville, KY 40292, USA
cChemical and Biological Engineering Department, Colorado School of Mines, Golden CO 80401, USA. E-mail: mcarreon@mines.edu
First published on 28th August 2014
Abstract
The catalytic decarboxylation and further conversions of oleic acid to paraffins, branched and aromatic hydrocarbons over Pt supported on small pore zeolites and hydrotalcite are demonstrated. The influence of the support, platinum source, and reaction temperature on the decarboxylation of oleic acid was investigated. An increase in reaction temperature increased the degree of decarboxylation and selectivity to heptadecane. Pt-SAPO-34 was a very effective catalyst. In addition to a high degree of decarboxylation, Pt-SAPO-34 displayed a high selectivity to heptadecane and dodecylbenzene among the products. Branched isomers, cracked (mostly <C17) paraffins, alkenes such as undecene and dodecene, and carboxylic acids such as nonanoic acid and decanoic acid were observed as side products. The further isomerization of the initially formed linear heptadecane (by decarboxylation of oleic acid) to branched isomers is suppressed in the narrow pores of SAPO-34 due to restricted transition state shape selectivity limitations in the pore system of SAPO-34. Catalyst acidity, dispersion of Pt and the pore diameter of the support played crucial roles in determining product selectivity.
1. Introduction
Biofuel production technologies have recently received considerable attention as CO2 emission of biofuels is lower compared to that of conventional fuels.1 Some of the sources of biofuels are natural oils and fats (e.g., animal fats, plant and seed oils). They are complex mixtures of triglycerides, which are fatty acid esters of glycerol. The carbon chain lengths of the fatty acids range from about C8 to C24, but C12, C16, and C18 fatty acids tend to be the most abundant.2Removal of oxygen from fatty acids (deoxygenation) leads to the formation of paraffinic hydrocarbons that can potentially serve as or be converted to direct replacements for traditional petroleum-derived liquid transportation fuels and paraffinic petrochemical feedstocks.3 Deoxygenation of unsaturated fatty acids is accomplished via hydrogenation of double bonds and further removal of the carboxyl group by releasing carbon dioxide and producing a paraffinic hydrocarbon (decarboxylation) or by releasing carbon monoxide and producing an olefinic hydrocarbon (decarbonylation).4–7 Intensive studies on liquid-phase deoxygenation of free fatty acids and fatty acid esters have been performed.6–15 Metals such as Ni, Ni/Mo, Ru, Pd, Pd/Pt, Pt, Ir, Os and Rh supported on different supports have been evaluated for their deoxygenation performance under elevated temperatures and pressures. The effect of the nature of the carrier gas on conversion and selectivity values was investigated by previous groups. Maier et al.16 deoxygenated aliphatic and aromatic carboxylic acids over Pd/SiO2 and Ni/Al2O3 catalysts at 330 °C in different environments (H2 or N2) in the gas phase and found that the presence of hydrogen is desirable to maintain stable catalytic activity during the decarboxylation reaction. In contrast, no reaction occurred under a nitrogen atmosphere. Snare et al.7 conducted liquid-phase deoxygenation experiments in a continuous flow reactor under Ar and H2 atmosphere and solvent-free conditions using a Pd/C catalyst. However, the amount of hydrocarbons formed, mainly olefins and aromatics, was below 10 mol%. Representative examples of catalysts employed for the conversion of stearic acid to heptadecane include selenium,17 complexes of Pd and Rh,18 and Pd–C.19 However, in these studies, the yield of heptadecane or stearic acid conversion was low. Some studies on the decarboxylation of the unsaturated oleic acid have also been reported.7,20–23 In most of the above studies, the support is a catalytically inert material (like carbon) or a relatively non-acidic component, such as silica or non-acidic alumina. To the best of our knowledge, the influence of stronger acidic supports on the modification of the carbon skeleton of linear C16–C18 paraffins (initial products of the decarboxylation of the fatty acids) by isomerization, cracking, cyclization, hydrocracking, etc. has not been explored. In contrast, the hydrocracking and isomerization reactions of hydrocarbons, especially those boiling in the gasoline range (C5–C10), over Pt supported on strongly acidic supports (like Pt-zeolites) have been extensively studied. The strength and surface density of the acidic sites as well as the pore diameter of the acidic support are known to influence the hydrocarbon distribution among the products. To satisfy the diesel blend requirements of diesel vehicles, hydrocarbons with different conformations (paraffin mixtures) should be produced from triglycerides and fatty acids. Since these normal paraffin (mainly n-C12 to n-C20) mixtures have a high cetane number, they exhibit excellent performance. On the other hand, the pour point of these paraffin mixtures is high, thus their structure should be modified to fulfill the requirements of diesel blending components (the pour point should be below 5 °C). For this purpose, the catalytic isomerization of normal to isoparaffins is often used to lower their pour points.24,25
Hydroisomerization reactions of normal paraffins are conducted over bi-functional catalysts. The degree of isomerization, the product yield and the composition are significantly affected by the properties of the used catalyst (acidity, surface area and pore size distribution) and the applied operational parameters (temperature and pressure). Isomerization occurs easily in the presence of different noble metal-containing zeolites (ZSM-5, ZSM-22), mesoporous structures (MCM-41, Al-MCM-41) and silica–alumina–phosphates (SAPO-11, SAPO-31, SAPO-41) as they have mild acidity.25 Some catalysts like SAPO-11, SAPO-31, SAPO-41 have been found to be favorable for the isomerization of long-chain paraffins.26 Studies on isomerization of paraffins over Pt-SAPO-11, Pd-SAPO-11, and Pt-SAPO-5 have been reported in the literature.25–29 In addition to acidity, the pore diameter of the catalyst also plays a crucial role in determining product selectivity.
If the deoxygenation of the fatty acid and subsequent isomerization/hydrocracking of the resulting C17–C18 linear paraffins to branched, gasoline-range C5–C10 hydrocarbons with lower pour points and higher octane numbers can be accomplished over a bi-functional catalyst in a single reactor, it will represent significant advancement in the process of conversion of lipid biomass materials to ‘drop-in’ hydrocarbon transport fuels, such as motor and aviation gasoline. In our previous work, we have demonstrated the catalytic decarboxylation and further conversion of oleic acid to branched and aromatic hydrocarbons in a single-step process over Pt-SAPO-11 and Pt/chloride Al2O3.30 Herein, we report the catalytic decarboxylation of oleic acid and further conversion of the resulting linear paraffins over catalysts of varying acidity and pore diameter, namely, Pt/SAPO-34, Pt/DNL-6, Pt/RHO and Pt/hydrotalcite in a single-process stage. Such an investigation has not been reported so far.
2. Experimental
2.1 Catalyst preparation and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P:Si:DEA:CTAB:H2O = 1:0.8:0.2:1:0.1:50. The final gel mixture was transferred into a stainless steel autoclave and held at 200 °C for 24 h. After 24 h, the autoclave was cooled down and the solid product was collected by centrifugation, washed three times with deionized water and dried at 110 °C overnight. Calcination was done at 600 °C for 4 h to remove the organic species (the calcination heating and cooling rates to remove the organic species were 1 and 2 °C min−1, respectively).
:0.3Cs2O:Al2O3:10SiO2:0.5R:100H2O (where R is the crown ether 18-C-6). Zeolite crystallization was done in an autoclave at 150 °C for 1 day. The zeolite RHO was recovered after filtration, washing with water and drying at 100 °C overnight. Then, it was calcined at 500 °C for 3 hours to remove the organic species (the calcination heating and cooling rates to the remove organic species were 1 and 2 °C min−1, respectively).
:P:Si:DEA:CTAB:H2O = 1:0.8:0.2:1:0.1:50. The final gel mixture was transferred into a stainless steel autoclave and held at 200 °C for 24 h. After 24 h, the autoclave was cooled down and the solid product was collected by centrifugation, washed three times with deionized water and dried at 110 °C overnight. Calcination was done at 600 °C for 4 h to remove the organic species (the calcination heating and cooling rates to remove the organic species were 1 and 2 °C min−1, respectively).
:0.3Cs2O:Al2O3:10SiO2:0.5R:100H2O (where R is the crown ether 18-C-6). Zeolite crystallization was done in an autoclave at 150 °C for 1 day. The zeolite RHO was recovered after filtration, washing with water and drying at 100 °C overnight. Then, it was calcined at 500 °C for 3 hours to remove the organic species (the calcination heating and cooling rates to the remove organic species were 1 and 2 °C min−1, respectively).
The four supports were impregnated with two different sources of Pt (potassium hexachloroplatinate and tetraammine Pt nitrate, denoted as salt 1 and 2, respectively) at 5 wt%. 0.126 g of the Pt source (salt 1 or salt 2) was dissolved in deionized water (0.7 cm3 per gram of support) and then impregnated on the support while the suspension was stirred. The mixture was further dried overnight at 100 °C followed by calcination at 400 °C for 5 h and then reduction under flowing hydrogen at 450 °C for 2 h to remove the ligands of the precursor and to reduce the metal to its active elemental state. The Pt ions in the impregnating solution will have access to the pores of all of the catalysts used in this study. Catalytic activity, however, is determined by the access of the reactant, oleic acid, to the Pt sites. In view of the large size of the oleic acid molecule (especially that of the cis isomer, the predominant fraction) and the small pore diameter of SAPO-34, RHO and DNL-6, most of the decarboxylation reactions (for which the active sites are the Pt sites) are expected to occur on the outer, exposed surface of these molecular sieve catalysts.
The catalysts were characterized by X-ray diffraction (XRD), nitrogen adsorption (BET), transmission electron microscopy (TEM) and scanning electron microscopy (SEM). XRD patterns were collected using a Bruker D8 Discover diffractometer at 40 kV and 40 mA with Cu Kα radiation. BET surface areas and N2 adsorption–desorption isotherms were collected with a MicroMeritics TriStar-3000 porosimeter at 77 K, using liquid nitrogen as the coolant. Before measurements, the catalyst was degassed at 150 °C for 3 h. Transmission electron microscopy (TEM) was performed to inspect the morphology of catalysts. TEM images were taken with a Tecnai F20 FEI TEM using a field emission gun, operating with an accelerating voltage of 200 kV. Scanning electron microscopy (SEM) was performed using a FE-SEM (FEI Nova 600) with an accelerating voltage of 6 kV.
2.2 Reaction procedure
Oleic acid (90%, Alfa Aesar) was used as the unsaturated fatty acid. Before reaction, the catalysts were preactivated under dry nitrogen in an oven for 3 h at 150 °C. The degassing temperature (150 °C) was chosen (from thermogravimetric studies of the catalyst) to ensure the removal of all adsorbed H2O. Three hours was found to be adequate to remove all of the adsorbed water. When preactivation was not performed, catalytic activity was low presumably due to the retarding effect of adsorbed H2O molecules. The decarboxylation reactions were conducted in a 250 ml stainless steel, high-pressure autoclave reactor (Parr model 4576A). Oleic acid and the catalyst with a mass ratio of 20:1 were loaded into the reactor. Before the reaction started, the reactor was flushed with H2 and the pressure was increased to the desired reaction pressure (usually 20 bar). Under constant stirring conditions, the reactor was heated at a rate of 10 °C min−1 to the reaction temperature (245 and 325 °C). The catalysts were reduced in hydrogen at 200 °C and a pressure of 20 bar for 2 h. One mole of H2 is needed for conversion of oleic acid to heptadecane. The reaction conditions (temperature (200–350 °C), H2 pressure and reaction time of 2 h) were chosen from our previous study on the decarboxylation of fatty acids30 to optimize the yield of hydrocarbons. At longer reaction times, cracked products (<C9) were observed in significant quantities. After the reaction, the catalyst was separated from the liquid product by filtration and washed with acetone for further characterization.
2.3 Product analysis
Besides the organic liquid and gas-phase hydrocarbons, a small amount of water was also observed in some cases. However, the amount was too small to be analyzed quantitatively. The liquid-phase product withdrawn from the reactor was analyzed using a gas chromatograph (GC, 7820 A) equipped with a HP-5ms column (with dimensions of 30 m × 250 μm × 0.25 μm) and a 5975 MSD detector. For the analysis, the samples were silylated with N,O-bis(trimethyl)-trifluoroacetamide (BSTFA, 98%, Acros Organics). After addition of the silylation agent, the samples were kept at 60 °C for 1 h. Silylation was incomplete at lower temperatures and shorter times. 0.2 μl of the sample was injected into the GC column (225 °C, 10.5 psi) with a split ratio of 20:1; the carrier gas (helium) flow rate was 1 ml min−1.22 The following temperature program of the gas chromatograph was used in the analysis: 100 °C for 5 min, 300 °C (20 °C min−1) for 2 min. Quantitative analysis was accomplished by generating and using calibration curves for each compound of interest. The products were identified and confirmed with a gas chromatograph-mass spectrometer (GC-MS).
To quantify the acid number, a known amount of the sample (about 0.1 g) is dissolved in a solvent (ethanol + petroleum ether) then titrated with a solution of sodium hydroxide (NaOH, 0.1 N) using phenolphthalein as a color indicator.
The acid number is calculated using this equation:
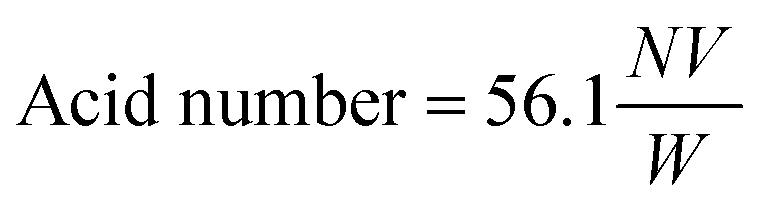
N = 0.1 (N)
V = volume of NaOH consumed (ml)
W = mass of the sample (g)
The degree of decarboxylation (%) was calculated using the acid number of oleic acid and the acid number of the product using the following equation:
| % decarboxylation = (acid number of oleic acid − acid number of the product)/acid number of oleic acid × 100% |
3. Results and discussion
The SEM images of RHO, DNL-6, SAPO-34 and hydrotalcite supports are shown in Fig. 1. Fig. 1a shows a representative SEM image of RHO showing spherical morphology with an average size of ~1.5 μm. Fig. 1b shows a representative SEM image of DNL-6 showing hexagonal-like morphology with an average size of ~2.5 μm. Fig. 1c shows a representative SEM image of SAPO-34 showing cubic-like morphology with an average size of 2.3 μm Fig. 1d shows a representative SEM image of hydrotalcite showing irregular ~3 μm agglomerates composed of needle-like shapes. | ||
| Fig. 1 SEM images of a) RHO, b) DNL-6, c) SAPO-34 and d) hydrotalcite supports. | ||
Fig. 2 shows the XRD patterns of all studied Pt-supported catalysts. Fig. 2a and b show the XRD patterns of Pt-RHO and Pt-DNL-6, respectively. Both are in agreement with the RHO topology.34Fig. 2c shows the XRD pattern of the chabazite structure, the typical topology of SAPO-34.36Fig. 2d shows the typical layered structure of the Mg–Al-based hydrotalcite corresponding to an interplanar spacing of ~0.7 nm.
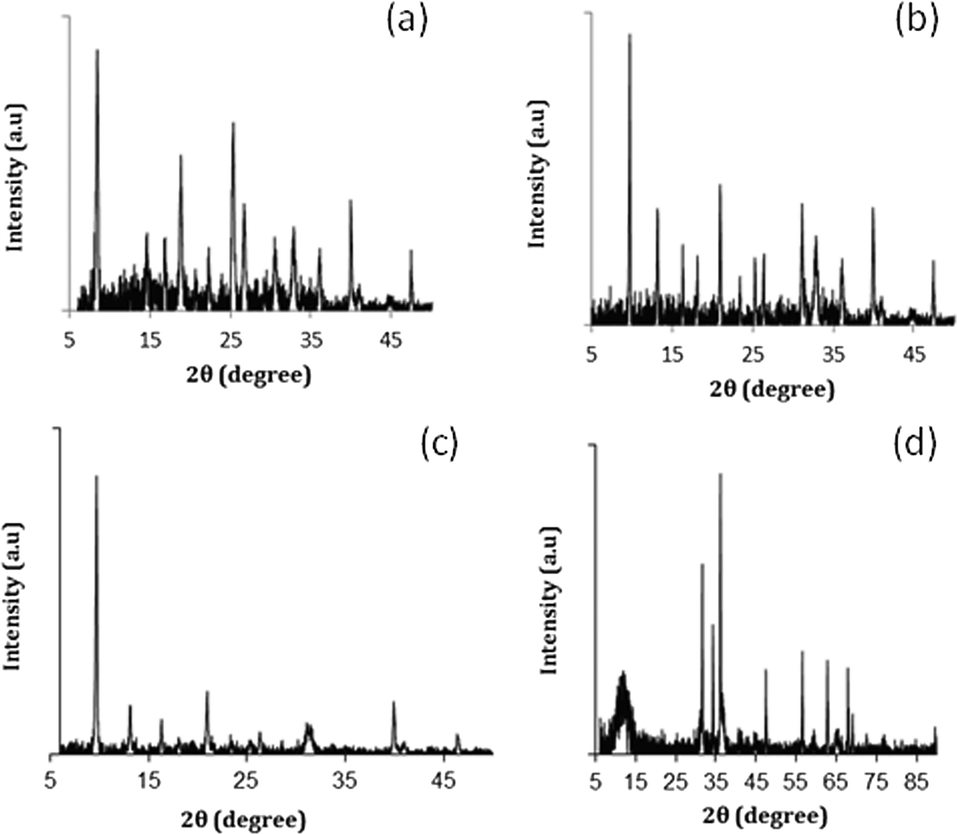 | ||
| Fig. 2 XRD patterns of a) Pt-RHO, b) Pt-DNL-6, (c) Pt-SAPO-34 and d) Pt-hydrotalcite. | ||
The BET specific surface area of the supports and Pt/supports are given in Table 1. As expected, in all cases, the surface area decreased upon incorporation of Pt. Interestingly, when salt 2 was used as the Pt source the decrease in the surface area was less pronounced, suggesting a better dispersion of Pt when salt 2 was employed. N2 BET measurements on zeolite RHO were unsuccessful (no measurable N2 adsorption was observed). This is not uncommon since the effective pore aperture of dehydrated zeolite RHO is ~2.9 Å,33 making it difficult for N2 (kinetic diameter ~3.6 Å) to be adsorbed within the zeolite pores.
| Sample | Surface area (m2 g−1) |
|---|---|
| RHO | NA |
| DNL-6 | 400 |
| Pt-DNL-6 (salt 1) | 250 |
| Pt-DNL-6 (salt 2) | 280 |
| SAPO-34 | 490.4 |
| Pt-SAPO-34 (salt 1) | 406.4 |
| Pt-SAPO-34 (salt 2) | 478.2 |
| Hydrotalcite | 55 |
| Pt-hydrotalcite (salt 1) | 49 |
| Pt-hydrotalcite (salt 2) | 53 |
Fig. 3 shows the decarboxylation of oleic acid over different catalysts as a function of temperature when salt 1 (chloroplatinic acid) was used to incorporate Pt on the supports. As expected, for all of these four catalysts (Pt-RHO, Pt-DNL-6, Pt-SAPO-34, Pt-hydrotalcite), a higher degree of decarboxylation was observed at 325 °C than at 245 °C. The same pattern was observed when these supports were impregnated with salt 2 (tetraammine platinum nitrate) (Fig. 4). The higher catalytic activity of Pt-based catalysts prepared from Pt(NH3)4[(NO3)]2 has been reported earlier.5,17,37 The use of cationic complexes (such as Pt[(NH3)4]2+) probably lead to better dispersion/anchoring to the anionic O2− centers on the catalyst surface and, consequently, higher catalytic activity than the use of anionic complexes of Pt (such as [PtCl6]4−). In the presence of H2, oleic acid is first hydrogenated to stearic acid which then undergoes facile decarboxylation. In the absence of H2, the hydrogenation route is not available and therefore thermal decarboxylation of the unsaturated acid is the only route available.
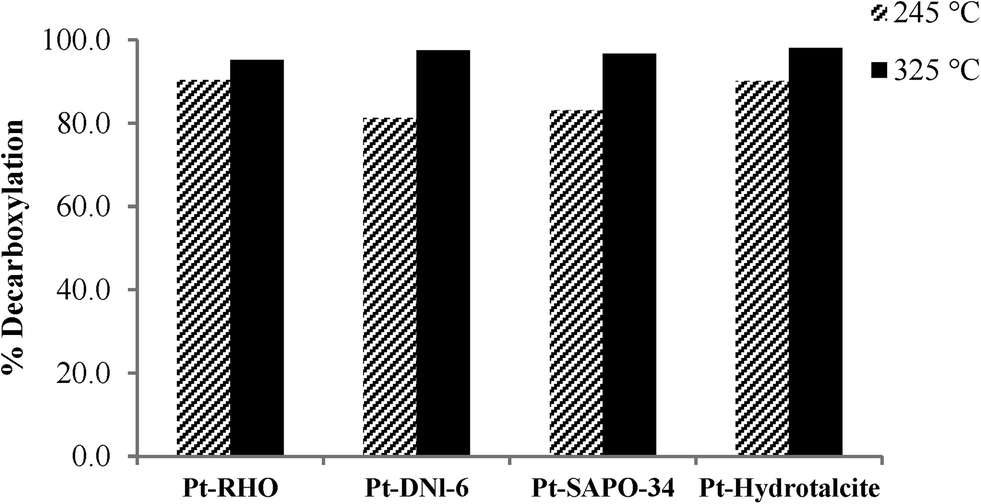 | ||
| Fig. 3 Decarboxylation of oleic acid over catalysts prepared using salt 1 at different reaction temperatures (reaction conditions: 40 ml of oleic acid, 2 g of catalyst, 2 h, and pressure of 20 bar at 245 and 325 °C). | ||
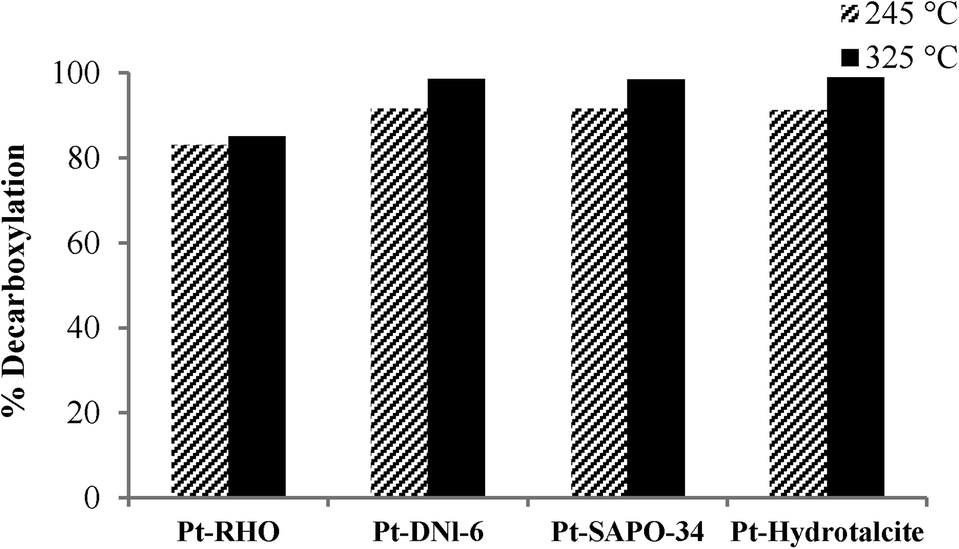 | ||
| Fig. 4 Decarboxylation of oleic acid over catalysts prepared using salt 2 at different reaction temperatures (reaction conditions: 40 ml of oleic acid, 2 g of catalyst, 2 h, and pressure of 20 bar at 245 and 325 °C). | ||
At 325 °C, when salt 1 was used as the Pt precursor, the degree of decarboxylation was similar for all four catalysts (above 90%). However, when salt 2 was used, Pt-DNL-6, Pt-SAPO-34 and Pt-hydrotalcite displayed a high degree of decarboxylation (around 98%). The degree of decarboxylation in the presence of RHO was only ~85%.
Table 2 shows the acid number of the reaction products (indicative of the concentration of unconverted fatty acids) for all these catalysts prepared by employing the two salts to impregnate the supports. The acid number at 325 °C is lower than at 245 °C for all cases. On the basis of the degree of decarboxylation and acid number, it should be expected that the amount of carboxylic acid groups remaining in the product after the reaction should be lower at higher temperature.
| Catalyst | Pt-RHO | Pt-DNL-6 | Pt-SAPO-34 | Pt-hydrotalcite | |||||
|---|---|---|---|---|---|---|---|---|---|
| T (°C) | 325 | 245 | 325 | 245 | 325 | 245 | 325 | 245 | |
| Acidity | Salt 1 | 9.7 | 19.3 | 4.9 | 37.6 | 6.6 | 33.9 | 4.0 | 19.8 |
| Salt 2 | 30.0 | 34.0 | 2.8 | 17 | 2.9 | 16.8 | 2.1 | 17.5 | |
For all of the catalysts, except for Pt-RHO, the acid number of the product (inversely proportional to the degree of decarboxylation) obtained when salt 2 was used as the Pt precursor is lower than that obtained when salt 1 was used as the precursor. For Pt-RHO, the degree of decarboxylation over the catalyst prepared using salt 1 was greater. The reasons behind this are not clear. Differences in (a) the size of the Pt-RHO crystals (Fig. 1) and (b) the Pt species present in aqueous solutions of salt 1 [PtCl6]2− and salt 2 [Pt(NH3)4]2+ may perhaps shed more light on this point. The average size of the crystals in Pt-RHO is ~1.5 μm, while those of the others are larger (2.3–3.0 μm). While the higher catalytic activity of Pt-based catalysts prepared from Pt(NH3)4(NO3)2 is known,5,17,37 the greater activity of salt 1-derived catalysts in the case of Pt-RHO is surprising. In principle, the high concentration of exchangeable cations (Na, Cs) of zeolite RHO will lead to a high dispersion of the anionic [PtCl6]2− complex rather than that of the cationic [Pt(NH3)4]2+ complex derived from salt 2. This effect is reinforced in the smaller crystals of Pt-RHO, leading to the observed high catalytic activity.
Stearic acid (and also other saturated carboxylic acid compounds) was formed by hydrogenation of oleic acid followed by decarboxylation to form heptadecane. The other components in the liquid product included branched paraffins formed by isomerization of the initially formed heptadecane, aromatics formed by cyclization and aromatization (such as dodecylbenzene) and lower molecular weight hydrocarbons (mostly C6–C16 paraffins) formed by cracking of the heptadecane. No oleic acid was observed in the products under all reaction conditions, suggesting 100% conversion of oleic acid. The amount of stearic acid and other carboxylic acid groups in the product (such as nonanoic acid, decanoic acid and undecanoic acid) was lower at 325 °C than that at 245 °C for all catalysts, which confirms that the highest degree of decarboxylation was observed at 325 °C. Some alkenes such as undecene, dodecene, tridecene, tetradecene, hexadecene, heptadecene and octadecene were also observed in the products. The detailed product distribution is given in Table 3.
| Catalyst | Product distribution (wt%) | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Salt | T (°C) | Hexane | Heptane | Octane | Nonane | Decane | Undecane | Dodecane | Tridecane | Tetradecane | Pentadecane | Hexadecane | Heptadecane | Octadecane | Nonadecane | UDBa | DDBb | Oleic acid | Stearic acid | Other carboxylicc acid compounds | Alkened | %gase | |
| a Undecylbenzene. b Dodecylbenzene. c Nonanoic acid, decanoic acid, undecanoic acid. d Undecene, dodecene, tridecene, tetradecene, hexadecene, heptadecene, octadecene. e Mass balance (CO2, CH4, others). | |||||||||||||||||||||||
| Pt-RHO | Salt 1 | 325 | 4.7 | 3.5 | 5.3 | 3.5 | 5.3 | 4.7 | 3 | 4.7 | 2.9 | 3.5 | 4.7 | 26.3 | 3.5 | 3 | 0 | 4.4 | 0 | 0.2 | 0.1 | 4.7 | 12 |
| Pt-RHO | Salt 1 | 245 | 5.3 | 4.7 | 4.1 | 2.9 | 4.7 | 4.1 | 2.4 | 3.5 | 4.1 | 3.5 | 2.9 | 23.4 | 3.5 | 2.4 | 0 | 7 | 0 | 0.9 | 5.3 | 5.3 | 10 |
| Pt-RHO | Salt 2 | 325 | 4 | 3.3 | 4.6 | 3.9 | 5 | 3.2 | 4.7 | 4 | 4.3 | 4.9 | 3.7 | 27 | 3.2 | 3.2 | 0 | 3.6 | 0 | 0.3 | 4 | 4.1 | 9 |
| Pt-RHO | Salt 2 | 245 | 3.8 | 2.9 | 4.1 | 4.3 | 4.9 | 3.7 | 3.9 | 4.5 | 5.6 | 4.3 | 3.7 | 22 | 4.3 | 3.4 | 0.1 | 5.8 | 0 | 0.4 | 4.2 | 3.7 | 10.4 |
| Pt-DNL-6 | Salt 1 | 325 | 4.1 | 4.1 | 4.7 | 2.4 | 5.3 | 5.9 | 7.6 | 5.3 | 2.9 | 4.7 | 5.7 | 16.2 | 4.7 | 5.3 | 0 | 5.3 | 0 | 0.1 | 0 | 4.7 | 11 |
| Pt-DNL-6 | Salt 1 | 245 | 5.3 | 4.1 | 3.5 | 6.3 | 4.1 | 5.3 | 5.9 | 3.5 | 4.7 | 2.3 | 5.1 | 10.8 | 4.1 | 4.1 | 0 | 5.3 | 0 | 7.6 | 4.1 | 2.9 | 11 |
| Pt-DNL-6 | Salt 2 | 325 | 4.1 | 2.9 | 4.1 | 4.2 | 2.9 | 2.5 | 3.5 | 3.3 | 2.4 | 2.9 | 2.4 | 47 | 2.4 | 1.8 | 0.5 | 2.9 | 0 | 0 | 0 | 0.2 | 10 |
| Pt-DNL-6 | Salt 2 | 245 | 4.8 | 4 | 5.5 | 4 | 3.4 | 2.8 | 3.4 | 4.5 | 5.7 | 4.9 | 3 | 30 | 3.6 | 3.9 | 0.1 | 3.3 | 0 | 0.4 | 2.3 | 0 | 10.4 |
| Pt-SAPO-34 | Salt 1 | 325 | 2.4 | 5.3 | 3.5 | 3.9 | 2.1 | 3.2 | 3.5 | 3.1 | 3.4 | 4.1 | 2.4 | 29.4 | 4.7 | 3.5 | 0 | 7.4 | 0 | 0.3 | 3.8 | 4 | 10 |
| Pt-SAPO-34 | Salt 1 | 245 | 2.7 | 5.1 | 3.8 | 2.7 | 4.7 | 3.9 | 3.5 | 3.9 | 4.6 | 4.3 | 3.4 | 23.5 | 4.7 | 3.6 | 0.2 | 7.8 | 0 | 0.5 | 4.2 | 2.9 | 10 |
| Pt-SAPO-34 | Salt 2 | 325 | 0.8 | 1.2 | 1.4 | 1.4 | 0.8 | 0.6 | 1.2 | 1.1 | 2.8 | 2.5 | 1.8 | 66.9 | 1 | 0.2 | 0 | 6 | 0 | 0.3 | 0 | 1 | 9 |
| Pt-SAPO-34 | Salt 2 | 245 | 2.6 | 1.5 | 2.3 | 2.2 | 3.4 | 2.2 | 2.4 | 2.7 | 2.1 | 3.9 | 3.2 | 50 | 1.2 | 0.9 | 0 | 6.2 | 0 | 0.7 | 1.1 | 0.9 | 10.5 |
| Pt-hydrotalcite | Salt 1 | 325 | 2.1 | 2.7 | 3.5 | 2.7 | 4.1 | 5.3 | 4.7 | 4.1 | 3.8 | 4.7 | 4.1 | 23.5 | 5.3 | 4.7 | 0.1 | 8.9 | 0 | 0.2 | 0 | 4.5 | 11 |
| Pt-hydrotalcite | Salt 1 | 245 | 2.1 | 3.7 | 3.8 | 2.7 | 5.4 | 5.3 | 4.7 | 4.1 | 4.5 | 4.7 | 4.1 | 20.6 | 5.3 | 4.7 | 0.1 | 7.9 | 0 | 0.5 | 0.1 | 4.7 | 11 |
| Pt-hydrotalcite | Salt 2 | 325 | 2.9 | 2.1 | 3.1 | 3.8 | 2.7 | 3.9 | 1.7 | 3.5 | 4.1 | 3.1 | 4.7 | 29.2 | 4.7 | 4.1 | 0 | 8.1 | 0 | 0 | 0 | 4.3 | 12 |
| Pt-hydrotalcite | Salt 2 | 245 | 1.8 | 2.4 | 3.5 | 4.1 | 2 | 2.8 | 4.1 | 4.6 | 4.7 | 3.5 | 4.1 | 27 | 4.7 | 3.5 | 0 | 7 | 0 | 0.6 | 3.5 | 4.1 | 12 |
Fig. 5 and 6 show the influence of reaction temperature and catalyst on selectivity to heptadecane, the linear C17 paraffin obtained by the decarboxylation of oleic acid when salts 1 and 2 were employed. In general, heptadecane selectivity increased with increasing temperature and was favored when Pt-SAPO-34 was used.
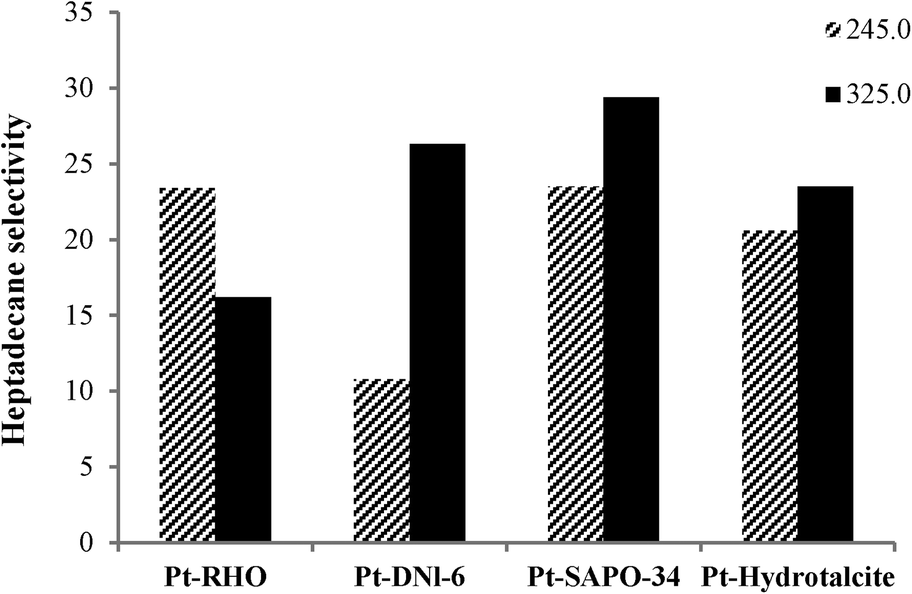 | ||
| Fig. 5 Effect of temperature and catalyst on heptadecane selectivity in the catalytic deoxygenation of oleic acid (Pt from salt 1 was impregnated on the supports; reaction conditions: 40 ml of oleic acid, 2 g of catalyst, 2 h and pressure of 20 bar). Heptadecane selectivity is in wt%. | ||
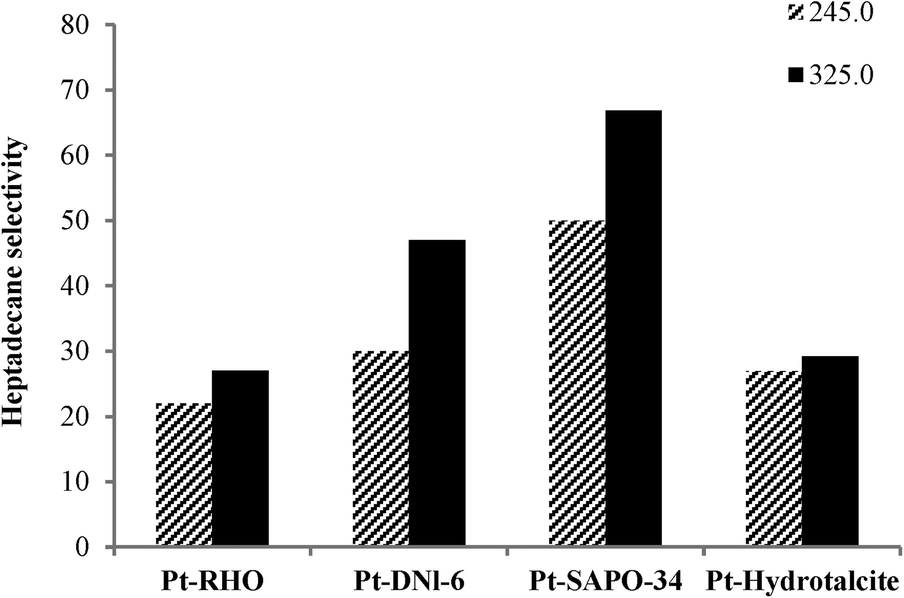 | ||
| Fig. 6 Effect of temperature and catalyst on heptadecane selectivity in the catalytic deoxygenation of oleic acid (Pt from salt 2 was impregnated on the supports; reaction conditions: 40 ml of oleic acid, 2 g of catalyst, 2 h and pressure of 20 bar). Heptadecane selectivity is in wt%. | ||
Although Pt-DNL-6, Pt-SAPO-34 and Pt-hydrotalcite displayed high degrees of decarboxylation (around 98%) (Fig. 1 and 2), selectivity to heptadecane was highest over Pt-SAPO-34. Pt-SAPO-34 shows also the highest selectivity to dodecylbenzene (which is one of the most significant industrial products in this reaction) among other catalysts (Table 3). This can be partially explained by the difference in acidity of the supports. For example, RHO has lower number of strong acid sites and higher number of weak acid sites than SAPO-34 (RHO shows an NH3 desorption peak at 170–200 °C which was assigned to weak acid sites, its second desorption peak is located at 480 °C. The area of the desorption peak for weak and strong acid sites was indicated to be around 150 and 75, respectively).38,39 For supported metal, bi-functional catalysts, such as those used in the present study, the selectivity for the initially formed linear paraffin, heptadecane (amongst the reaction products), is inversely proportional to the ability of the catalyst to isomerize it further to branched paraffins over the acidic sites of the support. In addition to acidity, the pore diameter of the support also plays a crucial role: supports with smaller pore diameters prevent further isomerization reaction of heptadecane since the large size of the transition state needed for isomerization cannot be accommodated in the constrained confines of the small pores (restricted transition state selectivity).40 In restricted transition state-type selectivity, certain reactions (such as the isomerization of linear to branched olefins/paraffins) are prevented because the transition state is too large for the cavities of the catalyst. SAPO-34 belongs structurally to the chabazite group of zeolites. It contains 8-membered ring pores with openings of 0.38 nm. DNL-6 is structurally related to zeolite RHO with a pore size of 0.36 nm (close to SAPO-34). Interestingly, both supports with very close pore sizes show the highest selectivity to heptadecane, suggesting the “molecular sieving” effect. Zeolite RHO also has a pore system comprising 8-membered rings. However, the pore openings can be as small as 0.29 nm. The other support, hydrotalcite, has a much larger pore diameter. The isomerization of heptadecane to bulkier branched isomers will hence be sterically more relatively difficult in the narrow pores of SAPO-34 or DNL-6 than that in the other two materials, accounting for its higher selectivity for linear heptadecane. In the case of RHO, our results suggest that the smaller pore size may impose diffusion limitations.
The product selectivity profile over Pt-SAPO-34 is shown in Fig. 7. Selectivity to heptadecane increased with increasing temperature, while selectivity to other alkane products (C6–C16, C18 and C19) decreased with increasing temperature (Fig. 7). A heptadecane selectivity of 66.9% and a dodecylbenzene selectivity of 8% were observed after 2 h in the presence of H2 at 325 °C over Pt-SAPO-34. For all of the catalysts employed in this study, a higher degree of decarboxylation and selectivity to heptadecane were observed over the catalysts prepared with salt 2. The higher selectivity for heptadecane of the catalyst obtained using salt 2 (tetraammine platinum nitrate) might be related to the higher dispersion of Pt observed when cationic complexes are used as precursors of Pt.5,17,36
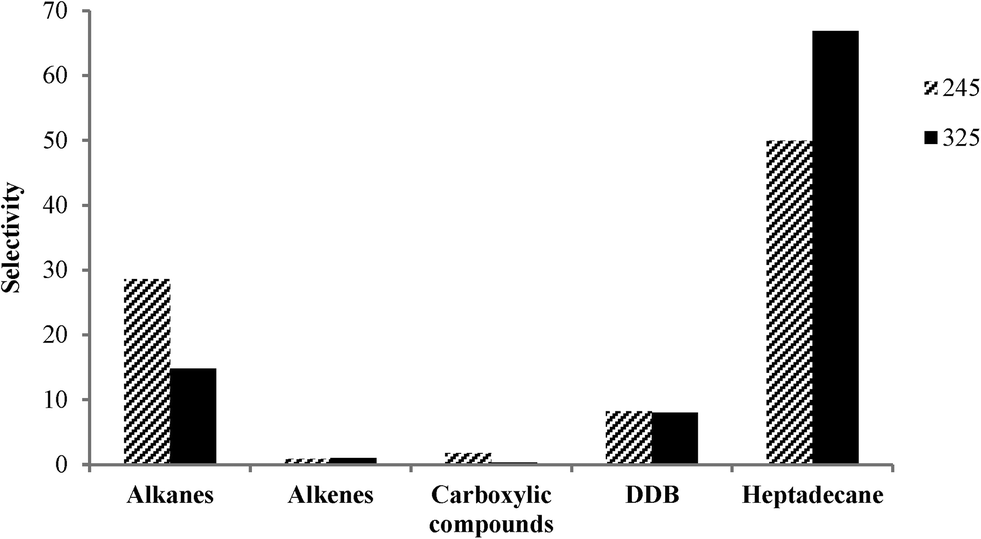 | ||
| Fig. 7 Effect of temperature on the selectivity of the final products in the catalytic deoxygenation of oleic acid over Pt-SAPO-34 (Pt from salt 2 was impregnated on the supports; reaction conditions: 40 ml of oleic acid, 2 g of catalyst, 2 h, and pressure of 20 bar). Selectivity is in wt%. | ||
Table 1 also indicates that heptadecane selectivity is influenced by the source of Pt; similar selectivity (~29%) is observed even for catalysts with different pore diameters when the Pt source is kept the same (salt 1), suggesting that the Pt source may be as important as the pore diameter in controlling selectivity values. The presence of chlorine in salt 1 perhaps modifies the nature of the pore system and thus reduces the importance of the pore diameter in influencing product selectivity. Thus, (1) the acidity of the support, (2) the dispersion of the Pt and (3) the nature of the Pt source are all crucial factors in controlling the catalytic activity and product selectivity in the conversion of oleic acid to hydrocarbons.
Fig. 8 shows the representative TEM images of the Pt-SAPO-34 catalysts employed in this study. In particular, a higher density of well-dispersed and smaller platinum nanoparticles on the SAPO-34 support was observed when salt 2 was used as the Pt source (Fig. 8b) as compared to salt 1 (Fig. 8a). In fact, Fig. 8a shows the presence of Pt clusters, suggesting poor metal dispersion.
 | ||
| Fig. 8 TEM images of 5% Pt-SAPO-34 catalysts prepared using a) salt 1 (low and high magnification) and b) salt 2 (low and high magnification). | ||
In general, for salt 2, the Pt particle histogram in Fig. 9a shows a higher fraction of small nanoparticles (in the 1–3 nm range) as compared to that for salt 1 (Fig. 9b).
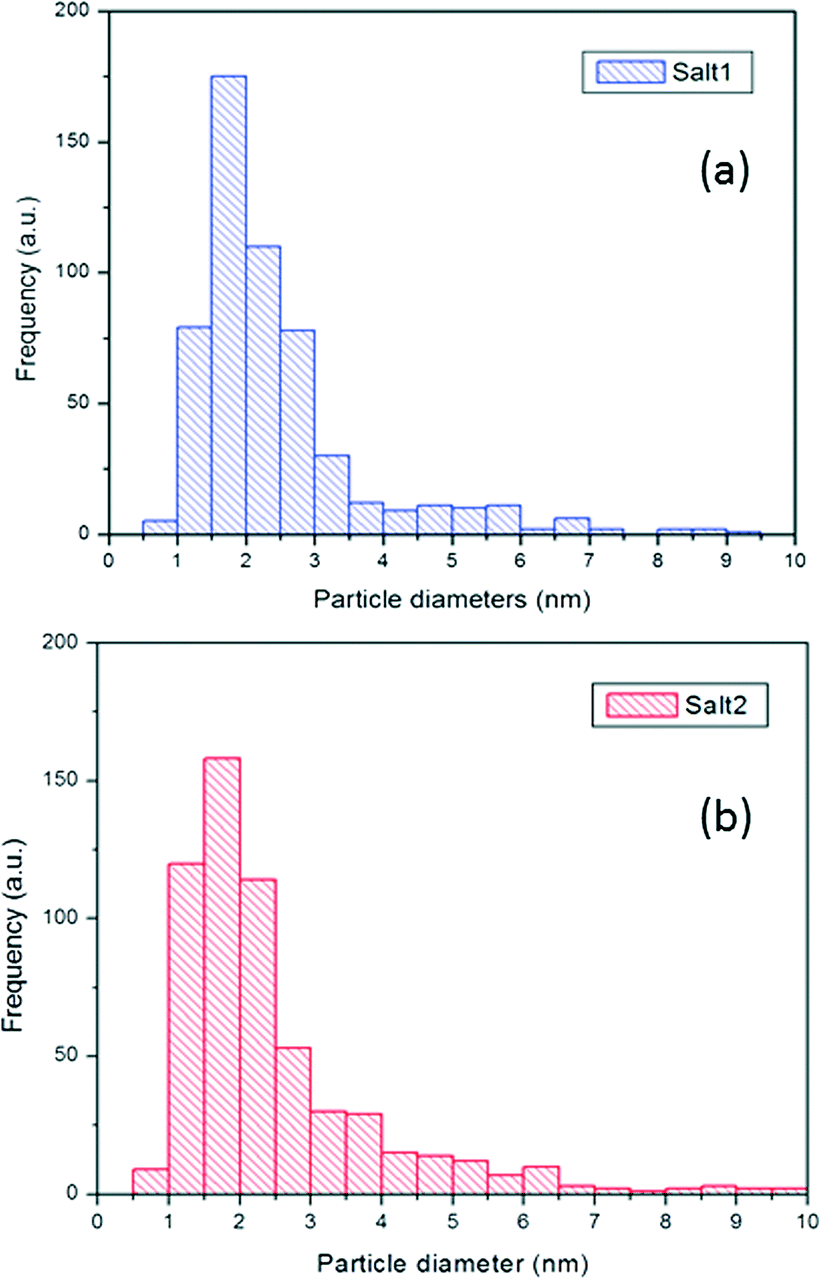 | ||
| Fig. 9 Pt particle size distribution histogram of 5% Pt-SAPO-34 prepared using a) salt 1 and b) salt 2. | ||
4. Conclusions
The decarboxylation and further conversion of oleic acid to gasoline/diesel-range hydrocarbons have been demonstrated over Pt/SAPO-34, Pt-DNL-6, Pt-RHO and Pt-hydrotalcite catalysts. The degree of decarboxylation and selectivity to the primary intermediate, linear heptadecane, are affected by reaction temperature and catalyst (support and nature of the Pt salt used for impregnation of the support). Higher temperatures increased the degree of decarboxylation (98%) and selectivity to heptadecane (up to ~66%). The initially formed paraffin intermediate undergoes further conversion to branched, cyclic and alkyl aromatic hydrocarbons. For example, dodecylbenzene, which is of high interest in the petrochemical industry as a raw material for detergents, could be obtained in high yields. In the presence of acidic supports with narrow pore diameters, such as SAPO-34, the degree of decarboxylation and selectivity to the linear paraffin heptadecane are high. The degree of dispersion of Pt within the porous support influenced the catalytic activity significantly. Higher degrees of decarboxylation and selectivity to heptadecane were observed over catalysts with more uniform and well-dispersed Pt metal nanoparticles such as those obtained when tetraammine platinum nitrate was used as the Pt precursor. Catalyst acidity, dispersion of Pt and the pore diameter of the support are the crucial factors determining the selectivity for the linear paraffin heptadecane in the product. Under more severe reaction conditions (higher temperatures), significant quantities of hydrocarbons boiling in the gasoline range are formed by hydrocracking reactions over these catalysts.References
- I. Simakova, O. Simakova, P. Mäki-Arvela and D. Murzin, Catal. Today, 2010, 150, 28–31 CrossRef CAS PubMed.
- J. Fu, X. Lu and P. Savage, ChemSusChem, 2011, 4, 481–486 CrossRef CAS PubMed.
- J. Fu, X. Lu, P. E. Savage, L. T. Thompson Jr. and X. Lu, ACS Catal., 2011, 1, 227–231 CrossRef CAS.
- P. Arvela, B. Rozmysłowicz, S. Lestari, O Simakova, K. Eränen, T. Salmi and D. Yu. Murzin, Energy Fuels, 2011, 25, 2815–2825 CrossRef.
- E. Laurent and B. Delmon, Appl. Catal., A, 1994, 109, 77–96 CrossRef CAS.
- M. Snare, I. Kubickova, P. Arvela, K. Eranen and D. Yu. Murzin, Ind. Eng. Chem. Res., 2006, 45, 5708–5715 CrossRef.
- M. Snare, I. Kubickova, P. Maki-Arvela, D. Chichova, K. Eranen and D. Yu. Murzin, Fuel, 2008, 87, 933–945 CrossRef CAS PubMed.
- R. Stern and G. Hillion, US Pat., 4, 695,411, 1985 Search PubMed.
- G. W. Huber and A. Corma, Angew. Chem., Int. Ed., 2007, 46, 7184–7201 CrossRef CAS PubMed.
- S. Lestari, P. Maki-Arvela, K. Eranen, J. Beltramini, G. Q. M. Lu and D. Y. Murzin, Catal. Lett., 2010, 134, 250–257 CrossRef CAS PubMed.
- H. Bernas, K. Eranen, I. Srimakova, A. R. Leino, K. Kordas, J. Myllyoja, P. Maki-Arvela, T. Salmi and D. Y. Murzin, Fuel, 2010, 89, 2033–2039 CrossRef CAS PubMed.
- M. Snare, I. Kubickova, P. Arvela, K. Eranen and D. Yu. Murzin, Catal. Today, 2005, 106, 197–200 CrossRef PubMed.
- M. Snare, I. Kubickova, P. Arvela, K. Eranen and D. Yu. Murzin, Catal. Org. React., 2006, 115, 415–425 Search PubMed.
- D. Yu. Murzin, I. Kubickova, M. Snare and P. Arvela, Eur. Pat. Appl., 05075068.6, 2005 Search PubMed.
- A. Sivasamy, K. Cheah, P. Fornasiero, F. Kemausuor, S. Zinoviev and S. Miertus, ChemSusChem, 2009, 2, 278–300 CrossRef CAS PubMed.
- W. F. Maier, W. Roth, I. Thies and P. V. Ragué Schleyer, Chem. Ber., 1982, 115, 808–812 CrossRef CAS PubMed.
- S. H. Bertram, Chem. Weekbl., 1936, 33, 457–459 CAS.
- T. A. Foglia and P. A. Barr, J. Am. Oil Chem. Soc., 1976, 53, 737–741 CrossRef CAS.
- S. Lestari, P. Maki-Arvela, H. Bernas, O. Simakova, R. Sjonholm, J. Beltrami, G. Q. M. Lu, J. Myllyoja, I. Simakova and D. Y. Murzin, Energy Fuels, 2009, 23, 3842–3845 CrossRef CAS.
- F. A. Twaiq, N. A. M. Zabidi and S. Bhatia, Ind. Eng. Chem. Res., 1999, 38, 3230–3237 CrossRef CAS.
- W. K. Craig and D. W. Soveran, US Pat., 4,992,605, 1991 Search PubMed.
- D. Lima, V. C. Soares, E. B. Ribeiro, D. A. Caravalho, E. C. V. Cardoso and F. C. Rassi, et al. , J. Anal. Appl. Pyrolysis, 2004, 71, 987–996 CrossRef CAS PubMed.
- M. Arenda, T. Nonnena, W. F. Hoeldericha, J. Fischerb and J. Groosb, Appl. Catal., A, 2011, 399, 198–204 CrossRef PubMed.
- T. Kasza, A. Holló, A. Thernesz and J. Hancsók, Chem. Eng. Trans., 2010, 21, 1225–1230 Search PubMed.
- J. Hancsók, S. Kovács, G. Pölczmann and T. Kasza, Top. Catal., 2011, 54, 1094–1101 CrossRef.
- J. M. Campelo, F. Lafont and J. M. Marinas, Appl. Catal., A, 1998, 170, 139–144 CrossRef CAS.
- P. Mériaudeau, V. A. Tuan, V. T. Nghiem, S. Y. Lai, L. N. Hung and C. Naccache, J. Catal., 1997, 169, 55–66 CrossRef.
- J. Walendziewski and B. Pniak, Appl. Catal., A, 2003, 250, 39–47 CrossRef CAS.
- O. V. Kikhtyanin, A. E. Rubanov, A. B. Ayupov and G. V. Echevsky, Fuel, 2010, 89, 3085–3092 CrossRef CAS PubMed.
- M. Ahmadi, E. E. Macias, J. B. Jasinski, P. Ratnasamy and M. A. Carreon, J. Mol. Catal. A: Chem., 2014, 386, 14–19 CrossRef CAS PubMed.
- R. Szostak, Molecular Sieves: Principles of Synthesis and Identification, Van Nostrand Reinhold, New York, 1989 Search PubMed.
- X. Su, P. Tian, J. Li, Y. Zhang, S. Meng, Y. He, D. Fan and Z. Liu, Microporous Mesoporous Mater., 2011, 144, 113–119 CrossRef CAS PubMed.
- T. Chatelain, J. Patarin, R. Farre, O. Petigny and P. Schulz, Zeolites, 1996, 17, 328 CrossRef CAS.
- M. Palomino, A. Corma, J. L. Jorda, F. Rey and S. Valencia, Chem. Commun., 2012, 48, 215 RSC.
- R. Frost, W. Martens and K. Erickson, J. Therm. Anal. Calorim., 2005, 82, 603–608 CrossRef CAS.
- S. Li, M. A. Carreon, Y. Zhang, H. H. Funke, R. D. Noble and J. L. Falconer, J. Membr. Sci., 2010, 352, 7–13 CrossRef CAS PubMed.
- A. T. Madsen, E. Ahmed, C. H. Christensen, R. Fehrmann and A. Riisager, Fuel, 2011, 90, 3433–3438 CrossRef CAS PubMed.
- H.-Y. Jeona, C.-H. Shina, H. J. Jungb and S. B. Hong, Appl. Catal., A, 2006, 305, 70–78 CrossRef PubMed.
- Z. Nawaz, X. Tang, Q. Zhang, D. Wang and W. Fei, Catal. Commun., 2009, 10, 1925–1930 CrossRef CAS PubMed.
- S. M. Csicsery, Pure Appl. Chem., 1986, 58(6), 841–856 CrossRef CAS.
Footnote |
| † Present address: 4, Lalit apts, Gulmohar Park, ITI road, Aundh, Pune-411007, India. |
| This journal is © The Royal Society of Chemistry 2015 |