
Striking difference between alkane and olefin metathesis using the well-defined precursor [![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e002.gif) Si–O–WMe5]: indirect evidence in favour of a bifunctional catalyst W alkylidene–hydride†
Si–O–WMe5]: indirect evidence in favour of a bifunctional catalyst W alkylidene–hydride†
N.
Riache
,
E.
Callens
*,
J.
Espinas
,
A.
Dery
,
M. K.
Samantaray
,
R.
Dey
and
J. M.
Basset
*
KAUST Catalyst Center, Physical Sciences and Engineering Division, King Abdullah University of Science and Technology, Thuwal, 23955-6900, Saudi Arabia. E-mail: emmanuel.callens@kaust.edu.sa; Jean-Marie.Basset@kaust.edu.sa
First published on 7th August 2014
Abstract
Metathesis of linear alkanes catalyzed by the well-defined precursor (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–O–WMe5) affords a wide distribution of linear alkanes from methane up to triacontane. Olefin metathesis using the same catalyst and under the same reaction conditions gives a very striking different distribution of linear α-olefins and internal olefins. This shows that olefin and alkane metathesis processes occur via very different pathways.
Si–O–WMe5) affords a wide distribution of linear alkanes from methane up to triacontane. Olefin metathesis using the same catalyst and under the same reaction conditions gives a very striking different distribution of linear α-olefins and internal olefins. This shows that olefin and alkane metathesis processes occur via very different pathways.
Introduction
Alkane metathesis, a promising reaction to convert light alkanes into more valuable higher molecular weight hydrocarbons, has aroused strong interest during the past decade.1,2 Two catalytic systems were developed, one employing a single-site multifunctional catalyst3 and the other using a tandem catalytic approach.4,5 The first catalytic system notably used groups V and VI transition metals supported on various metal oxides synthesized by surface organometallic chemistry. It was found to be an efficient precatalyst for the metathesis of light alkanes (C2–C4).1,6,7 For instance, supported Ta mono- or polyhydrides catalyze n-butane metathesis to C1–C3 light hydrocarbons as well as liquid hydrocarbon fuels (C5–C10) in a pressurized fixed-bed reactor.8 In such a supported system, it has been observed that the initial C–H bond activation occurs on the metal hydride with hydrogen evolution.9 Next, α- or β-H elimination from the resulting alkyl species would give the corresponding carbene or olefin. To account for the overall features of the reaction, a propagative bifunctional alkylidene/hydride species was proposed.10 The metal alkylidene converts alkene intermediates via a metallacyclobutane, while the W-hydride is needed to release H2 for the hydrogenation of newly formed olefins to new alkanes.10 Recently, we disclosed the synthesis of a well-defined silica-supported catalyst [Si–O–WMe5] 1 (Fig. 1).11 We have shown that 1 is thermally transformed into a W-methyl carbyne species 2 at 150 °C (Fig. 1). Moreover, in the presence of an olefin, an equilibrium with this W-carbyne and the corresponding bis-carbene 3 (needed for olefin metathesis) is also observed (Fig. 1).12 Complex 1 was found to be an efficient precatalyst for propane11 and cycloalkane metathesis.13 As mentioned for W-polyhydride species, 1 is able to promote several elementary steps: σ-bond metathesis on the W-methyl, α- and β-H elimination from the resulting W-alkyl group, olefin metathesis via the carbene functionality and double bond hydrogenation via a classical 1,2 insertion mechanism. Thus, we envisaged employing this multifunctional system for higher alkane metathesis and comparing its behaviour with the corresponding pure α-olefin metathesis.
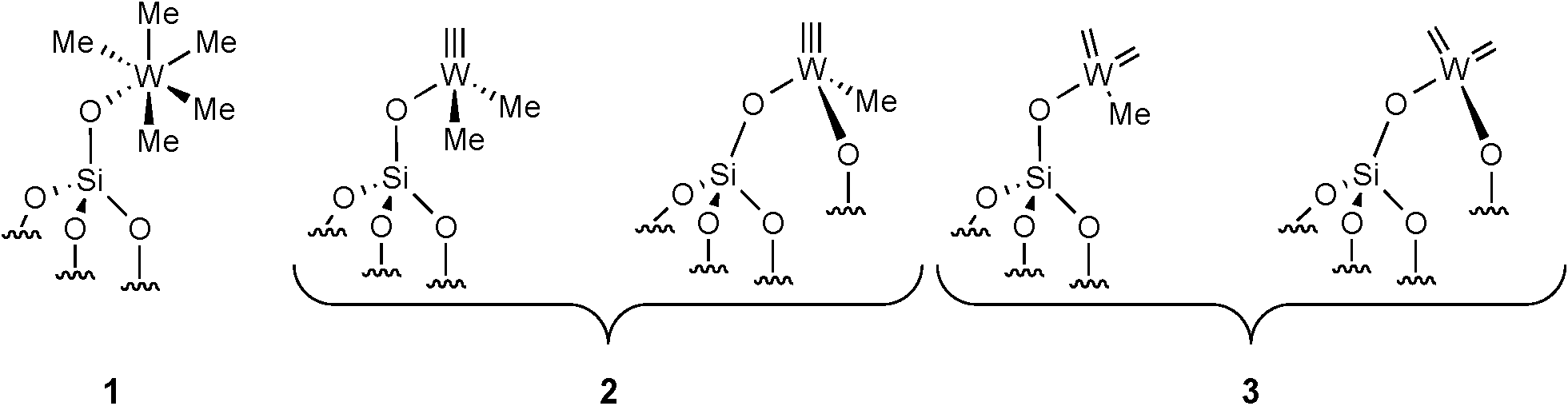
Herein, we wish to report the metathesis of different linear alkanes and their corresponding α-olefins catalyzed by 1. Our results support the multifunctional characteristics of this supported catalyst with a W-carbene hydride playing a crucial role in this transformation.
Results and discussion
In a batch reactor at 150 °C, metathesis of n-decane produces a broad distribution of linear alkanes ranging from methane to triacontane (C30). These linear alkanes were assigned (by GC and GC-MS) according to their retention time and fragmentation pattern by comparison with available references.14 Traces of branched alkanes (molar fractions <1%) were also observed, while no cyclic or olefinic products were detected by GC-MS and NMR spectroscopy. In a typical catalytic run, a conversion of 38% is reached with 142 TONs after a 5-day period. The product distribution curve of the molar quantity of n-decane metathesis products versus carbon number (from C5) is shown in Fig. 2.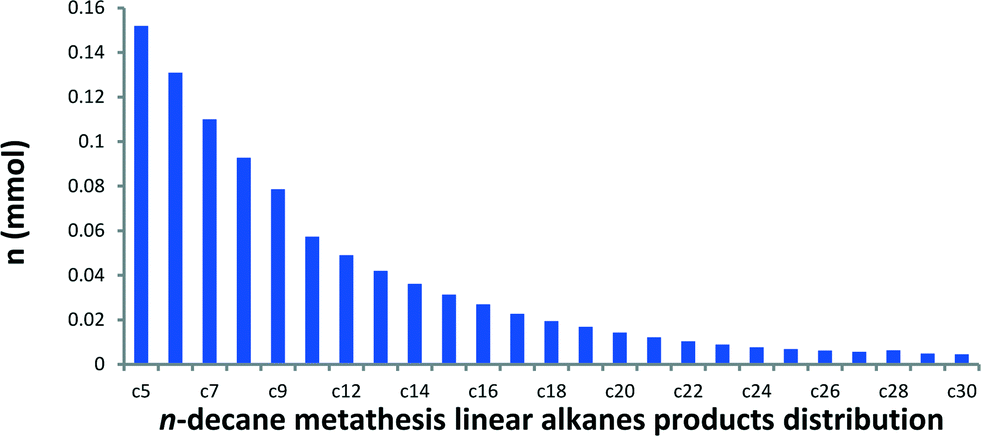 | ||
| Fig. 2 Distribution of n-decane metathesis products from C5 to C30 catalyzed by 1. Reaction conditions: batch reactor, 1 (50 mg, 6.7 μmol, W loading: 2.48 wt%), n-decane (0.5 mL, 2.57 mmol), 120 h, 150 °C. Conversion = 38%, TON = 142. The turnover number (TON) is the number of moles of n-decane transformed per mole of W. | ||
A narrow distribution centred on n-decane is not observed in this liquid/solid interface catalytic system in sharp contrast with propane metathesis.1 Actually, this observed selectivity is closer to the one reported with a fully heterogeneous tandem system.15,16 It is worth noting that our catalytic system is also active in the transformation of other linear alkanes, from hexane to nonane. Metathesis of each Cn alkane afforded a similar distribution of higher and lower linear hydrocarbons (Fig. 3, for corresponding chromatograms, see Fig. S1†). The molar ratio of ∑(Cn+1,Cn+5)/Cn is 0.29 for octane and 0.30 for decane metathesis at similar conversions. At lower conversions, molar ratios of 0.74 for hexane and 0.8 for nonane were obtained. These results indicate that the distribution is independent of the alkane carbon number.
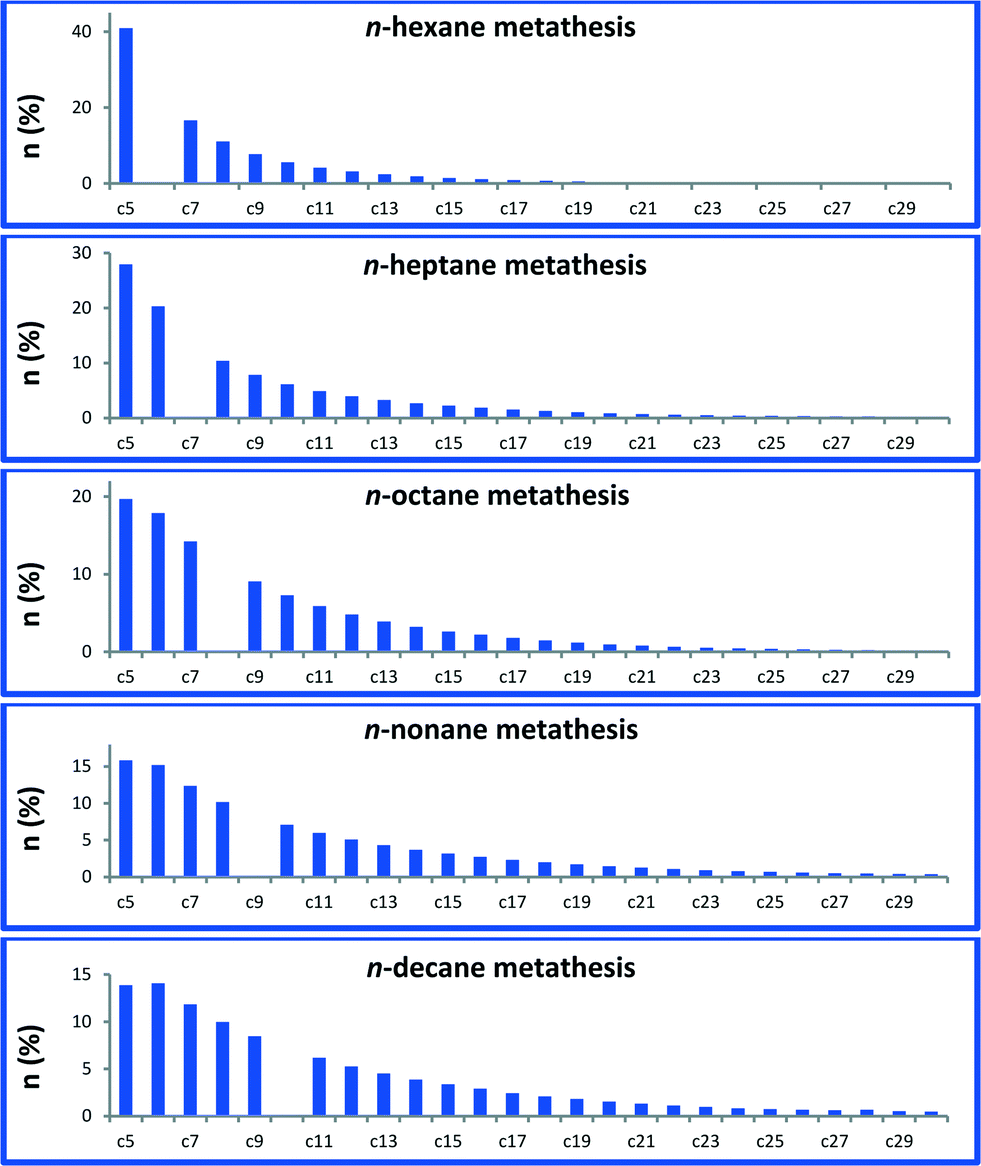 | ||
| Fig. 3 Alkane metathesis products distribution from C5 to C30 catalyzed by 1. Conversion = 30–45%; TON = 150–300. Reaction conditions: batch reactor, 1 (50 mg, 6.7 μmol, W loading: 2.48 wt%), n-alkane (0.5 mL, 3.83–2.57 mmol), 120 h, 150 °C. The turnover number (TON) is the number of moles of a given alkane transformed per mole of W. | ||
In addition, n-decane metathesis products were examined as a function of reaction time. The plot of the molar fraction of linear alkane products at 1 h, 4 h and 5 days is shown in Fig. 4. The initial distribution of alkanes remains similar with time, suggesting no distinction between kinetic and thermodynamic products for these reaction times. Furthermore, the metathesis of a mixture of octane and decane did not follow the alkane selectivity distribution (Cn−1/Cn+1 as major products) observed for the gas phase reaction (see Fig. S2†).
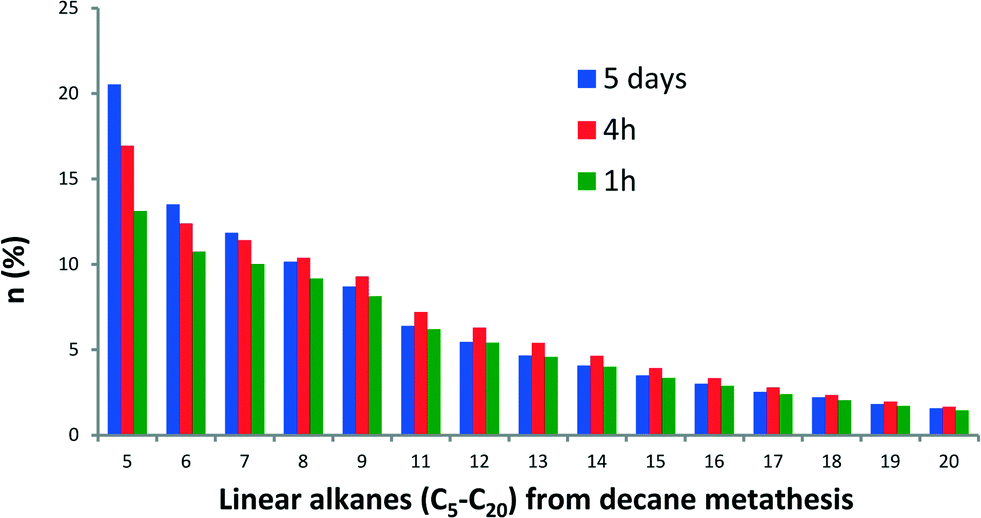 | ||
Fig. 4 Molar fraction of decane metathesis products as a function of time catalyzed by 1. Reaction conditions: batch reactor, 1 (50 mg, 5.73 μmol, W loading: 2.11 wt%), decane (0.5 mL), ( ) 5 days; ( ) 5 days; ( ) 4 h and ( ) 4 h and ( ) 1 h, 150 °C. ) 1 h, 150 °C. | ||
Since it has been demonstrated that olefins are key intermediates in alkane metathesis transformation, we also studied the direct metathesis of solely α-olefins catalyzed by 1 to compare their reactivity with their homologous alkanes. Although 1-decene is the thermodynamically less favoured olefin, we have chosen 1-decene as a comparison for the following reason: we have shown that in alkane metathesis linear paraffin is the major product (branched alkanes <1%), which means that n-alkyls are preferred over secondary alkyls. Therefore, the simplest olefin to be considered is the one resulting from β-H elimination of linear alkyl chains. Reaction of 1-decene catalyzed by 1 at room temperature affords octadecane as major metathesis product (77% conversion) (see Fig. S3 and S4†). Under the alkane metathesis reaction conditions, 1-decene is transformed into olefins of shorter (C2–C9) and longer carbons (C11–C20) (Fig. 5). A higher conversion of 1-decene (90%) is obtained after only 0.5 h with 1650 TONs. An initial turnover frequency of 150 min−1 is obtained. These TONs could reach 5500 by increasing the alkene/1 ratio. With time, GC chromatograms exhibit two clearly distinguishable regions: one comprising olefins from C7 to C12 and another from C13 to C20 as major products (Fig. 5). We identified that these two regions should correspond to regioisomers of given alkenes (C7 to C11) and to higher internal olefins (C15 to C18). The self-metathesis of 1-decene is followed by secondary cross metathesis of newly formed olefin from isomerization of the 1-decene to internal isomers of decene. With this olefin distribution from 1-decene, we examined other α-olefins from 1-hexene to 1-nonene. Olefin metathesis chromatograms show similar distribution curves as shown in Fig. S5 and S6.† Olefin metathesis chromatograms show similar distribution curves but with a limitation in the olefin carbon number (C2n+2) as shown in Fig. S4 and S5.†
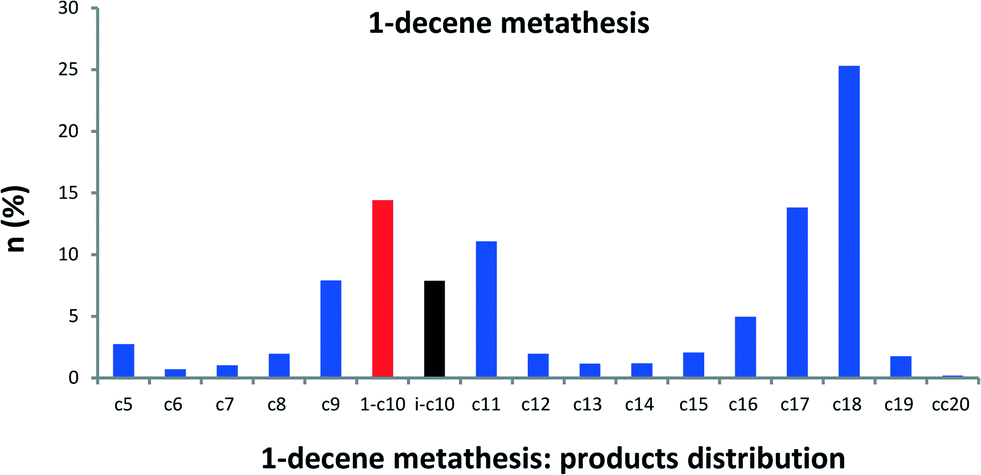 | ||
| Fig. 5 Molar fraction of α-decene (red) metathesis products (blue) catalyzed by 1 (1-decene isomers in black). Conversion = 90%; TON = 1650. Reaction conditions: batch reactor, 1 (25 mg, 1.44 μmol, W loading: 1.06 wt%), 1-decene (0.5 mL, 2.64 mmol), 0.5 h, 150 °C. The turnover number (TON) is the number of moles of 1-decene transformed per mole of W. | ||
Next, we monitored the reaction of 1-decene metathesis versus time. A plot of molar fraction of the medium at different periods of time is shown in Fig. 6 (for GC chromatograms, see Fig. S7†). At t = 0 h (defined by the time where the reactor temperature reaches 150 °C) the predominant reaction is the self-metathesis of 1-decene leading to 9-octadecene and ethylene (Fig. 6 and S7†). 51% of reacted 1-decene is transformed into 9-octadecene as the primary product. After 15 min, the peaks of C9 and C11 olefins become apparent due to a competitive isomerization/metathesis process. Those olefins (secondary metathesis products) should result from the cross-metathesis of 1-decene and decene isomers or self metathesis product isomers. The C17 olefin peak formed from secondary metathesis of 1-decene with 1-nonene is also observed. After 3 h, the conversion reaches an apparent plateau but the reaction has not reached thermodynamic equilibrium. At this point, the self-metathesis of 1-decene is overcome by secondary reactions. In the early stage, 1-decene accounts for 90% of the total fraction of C10 while it decreases to 30% after 3 h (Fig. 7). These substituted C10 isomers would react consecutively to form secondary olefin metathesis products as depicted in Fig. S10.† The plot of TONs and conversion versus time are given in Fig. 8. A final conversion of 97% is reached after several days, whereas after only 3 h, 95% of the substrate is converted. Final TONs of 1790 are obtained (several days reaction period). In addition, 5-decene metathesis was examined under the same reaction conditions. This experiment led to a Gaussian distribution of olefins with only C20 as the higher alkene in contrast to >C30 for decane (Fig. S8†). A cross-metathesis process of 1-decene and 5-decene (ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) catalytic run was conducted (Fig. S9†). We found that in the early stage no significant amount of C18 (homo coupling of 1-decene product) was observed supporting a preferential olefin metathesis process between 1-decene and 5-decene (see the ESI†).
:1 v/v) catalytic run was conducted (Fig. S9†). We found that in the early stage no significant amount of C18 (homo coupling of 1-decene product) was observed supporting a preferential olefin metathesis process between 1-decene and 5-decene (see the ESI†).
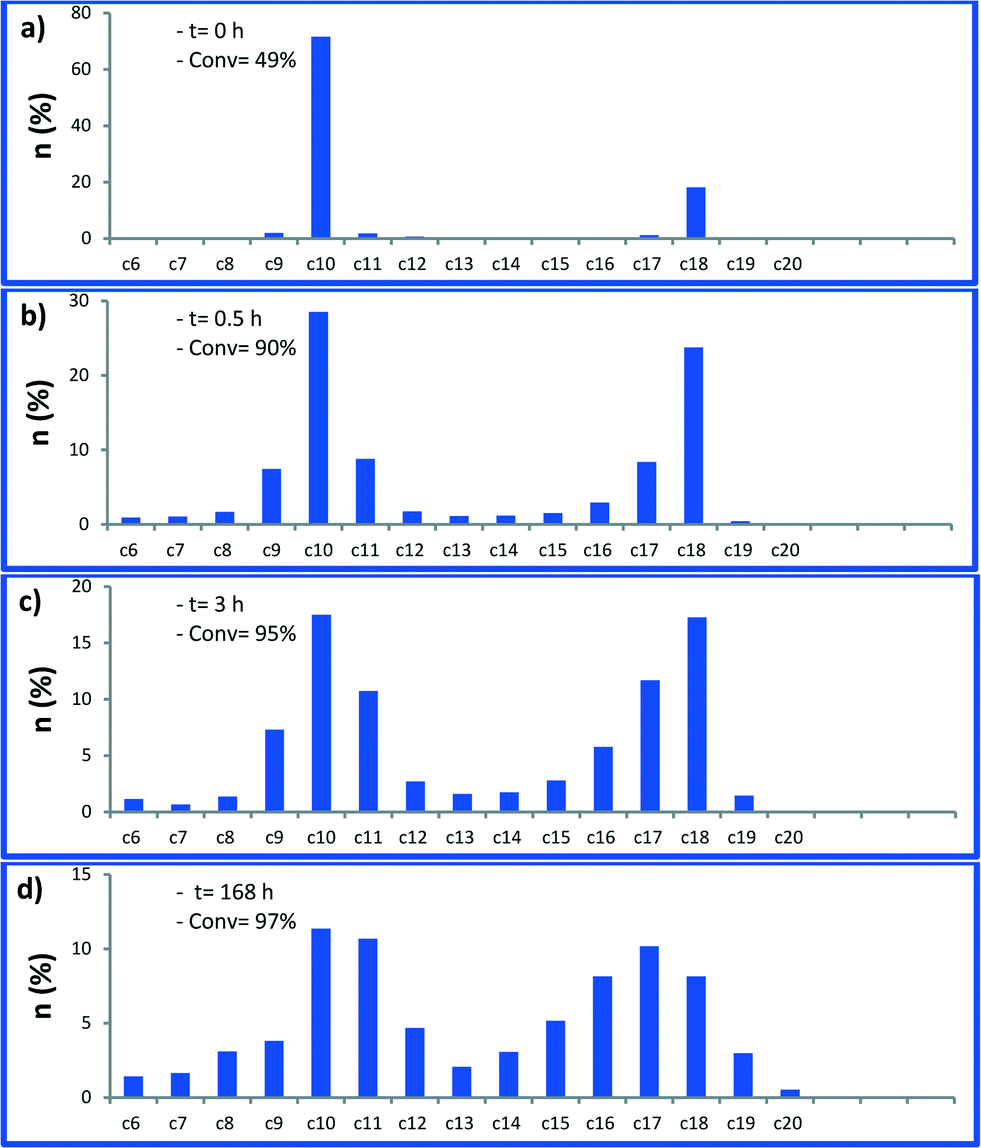 | ||
| Fig. 6 Molar fraction of 1-decene metathesis products as a function of time catalysed by 1, a) 0 h, b) 0.5 h, c) 3 h, d) 168 h. Reaction conditions: batch reactor, 1 (25 mg, 1.44 μmol, W loading: 1.06 wt%), 1-decene (0.5 mL, 2.64 mmol), 150 °C. For clarity, C10 represents all decene regioisomers. | ||
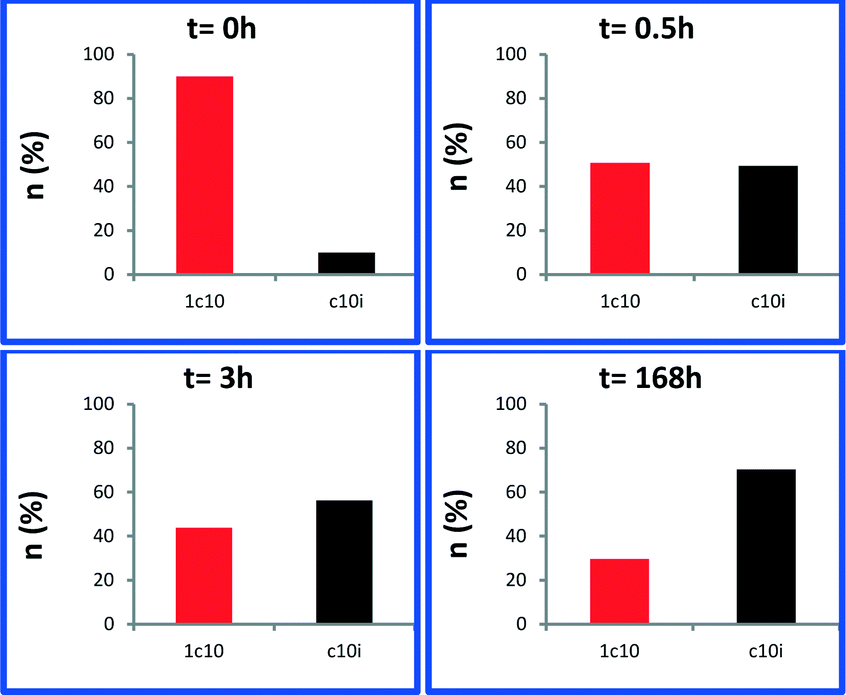 | ||
| Fig. 7 Molar fraction of 1-decene (red) and isomers of 1-decene (black) at different time periods: 0 h, 0.5 h, 3 h and 163 h. Reaction conditions: batch reactor, 1 (25 mg, 1.44 μmol, W loading: 1.06 wt%), 1-decene (0.5 mL, 2.64 mmol), 150 °C. | ||
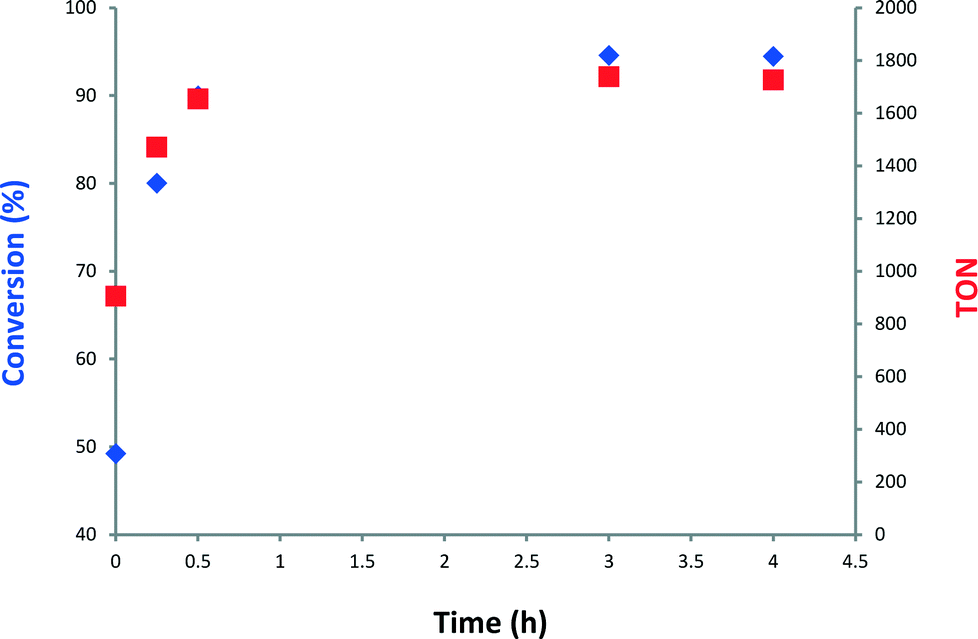 | ||
Fig. 8 Plot of conversion ( ) and TON ( ) and TON ( ) versus time of 1-decene metathesis catalyzed by 1. Reaction conditions: batch reactor, 1 (25 mg, 1.44 μmol, W loading: 1.06 wt%), 1-decene (0.5 mL, 2.64 mmol), 150 °C. ) versus time of 1-decene metathesis catalyzed by 1. Reaction conditions: batch reactor, 1 (25 mg, 1.44 μmol, W loading: 1.06 wt%), 1-decene (0.5 mL, 2.64 mmol), 150 °C. | ||
Additionally, the terminal sp3 C–H bond of 1-decene could theoretically be active for CH activation via a sigma-bond elementary step, as observed for n-decane, to generate either a diene or a W-alkylidene with a terminal olefin. Thus, we examined the direct reaction of 1,9-decadiene catalyzed by 1. In this case, we observed cyclic olefins (cyclohexene and cycloheptene) resulting from intramolecular ring-closing metathesis of dienes. The formation of such 6- and 7-membered carbocycles has been reported for cyclooctane metathesis in which lower cyclic alkanes (from cyclopentane to cycloheptane) were observed. Since 1-decene with no cyclic products was detected, the in situ formation of a diene seems unlikely under these reaction conditions. Olefin metathesis/isomerization must be the predominant elementary step at the expense of sp3 carbon activation when using a terminal olefin as a substrate.
We also compared the 1-decene metathesis of a multifunctional catalyst composed of a typical Grubbs' first-generation Ru-based catalyst impregnated on alumina under our reaction conditions.17,18 This catalyst possesses the alkylidene function without coordinated alkyl groups. After 24 h, the GC chromatogram indicates mainly the self-metathesis of 1-decene with only traces of isomers (see Fig. S11†).
In order to compare the product distribution of metathesis of n-decane and 1-decene, hydrogenation of an olefin mixture resulting from 1-decene metathesis with catalyst 1 was also carried out in methanol in the presence of Pd/C under atmospheric pressure of hydrogen. The hydrogenated products are linear alkanes (Fig. 9). It is worth noting that this alkane distribution resulting from olefin metathesis differs totally from the decane metathesis with the same catalyst. In the latter, we observe products up to C30 (Fig. 9). This could be attributed to the different amounts of hydrogen released in a catalytic cycle in these two systems. Interestingly, addition of a hydrogen acceptor tert-butylethylene (tbe) with pre-catalyst 1 (W/tbe molar ratio: 1/13) led to no activity in n-decane metathesis over a 3 day period, and the n-decane metathesis products were not observed, while with the same ratio of W/tbe, metathesis of 1-decene occurs.
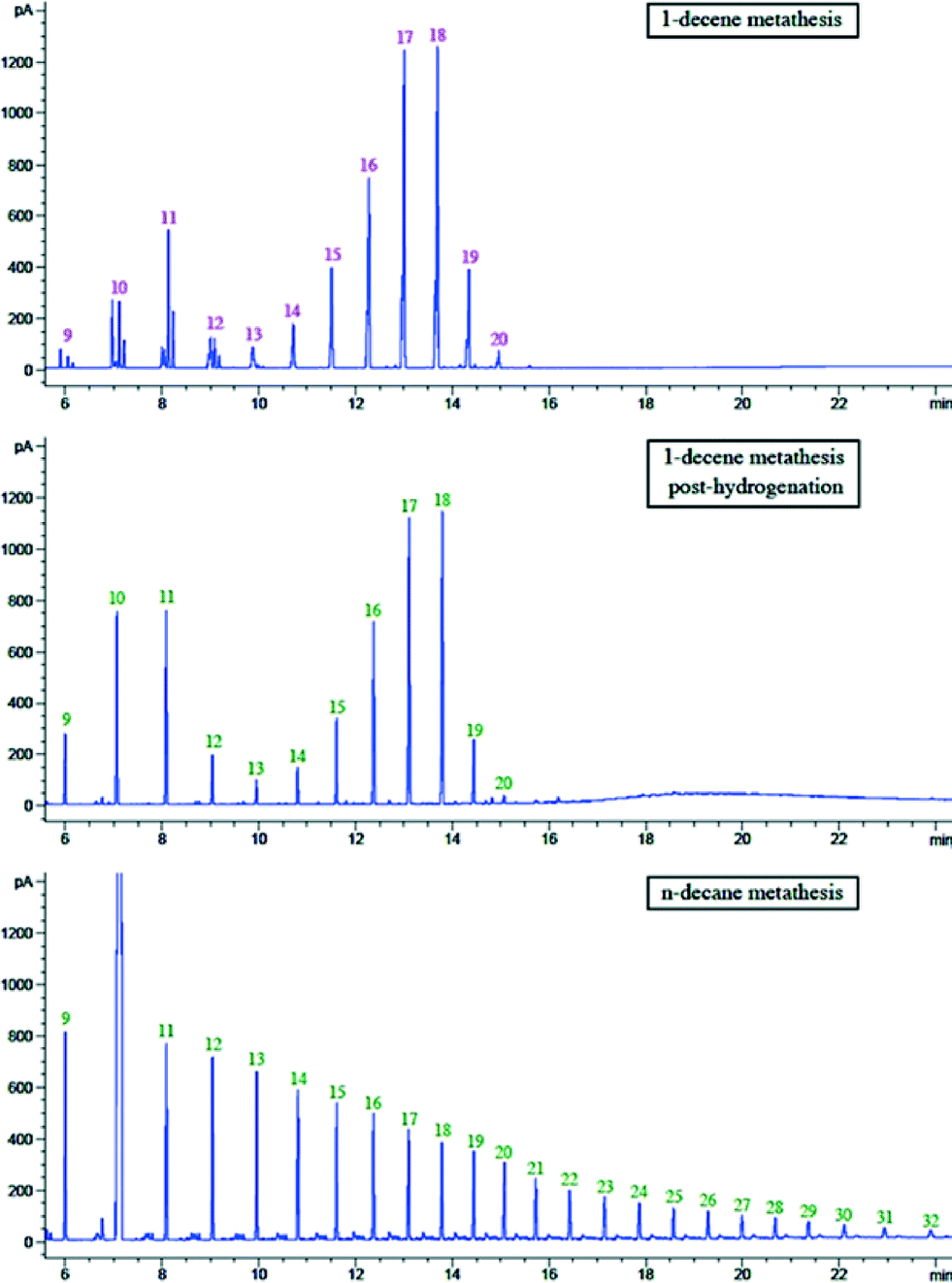 | ||
| Fig. 9 GC chromatograms of 1-decene metathesis, hydrogenation of 1-decene metathesis mixture and n-decane metathesis. | ||
This result is proof of a strong mechanistic difference between alkane and olefin metathesis. For the alkane metathesis, the olefin formed by succession of σ-bond metathesis and β-H elimination steps does not decoordinate rapidly from the W catalyst as would be expected on a d0 system. In the contrary, it would immediately undergo [2 + 2] cycloaddition on the coordinated carbene. After cycloreversion of the metallacycle, there is a fast reaction of the resulting olefin with the coordinated hydride to form an alkyl group. This implies the importance of the low steady-state olefin concentration in alkane metathesis.
We propose the following mechanism pathway to account for the formation of heptacosane (C27) starting from n-decane (Fig. 10). An initial σ-bond metathesis generates methane and the W bis-decyl species.19–21 A classical β-H elimination gives the W-H and the coordinated olefin. Next, the well-established α-hydrogen transfer from the alkyl to alkylidyne forms the W-carbene.22–24 A [2 + 2] cycloaddition followed by the cycloreversion leads to the internal alkene and the W-bis-carbene. Successive insertion/elimination steps (by chain walking) give the terminal alkene, which reacts to a new W-alkylidene.25,26 The CH activation of the pendant W-hydride with n-decane followed by β-H elimination provides 1-decene. A second 1-decene metathesis with newly formed W-alkylidene and hydrogenolysis affords heptacosane (C27). For the olefin metathesis, H-transfer of the W-methylidyne generates the corresponding W-bis-carbene. Next, successive [2 + 2] cycloaddition of olefin and [2 + 2] cycloreversion steps lead to W-alkylidene. The latter reacts with an additional olefin to release the substituted olefin. The major difference between the olefin and the alkane mechanisms is the amount of hydrogen produced which is higher for alkanes.
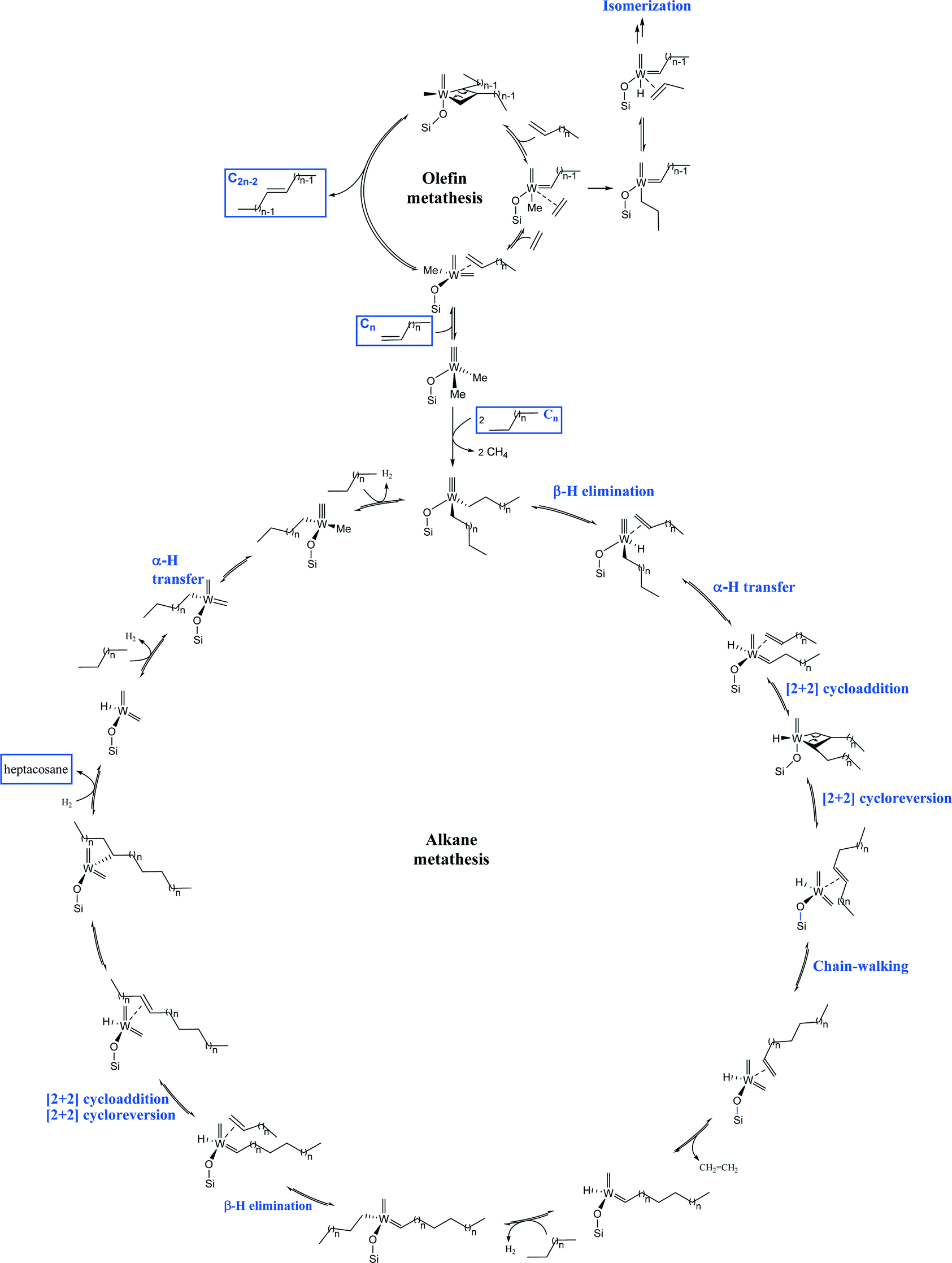 | ||
| Fig. 10 Plausible mechanism for linear decane metathesis and α-decene metathesis from common pre-catalyst 2 (n = 7 for n-decane and 1-decene metathesis). | ||
Conclusion
We have demonstrated the efficiency of 1 and 2 in n-alkane metathesis leading to distribution of linear alkanes in the range of C1 to C30. By comparing the homologous terminal alkenes, we found a strong difference of product distribution between n-decane and 1-decene metathesis. This shows the striking difference between alkane metathesis and olefin metathesis. This difference is attributed to the very small amount of olefin produced freely on the single-site catalyst due to the supposedly very fast 2 + 2 intramolecular cycloaddition of the formed olefin to the metallocarbene and then followed again by fast reinsertion of the resulting olefin to the metal hydride. Considering the tandem catalytic system, our single-site system is certainly a convenient protocol to upgrade hydrocarbons.Acknowledgements
Research reported in this publication was supported by the King Abdullah University of Science and Technology.Notes and references
- J. M. Basset, C. Coperet, D. Soulivong, M. Taoufik and J. T. Cazat, Acc. Chem. Res., 2010, 43, 323–334 CrossRef CAS PubMed.
- M. C. Haibach, S. Kundu, M. Brookhart and A. S. Goldman, Acc. Chem. Res., 2012, 45, 947–958 CrossRef CAS PubMed.
- V. Vidal, A. Theolier, J. Thivolle-Cazat and J. M. Basset, Science, 1997, 276, 99–102 CrossRef CAS.
- A. S. Goldman, A. H. Roy, Z. Huang, R. Ahuja, W. Schinski and M. Brookhart, Science, 2006, 312, 257–261 CrossRef CAS PubMed.
- R. L. Burnett and T. R. Hughes, J. Catal., 1973, 31, 55–64 CrossRef CAS.
- C. Coperet, Chem. Rev., 2010, 110, 656–680 CrossRef CAS PubMed.
- E. Le Roux, M. Taoufik, C. Coperet, A. de Mallmann, J. Thivolle-Cazat, J. M. Basset, B. M. Maunders and G. J. Sunley, Angew. Chem., Int. Ed., 2005, 44, 6755–6758 CrossRef CAS PubMed.
- K. C. Szeto, L. Hardou, N. Merle, J. M. Basset, J. Thivolle-Cazat, C. Papaioannou and M. Taoufik, Catal. Sci. Technol., 2012, 2, 1336–1339 CAS.
- F. Quignard, A. Choplin and J. M. Basset, J. Chem. Soc., Chem. Commun., 1991, 1589–1590 RSC.
- J. M. Basset, C. Coperet, L. Lefort, B. M. Maunders, O. Maury, E. Le Roux, G. Saggio, S. Soignier, D. Soulivong, G. J. Sunley, M. Taoufik and J. Thivolle-Cazat, J. Am. Chem. Soc., 2005, 127, 8604–8605 CrossRef CAS PubMed.
- M. K. Samantaray, E. Callens, E. Abou-Hamad, A. J. Rossini, C. M. Widdifield, R. Dey, L. Emsley and J. M. Basset, J. Am. Chem. Soc., 2014, 136, 1054–1061 CrossRef CAS PubMed.
- E. Callens, E. Abou-Hamad, N. Riache and J. M. Basset, Chem. Commun., 2014, 50, 3982–3985 RSC.
- N. Riache,
et al., Cyclooctane metathesis catalyzed by silica supported tungsten pentamethyl (SiO)W(Me)5: distribution of macrocyclic alkanes, Chem. – Eur. J., 2014 DOI:10.1002/chem.201403704 , in press.
- NIST Standard Reference Database, http://webbook.nist.gov/chemistry/ Search PubMed.
- S. Kundu, Y. Choliy, G. Zhuo, R. Ahuja, T. J. Emge, R. Warmuth, M. Brookhart, K. Krogh-Jespersen and A. S. Goldman, Organometallics, 2009, 28, 5432–5444 CrossRef CAS.
- Z. Huang, E. Rolfe, E. C. Carson, M. Brookhart, A. S. Goldman, S. H. El-Khalafy and A. H. R. MacArthur, Adv. Synth. Catal., 2010, 352, 125–135 CrossRef CAS PubMed.
- M. Bru, R. Dehn, J. H. Teles, S. Deuerlein, M. Danz, I. B. Muller and M. Limbach, Chem. – Eur. J., 2013, 19, 11661–11671 CrossRef CAS PubMed.
- B. Van Berlo, K. Houthoofd, B. F. Sels and P. A. Jacobs, Adv. Synth. Catal., 2008, 350, 1949–1953 CrossRef CAS PubMed.
- P. L. Watson, J. Am. Chem. Soc., 1983, 105, 6491–6493 CrossRef CAS.
- C. Coperet, O. Maury, J. Thivolle-Cazat and J. M. Basset, Angew. Chem., Int. Ed., 2001, 40, 2331 CrossRef CAS.
- F. Rascon and C. Coperet, J. Organomet. Chem., 2011, 696, 4121–4131 CrossRef CAS PubMed.
- K. G. Caulton, M. H. Chisholm, W. E. Streib and Z. L. Xue, J. Am. Chem. Soc., 1991, 113, 6082–6090 CrossRef CAS.
- L. A. Morton, S. J. Chen, H. Qiu and Z. L. Xue, J. Am. Chem. Soc., 2007, 129, 7277–7283 CrossRef CAS PubMed.
- Z. L. Xue and L. A. Morton, J. Organomet. Chem., 2011, 696, 3924–3934 CrossRef CAS PubMed.
- C. M. Killian, D. J. Tempel, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 11664–11665 CrossRef CAS.
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cy00663a |
| This journal is © The Royal Society of Chemistry 2015 |