
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of Pd nanoparticles supported on zeolites for hydrogen production from formic acid dehydrogenation
M.
Navlani-García
a,
M.
Martis
b,
D.
Lozano-Castelló
*a,
D.
Cazorla-Amorós
a,
K.
Mori
bc and
H.
Yamashita
*bc
aDepartamento de Química Inorgánica e Instituto Universitario de Materiales, Universidad de Alicante, Ap. 99, E-03080, Alicante, Spain. E-mail: d.lozano@ua.es; Fax: +34 96590 3454; Tel: +34 96590 3946
bDivision of Materials and Manufacturing Science, Graduate School of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: yamashita@mat.eng.osaka-u.ac.ps
cElements Strategy Initiative for Catalysts and Batteries (ESICB), Kyoto University, Katsura, Kyoto 615-8520, Japan
First published on 20th August 2014
Abstract
Catalysts based on palladium nanoparticles supported on different zeolites (BETA, ZSM-5 and Y) were prepared and their catalytic performance in formic acid dehydrogenation was studied. The effects of the zeolite structure and porous texture on the catalytic activity were investigated by comparing the behaviour of these samples. The results revealed that the samples based on BETA zeolite are promising catalysts for this application.
Introduction
Nowadays, the demand for energy is getting more and more attention, and in this sense, hydrogen is considered one of the most promising clean energy carriers since it has several advantages with respect to fossil fuels. The main benefits of using H2 as an energy vector lie in the fact that it is an inexhaustible fuel and the only product of its combustion with oxygen is water.1 However, one of the major drawbacks to the use of hydrogen as fuel is its storage. For this reason, a lot of effort is being made in order to get a suitable material which is able to fulfill the hydrogen storage requirements. Several kinds of materials have been considered for hydrogen storage, either physical or chemical storage. In relation to the physical methods, hydrogen can be stored as a compressed gas or as a cryogenic liquid,2 as well as by physical adsorption on the surface of porous solid materials, with carbon materials,3–5 organic polymers,6,7 zeolites,8,9 metal–organic frameworks (MOFs),10,11 and clathrate hydrates12,13 as the main adsorbents. Concerning the chemical storage, hydrogen storage can take place in solid-phase (hydrides, alanates, amides and imides, among others)14,15 or liquid-phase components (hydrazine, alcohols and formic acid (FA), among others).16,17FA, which has gravimetric and volumetric hydrogen capacities of 4.4 wt.% and 52 g l−1,18 respectively, has recently received attention as a hydrogen source since it is nontoxic and liquid at room temperature (density, 1.22 g cm−3).19 It is well known that FA decomposition can take place following two different pathways:
| Dehydrogenation: HCOOH ↔ H2 + CO2, ΔG = − 48.4 kJ mol−1 | (1) |
| Dehydration: HCOOH ↔ CO + H2O, ΔG = − 28.5 kJ mol−1 | (2) |
As one of the main goals of hydrogen production is its use in fuel cells, the dehydration reaction must be avoided in order to obtain H2 streams free of CO (or at least with CO concentration below 10 ppm) because this gas acts as a poison for the Pt electrodes used in fuel cells,20 and that is the reason why several approaches have been made to obtain a suitable catalyst for the selective dehydrogenation of FA. Among all the catalysts studied in this application, those based on palladium nanoparticles seem to be very promising, since a relatively high amount of H2 was generated and high selectivity was achieved at room temperature.17,21,22 Metal nanoparticles (NPs) are widely applied in catalysis due to their extremely small size and high surface-to-volume ratio, which make them very interesting candidates for use as active phases in several catalysts. However, when this kind of metal nanoparticles is used, some problems related to NP aggregation may occur and this might reduce both the catalytic activity and the selectivity.23 Hence, one possible solution for this problem could be the use of a stabilising agent which would prevent sintering of the metal nanoparticles. The use of several stabilising agents, such as dendrimers,24 tetraalkylammonium salts,25 or ionic liquids,26 has already been reported, but polymers are one of the most used stabilising agents.27,28 Among these polymers, polyvinylpyrrolidone (PVP) is one of the most common29 combined with different synthesis strategies, such as electrochemical methods30 or reduction-by-solvent method.31
Several kinds of materials have been used as catalyst supports of metallic nanoparticles in FA decomposition (i.e. macroreticular resin,21 graphene oxide,32 amines,33 MOFs22 and so on). Although zeolites constitute one of the most important groups of heterogeneous catalysts supports with large-scale applications,34 to our knowledge, their use in this application has not been reported yet. These materials are crystalline aluminosilicates having unique properties, such as thermal stability, relatively high surface area, intrinsic acidity, defined microporosity and ion exchange capacity, which make them suitable materials in different applications.
There are many different framework structures. The supports studied in this work are BETA zeolite (Framework Type *BEA with 3-dimensional channels and a pore system consisting of 12-membered rings with two opening channel diameters of 0.56 × 0.56 and 0.77 × 0.66 nm), ZSM-5 zeolite (Framework Type MFI with 3-dimensional channels and a pore system consisting of 10-membered rings with opening channel diameters of 0.51 × 0.55 and 0.53 × 0.56 nm) and Y zeolite (Framework Type FAU with 3-dimensional channels and a pore system consisting of 12-membered rings with an opening channel diameter of 0.74 × 0.74 nm).35,36 Among them, it is well known that BETA zeolite is a promising heterogeneous catalyst for several liquid-phase reactions.37,38
Considering all the abovementioned, the goal of the present work is to prepare catalysts based on PVP-stabilised palladium nanoparticles supported on different zeolites (BETA, ZSM-5 and Y) and to study their performance in the formic acid dehydrogenation reaction at low temperature.
Experimental
Synthesis and purification of nanoparticles
Palladium nanoparticles were synthesised in an inert argon atmosphere by following the reduction by solvent method as reported by our research group elsewhere.31,39 The palladium precursor was Pd(ac)2 (98%, Sigma-Aldrich) and the stabilising agent used was polyvinylpyrrolidone (PVP, 40 K, Sigma-Aldrich). The metal nanoparticles were synthesised by the following experimental procedure.For solution 1, 1.114 g (10 mmol) of PVP (stabilising agent) were added to 120 ml of ethylene glycol in a three-necked round-bottomed flask and the solution was stirred at 80 °C for 2 h using a magnetic stirrer and an oil bath.
For solution 2, 0.2245 g (1 mmol) of Pd(ac)2 were dissolved in 50 ml of dioxane by stirring for 2 h at room temperature using a magnetic stirrer. The colour of this solution was light orange.
After the solution preparation, solution 1 was cooled down to 0 °C in an ice bath and 3 ml of 1 M NaOH solution was added under stirring in order to adjust the pH of the resulting mixture. Then, solution 2 was poured into solution 1 under vigorous stirring in order to ensure homogenisation and the final mixture was heated up to 100 °C. The solution changed its colour from light orange to dark brown, which indicated that the metallic colloid had formed.40 Then, heating was maintained for 2 h, after which the bath was removed and the colloidal suspension was cooled down to room temperature.
Once the colloids were prepared, the palladium nanoparticles were purified and redispersed in methanol. The purification was carried out by pouring an aliquot of nanoparticle colloid into a glass bottle, adding an excess of acetone and shaking the solution. As we have previously reported,39 the treatment with acetone caused the extraction of the protecting polymer to the acetone phase and flocculation of the palladium nanoparticles. Then, the supernatant organic phase was removed and the purified nanoparticles were redispersed by stirring in a known amount of methanol, so that the final suspension concentration was perfectly known.
Catalysts support preparation
Four catalyst supports consisting of the acid zeolites BETA, ZSM-5 and Y were studied. The acid zeolites BETA and ZSM-5 (H-BETA and H-ZSM-5) were obtained directly by conversion of commercial zeolites (NH4-BETA and NH4-ZSM-5 with Si/Al = 12.5 and 15.0, respectively, supplied by Zeolyst International) by heat treatment in air at 450 °C for 6 h, with a heating rate of 5 °C min−1. On the other hand, the acid counterpart of the Y zeolite was obtained by ion exchange of the commercial sodium form (Na-Y, with Si/Al = 2.5, supplied by Sigma Aldrich). For this purpose, the sodium zeolite was ion exchanged with 1 M NH4NO3, using a zeolite/solution ratio of 0.25 g/100 ml and maintaining stirring in a thermostatic bath at 60 rpm at 30 °C for 24 h. Afterwards, the NH4-Y zeolite was heat treated in air at 550 °C for 4 h with a heating rate of 1 °C min−1 in order to obtain the final H-Y support. After ion exchange, the sample was filtered, washed with distilled water and dried at 110 °C.Catalysts preparation
The catalysts studied in this work were prepared by impregnation of the zeolitic supports with the palladium nanoparticle colloid following a previously reported methodology.31 The impregnation was carried out by mixing a known amount of each support with an adequate amount of the purified nanoparticle colloid suspension in order to obtain a final metallic loading of 1 wt.%. The mixtures were vigorously stirred for 48 h using a magnetic stirrer to guarantee similar metal loading and distribution in all the catalysts and then the samples were heat treated at 60 °C to remove the solvent (methanol). Finally, the catalysts were washed several times with a cold mixture of H2O/EtOH (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, v/v) and dried at 60 °C for 12 h. The resulting catalysts were denoted as Pd/H-BETA, Pd/H-ZSM-5 and Pd/H-Y.
:50, v/v) and dried at 60 °C for 12 h. The resulting catalysts were denoted as Pd/H-BETA, Pd/H-ZSM-5 and Pd/H-Y.
Catalysts characterisation
The textural characterisation of the zeolitic supports was carried out by means of adsorption of N2 at −196 °C and CO2 at 0 °C (Autosorb 6, Quantachrome). Prior to the adsorption measurements, the supports were outgassed in situ under vacuum at 250 °C for 4 h in order to remove any adsorbed impurities. Apparent surface area values were calculated from nitrogen adsorption isotherms using the BET equation (SBET). Total micropore volume (VDR(N2)) and narrow micropore volume (VDR(CO2)) were calculated by applying the Dubinin–Radushkevich (DR) equation to the N2 adsorption data at −196 °C and the CO2 adsorption data at 0 °C, respectively. Mesopore volume (Vmeso(N2)) was also calculated from the N2 isotherm taking into account the volume adsorbed between the partial pressures 0.9 and 0.2 in the desorption branch. External zeolite surface area was calculated by applying the t-plot method.Transmission electron microscopy (TEM) images were obtained by using a JEOL (JEM-2010) equipped with an EDS analyzer (Oxford, model INCA Energy TEM 100) operating at 200 kV with a space resolution of 0.24 nm. In order to prepare the samples for the TEM analysis, a small amount of the catalyst was suspended in methanol and sonicated in an ultrasonic bath for a few minutes. After that, a drop of this suspension was deposited onto a 300-mesh Lacey copper grid and dried at room temperature. TEM images were also captured for the palladium colloid in order to compare the NP diameter and particle size distribution before and after the impregnation step.
The dispersion of the palladium nanoparticles on the catalyst (D), which is an estimation of the amount of metal exposed in the nanoparticles, was calculated from the following equation, assuming a spherical shape of the particles (d, particle diameter):41
| D (%) ≈ (0.9)/d (nm) |
Inductively coupled plasma-optical emission spectroscopy (ICP-OES) was the selected technique to determine the palladium loading. For this purpose, a Perkin Elmer Optima 4300 system was used. The palladium extraction was carried out by oxidative treatment with aqua regia for 48 h. Then, the samples were filtered and diluted.
Pd K-edge XAFS spectra were recorded using a fluorescence-yield collection technique at the beam line 01B1 station with an attached Si (311) monochromator at SPring-8, JASRI, Harima, Japan (prop. no. 2013B1041). The EXAFS data were normalized by fitting the background absorption coefficient around the energy region higher than the edge of about 35–50 eV, with the smoothed absorption of an isolated atom. The EXAFS data were examined using the Rigaku EXAFS analysis program.
Moreover, in order to analyse the interaction between the formic acid and the different zeolites, DRIFT spectra of zeolitic supports and catalysts before and after formic acid adsorption were recorded by using a FTIR spectrometer (Jasco, model FT/IR-4100). Prior to the DRIFT measurement, the adsorption of formic acid was carried out at room temperature. For this purpose, the samples were placed in a dry chamber (free of humidity) with a vessel containing formic acid for 72 h. The samples were weighted before and after the formic acid adsorption, thus the increase in weight indicated that in all cases the formic acid adsorption had taken place due to its high vapor pressure. Afterwards, samples were placed in the spectrometer chamber in the presence of a dry air flow (CO2 and H2O free) and the spectra were recorded at room temperature.
Catalytic tests of formic acid dehydrogenation
In order to analyse the catalytic performance of the different samples, 50 mg of each powder sample and 9.6 ml of water were mixed in a Pyrex reaction vessel (30 ml) which was sealed with a rubber septum. Afterward, samples were treated in an ultrasound bath for 15 min for complete dispersion of the zeolitic samples. Then, the mixture was bubbled with argon gas for 30 min to ensure an inert atmosphere. Once this mixture was prepared, 0.39 ml of FA was added into the vessel and it was placed in an oil bath at 50 °C with magnetic stirring for 3 h. The subsequent H2 and CO2 generation was monitored by using a Shimadzu GC14B equipped with an MS5 A column (H2) and a Shimadzu GC (CO2). For this purpose, injections in both GC were made and the resulting peak areas were converted into H2 and CO2 concentrations by using a calibration with the corresponding standard gases.Results and discussion
Characterisation
Fig. 1 shows the N2 adsorption–desorption isotherms at −196 °C for all the zeolitic supports. ZSM-5 and Y zeolites present type I isotherms according to the IUPAC classification,42 showing a high uptake of nitrogen at very low relative pressures, which is characteristic of microporous materials. BETA zeolite has a type I+IV isotherm,42 with a high uptake of nitrogen at very low relative pressures and a hysteresis loop because of the presence of micro- and mesoporosity.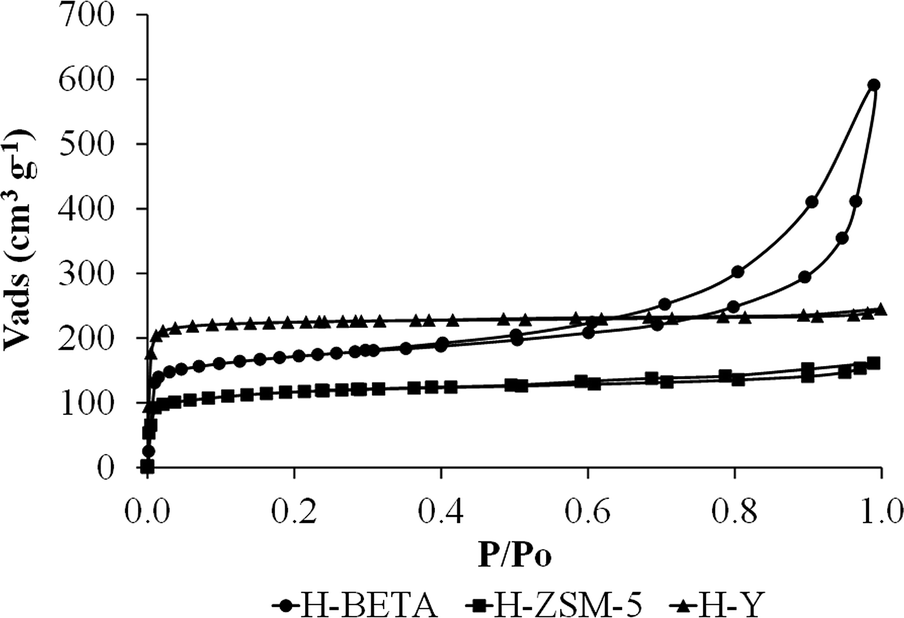 | ||
| Fig. 1 N2 adsorption–desorption isotherms at −196 °C corresponding to H-BETA, H-ZSM-5 and H-Y zeolites. | ||
Table 1 summarizes the BET surface area (SBET), the total micropore volume (VDR(N2)), the narrow micropore volume (VDR(CO2)), the mesopore volume (Vmeso(N2)) and the external zeolite surface area of the catalyst support. As can be observed from Table 1, the zeolite specific surface area (SBET) follows the order Y > BETA > ZSM-5. The total micropore volume (VDR(N2)) calculated from the nitrogen isotherm and the narrow micropore volume (VDR(CO2)) calculated from the CO2 isotherm follow the same tendency. The mesopore volume (Vmeso) is much higher for the BETA zeolite than for the other supports, which is in good agreement with the shape of the nitrogen isotherms, where a more important hysteresis loop was observed for that zeolite. Moreover, a very important difference was observed for the external surface area determined for the catalyst support since the H-BETA zeolite showed a much higher external surface than those observed for H-ZSM-5 and H-Y as a consequence of its characteristic surface roughness.43,44
| Sample | S BET (m2 g−1) | V DR(N2) (cm3 g−1) | V DR(CO2) (cm3 g−1) | V meso (cm3 g−1) | External surface area (m2 g−1) |
|---|---|---|---|---|---|
| H-BETA | 590 | 0.27 | 0.25 | 0.21 | 122 |
| H-ZSM-5 | 370 | 0.16 | 0.15 | 0.03 | 42 |
| H-Y | 725 | 0.35 | 0.41 | 0.01 | 52 |
Fig. 2 shows a TEM micrograph of the palladium nanoparticle colloid and the histogram with the particle size distribution for this colloid. The analysis of the TEM images for the colloid indicated that the Pd NPs show a small average particle size (d = 2.1 ± 0.3 nm) and a very narrow size distribution, as was previously reported by our research group.31,45 Moreover, as can be observed, palladium nanoparticles present a spherical shape in the as-prepared colloid.
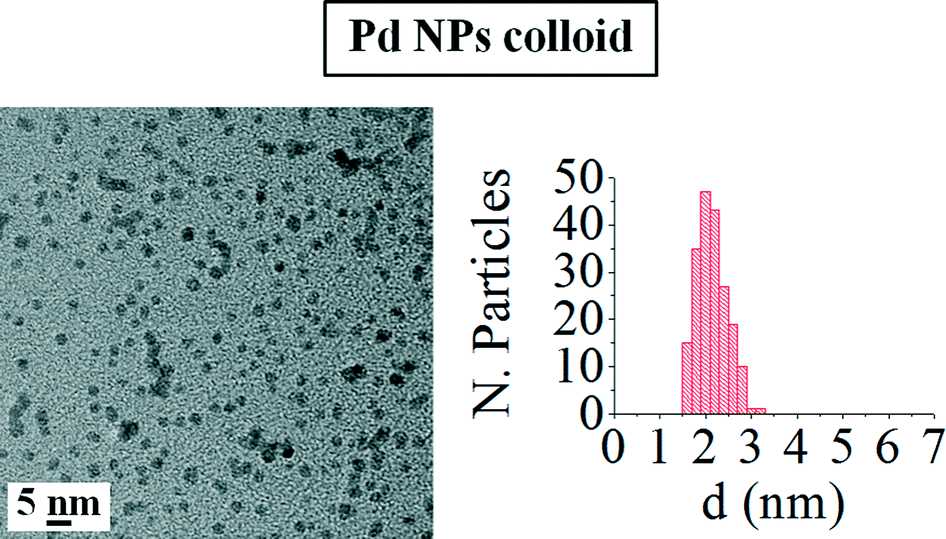 | ||
| Fig. 2 TEM image and histogram with the nanoparticle size distribution of the palladium colloid. | ||
Fig. 3 includes the TEM images of the as-prepared catalysts and the pertinent histograms with the particle size distributions. From the TEM images observation, it can be concluded that the NPs supported on the different zeolites show a very narrow size distribution and aggregates of palladium nanoparticles are not formed as a consequence of the impregnation or drying steps. However, a slight loss of their spherical shape can be appreciated, which might be favoured by the interaction between the metal and the zeolite surface.
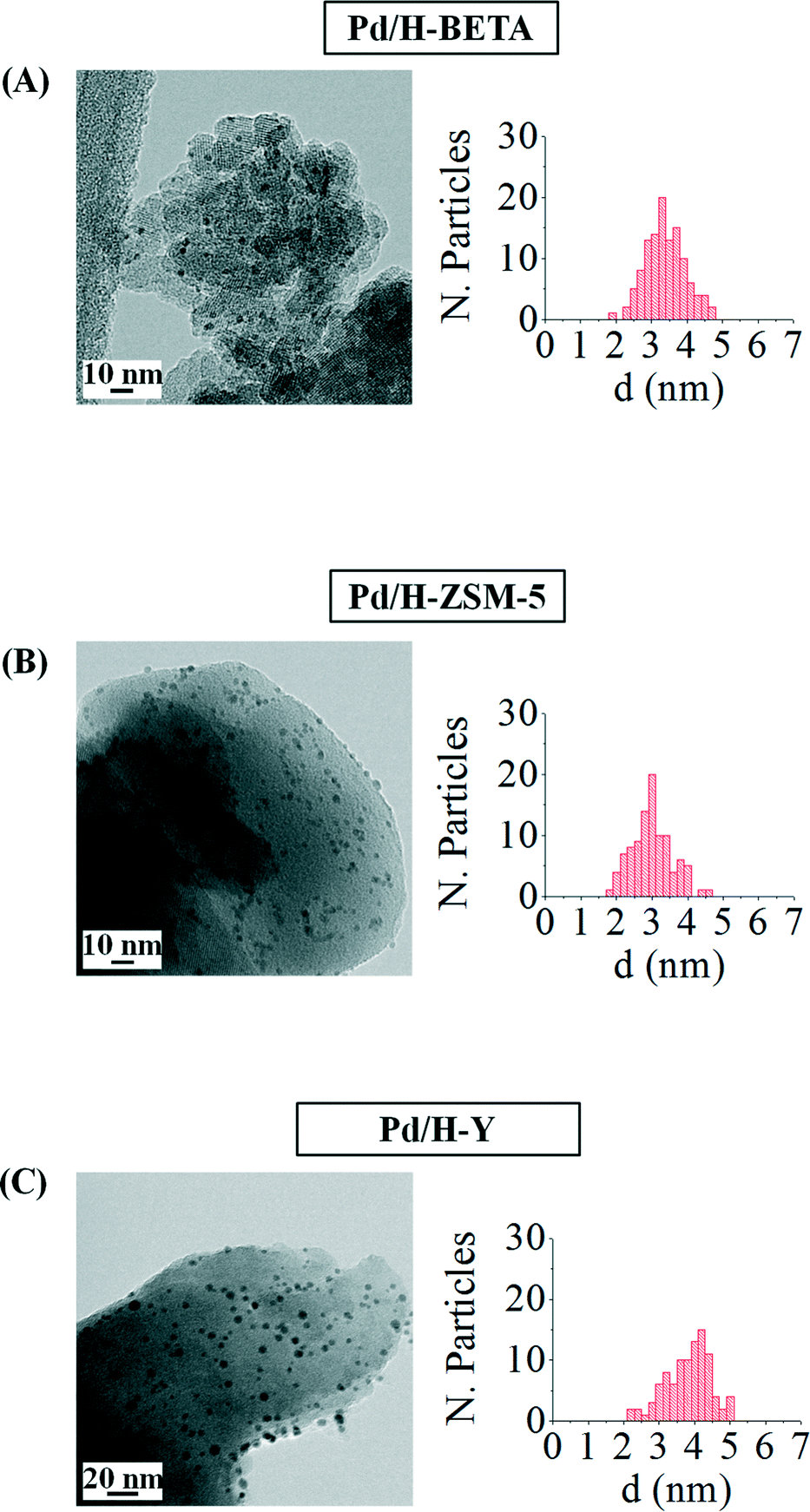 | ||
| Fig. 3 TEM images and the corresponding histograms of samples (A) Pd/H-BETA, (B) Pd/H-ZSM-5 and (C) Pd/H-Y. | ||
Table 2 summarizes the nanoparticle sizes for all the studied samples, the dispersion calculated from the TEM images and the metal loading determined by means of ICP. Metal loading, determined by means of ICP-OES (see Table 2), achieved after 48 h of impregnation is very close to 1 wt.% for all the samples.
| Sample | d TEM (nm) | D TEM (%) | Pd loading ICP (wt.%) |
|---|---|---|---|
| Pd/H-BETA | 3.4 ± 0.5 | 26.5 | 1.0 |
| Pd/H-ZSM-5 | 3.0 ± 0.6 | 30.0 | 0.9 |
| Pd/H-Y | 4.1 ± 0.9 | 22.0 | 0.9 |
Table 2 shows that an important particle size increase is produced when the palladium nanoparticles are deposited onto the different zeolites, especially in the case of the sample Pd/H-Y, where the particle size almost doubles the size measured in the metal colloid (d = 2.1 ± 0.3 nm). This size increase could be a consequence of the impregnation process, where some nanoparticle deformation can take place leading to larger metal nanoparticles. Considering the nanoparticle size and the zeolite pore openings, it could be said that in all the samples the palladium nanoparticles are located on the external zeolite surfaces.
Fig. 4 shows the Fourier transform (FT) k3-weighted Pd K-edge extended X-ray absorption fine structure (EXAFS) of these samples. All samples display a main peak centered at 2.65 Å, which is assigned to a single Pd–Pd bond, suggesting that Pd nanoparticles exist in a metallic form.
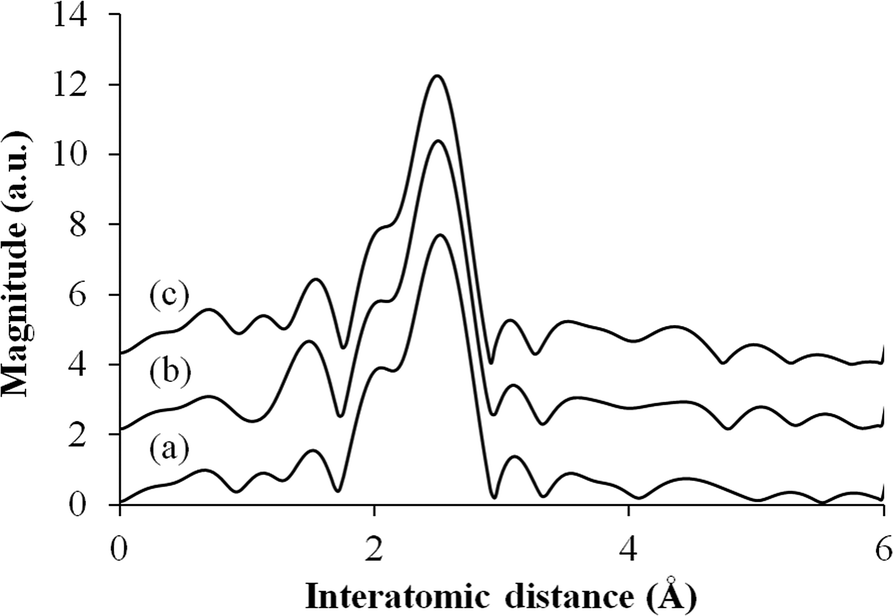 | ||
| Fig. 4 Pd K-edge FT-EXAFS spectra for (a) Pd/H-BETA, (b) Pd/H-Y and (c) Pd/H-ZSM-5. | ||
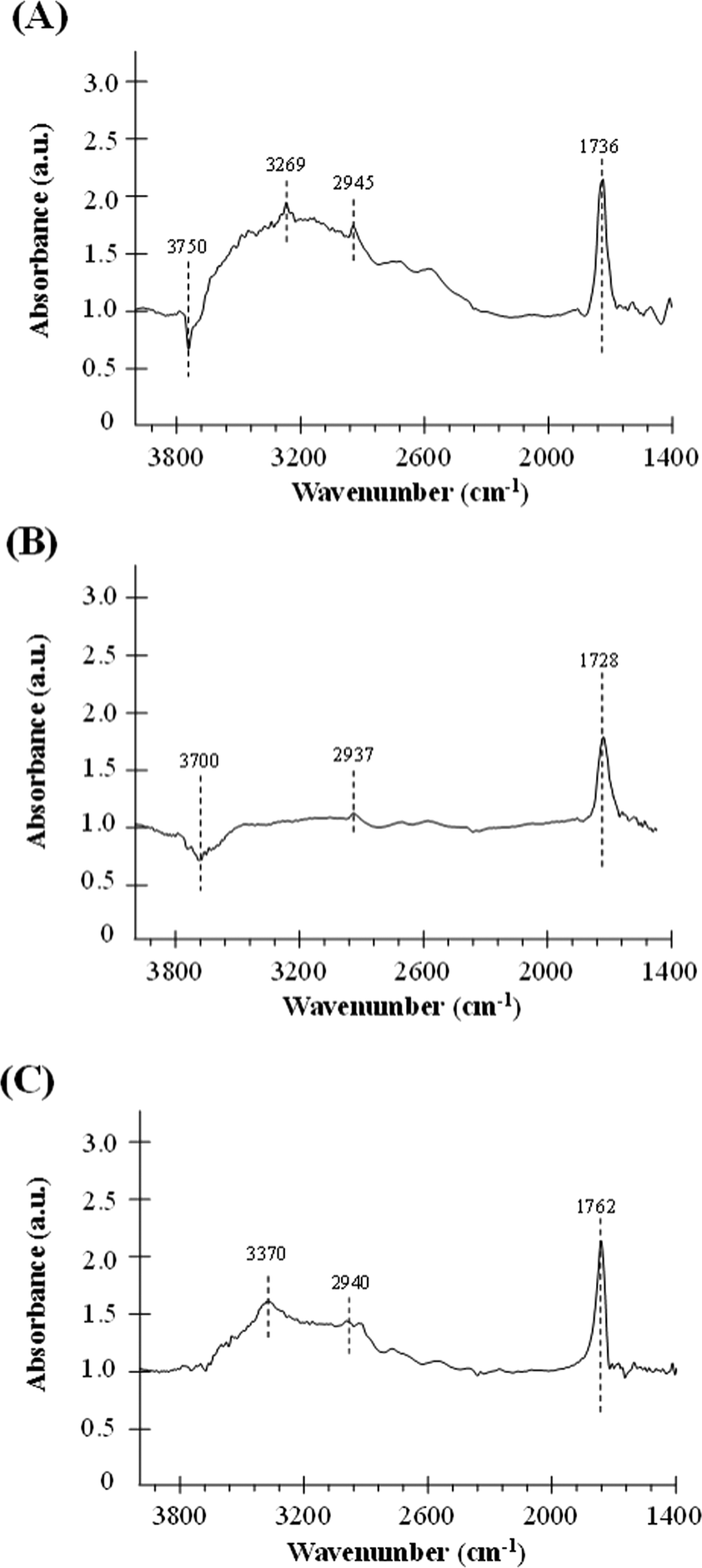
For a better understanding of the catalytic performance in the formic acid decomposition reaction, the interaction between formic acid and the different samples has been studied by DRIFT experiments. DRIFT spectra were recorded before and after formic acid adsorption, and Fig. 5 contains the DRIFT spectra for the catalysts Pd/H-BETA, Pd/H-ZSM-5 and Pd/H-Y after subtraction of the spectrum before formic acid adsorption. Therefore, the appearance of a positive peak means that this peak appears as a consequence of the interaction between the sample and the formic acid, while the negative peaks indicate that these peaks have been removed from the original sample spectrum after this adsorption. Moreover, it should be highlighted that only the spectral region from 1400 to 4000 cm−1 is shown, since, although Pd surfaces give rise to an absorption band at ~1350 cm−1, in our case the peaks obtained in this region led to a very complicated spectrum in which the contribution of formic acid was not clearly elucidated. This was due to the fact that the studied palladium nanoparticles are not only constituted by Pd but also by a polymeric capping agent, polyvinylpyrrolidone (PVP), which shows typical vibrational bands between 1300 and 1660 cm−1,45 and these bands overlap with those caused by the formic acid adsorption on the palladium surface.
 | ||
| Fig. 5 DRIFT spectra (after subtraction of the spectrum before formic acid adsorption) for the catalysts (A) Pd/H-BETA, (B) Pd/H-ZSM-5 and (C) Pd/H-Y. | ||
It can be observed from Fig. 5 that for all the samples, after formic acid adsorption, two new peaks at about ~1700 cm−1 and ~2940 cm−1 appear, which correspond to the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and ν(CH) of the formic acid, respectively.46 Comparing the relative intensities of these peaks, it can be seen that the lowest intensity peaks correspond to the catalyst based on ZSM-5 zeolite, which indicates that this material has the lowest formic acid adsorption capacity in concordance with its lower specific surface area (see Table 1). Other relevant spectroscopic features observed after the formic acid adsorption are in the OH group region. In this sense, the exposure of the surface of the samples based on BETA and ZSM-5 zeolites results in the adsorption of the formic acid molecules on the zeolite surface with a loss of surface –OH groups, which is confirmed by the negative intensities of the peak attributed to the O–H stretching of free silanols mainly located on the external surface, which is centered at 3750 and 3700 cm−1, respectively.47,48 Several studies analysing the adsorption of formic acid on different materials have already been reported in the literature.46,49 Regarding the adsorption on OH groups, in a theoretical study carried out by Iuga et al.50 where the adsorption of formic acid molecules on different silicate surface models were included, it was reported that the most stable configuration is the one in which the carbonyl O atom interacts with a silanol group, while the hydroxyl H atom of HCOOH interacts with the O atom of the siloxane Si–O–Al bridge by forming a hydrogen bond in both cases. Thus, if this adsorption geometry is extrapolated to the formic acid adsorption carried out in the present work, the zeolite surface –OH groups would be hydrogen-bonded to the formic acid and that could explain the broad band centered at 3269 and 3370 cm−1 for samples based on BETA and Y zeolites, respectively.46Fig. 6 shows a schematic representation of the interaction between formic acid and zeolites.46
O) and ν(CH) of the formic acid, respectively.46 Comparing the relative intensities of these peaks, it can be seen that the lowest intensity peaks correspond to the catalyst based on ZSM-5 zeolite, which indicates that this material has the lowest formic acid adsorption capacity in concordance with its lower specific surface area (see Table 1). Other relevant spectroscopic features observed after the formic acid adsorption are in the OH group region. In this sense, the exposure of the surface of the samples based on BETA and ZSM-5 zeolites results in the adsorption of the formic acid molecules on the zeolite surface with a loss of surface –OH groups, which is confirmed by the negative intensities of the peak attributed to the O–H stretching of free silanols mainly located on the external surface, which is centered at 3750 and 3700 cm−1, respectively.47,48 Several studies analysing the adsorption of formic acid on different materials have already been reported in the literature.46,49 Regarding the adsorption on OH groups, in a theoretical study carried out by Iuga et al.50 where the adsorption of formic acid molecules on different silicate surface models were included, it was reported that the most stable configuration is the one in which the carbonyl O atom interacts with a silanol group, while the hydroxyl H atom of HCOOH interacts with the O atom of the siloxane Si–O–Al bridge by forming a hydrogen bond in both cases. Thus, if this adsorption geometry is extrapolated to the formic acid adsorption carried out in the present work, the zeolite surface –OH groups would be hydrogen-bonded to the formic acid and that could explain the broad band centered at 3269 and 3370 cm−1 for samples based on BETA and Y zeolites, respectively.46Fig. 6 shows a schematic representation of the interaction between formic acid and zeolites.46
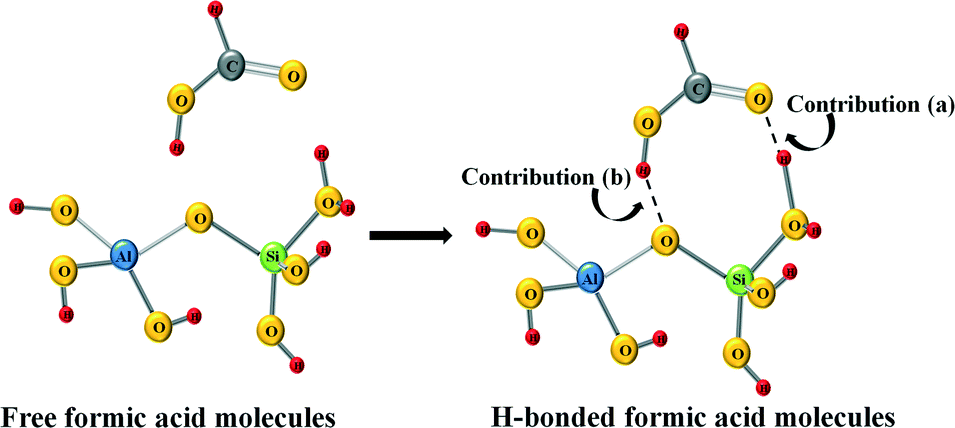 | ||
| Fig. 6 Schematic representation of the interaction between formic acid and zeolites. | ||
In summary, the changes observed in the OH region are a consequence of two different contributions. On the one hand, the adsorbed formic acid molecules on the zeolite surface produce the break of some –OH bonds of the free silanols (see contribution (a) in Fig. 6), which reflects in the negative bands at 3750 and 3700 cm−1 for Pd/H-BETA and Pd/H-ZSM-5, respectively. On the other hand, the formic acid molecules can interact with the zeolite through the hydroxyl H atom of the HCOOH molecule, which interacts with the O atom of the Si–O–Al bridge (see contribution (b) in Fig. 6), which reflect in the positive broad band centered at 3269 and 3370 cm−1 for samples Pd/H-BETA and Pd/H-Y zeolites, respectively.
Bearing all these considerations in mind and considering the relative intensities of the peaks and bands of the different spectra, it could be elucidated that the strength of the interaction between the formic acid molecules and the zeolite surface is different for the studied samples. It seems that this interaction is favoured in the case of the sample based on BETA and Y zeolites and is less important for the ZSM-5 zeolite-based sample.
Catalytic tests of formic acid dehydrogenation
Dehydrogenation of formic acid was performed at 50 °C for all the as-prepared catalysts and the progress of the reaction was monitored by quantifying the hydrogen output after fixed times.Fig. 7 shows the amount of hydrogen produced from the formic acid decomposition at 50 °C by using the acid catalysts Pd/H-BETA, Pd/H-ZSM-5 and Pd/H-Y.
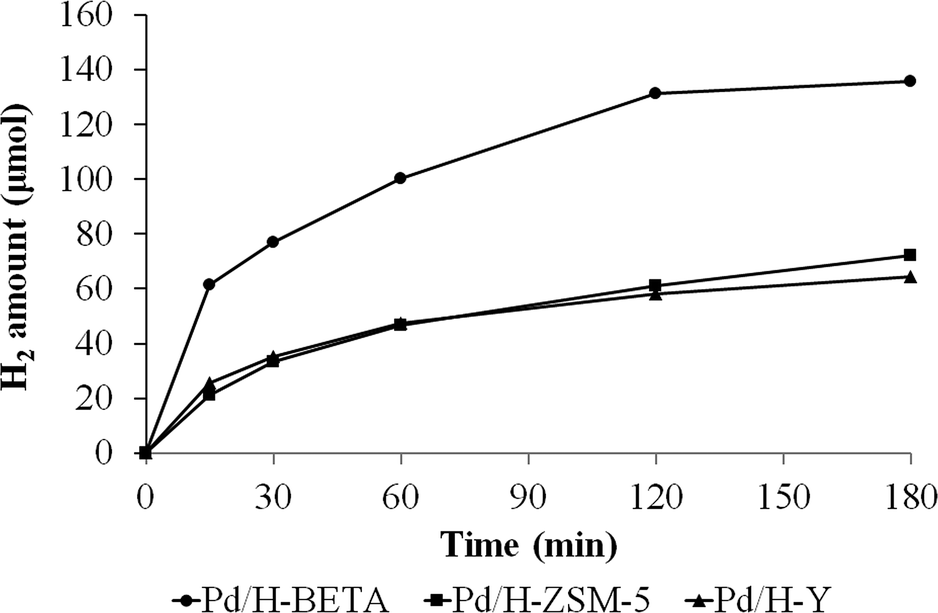 | ||
| Fig. 7 Evolution of the hydrogen production from formic acid dehydrogenation at 50 °C using Pd/H-BETA, Pd/H-ZSM-5 and Pd/H-Y catalysts. | ||
Fig. 7 shows that the catalytic activity is different for all the samples. In this sense, the sample Pd/H-BETA shows the best catalytic performance, generating 135.6 μmol of H2 after 3 h of reaction at 50 °C. Samples Pd/H-ZSM-5 and Pd/H-Y displayed similar H2 production profiles, although the final H2 generated after 3 h is slightly higher in the case of sample Pd/H-ZSM-5 (72.4 and 64.2 μmol of H2, respectively). In order to confirm that the dehydrogenation of formic acid does not proceed on bare zeolites, control experiments with the bare zeolitic supports (H-BETA, H-ZSM-5 and H-Y) were carried out, and as expected, no catalytic activity was observed for any of them.
Taking into account that all the catalysts have similar metal content and similar nanoparticle size, it seems that the zeolite structure and porous texture play important roles in the catalytic performance. In this sense, since the palladium nanoparticles are located on the external zeolite surface, the distribution of the nanoparticles and the stabilising effect of the zeolite are determined by their external zeolite surface. Thus, the higher external surface of the BETA zeolite (see Table 1), which is a consequence of its characteristic surface roughness, leads to a better palladium nanoparticle distribution in comparison with those obtained for ZSM-5 and Y zeolites, which reflects in the paramount catalytic performance displayed by sample Pd/H-BETA.
Moreover, the DRIFT spectra analysis suggests that there is an interaction between the acid sites of the zeolites and the formic acid molecules, which seems to be favoured in the case of sample Pd/H-BETA in comparison with Pd/H-ZSM-5 and Pd/Y samples. This interaction could lead to an activation of the formic acid molecules and the subsequent catalytic activity improvement. The interaction between acid sites and formic acid molecules could also increase the surface concentration of formic acid, which would be more important in the case of the Pd/H-BETA sample.
To check the effect of temperature on the catalytic performance of the Pd/H-BETA, the same reaction was carried out at 30 °C. As expected, the hydrogen production achieved after 3 h is slightly lower compared with the one obtained when the reaction was followed at 50 °C (see Table 3) but still quite high.
| Sample | Pd loading ICP (wt.%) | Temperature (°C) | H2 produced (μmol, t = 3 h) |
|---|---|---|---|
| Pd/H-BETA | 1.0 | 50 | 135.6 |
| Pd/H-BETA | 1.0 | 30 | 99.1 |
| Pd/H-BETA(0.5) | 0.5 | 50 | 111.1 |
For comparison purposes with previous results obtained by our group, a catalyst based on Pd/H-BETA with a lower palladium loading (0.5 wt.%; sample denoted as Pd/H-BETA(0.5)) was prepared and tested in the formic acid decomposition reaction (see results in Table 3). Comparing these results with those previously reported by our research group, the outstanding catalytic performance of the sample based on the BETA zeolite is demonstrated. In this sense, when the catalyst was based on Pd nanoparticles supported on MOF,22 the amount of hydrogen produced after 3 h of reaction (under almost the same experimental conditions) was much lower than the amount of hydrogen generated by using sample Pd/H-BETA(0.5) (48.1 and 99.1 μmol, respectively). Moreover, the hydrogen produced by using the sample Pd/H-BETA(0.5) is slightly higher than that produced by using a more complex material based on Pd nanoparticles within a macroreticular basic resin with the same metal loading (111.1 and 96 μmol, respectively) and very similar to those achieved when the catalytic activity of this macroreticular basic resin is enhanced by adding Ag in Pd/Ag molar ratios of 1:1 (Pd 0.5 wt.% and total metal loading 1.0 wt%).21
The turnover frequency (TOF, h−1) defined as the amount of hydrogen produced per mole of surface metal was 59.2 h−1 for the sample Pd/H-BETA(0.5) at 50 °C, which is higher than those obtained in the literature for different catalytic systems such as Ag@Pd/C (49 h−1, 40 °C)19 or Pd–Au/C (27 h−1, 92 °C).51
Considering the outstanding results obtained with the sample Pd/H-BETA and taking into account the simplicity of the Pd/zeolite system studied, this material can be considered as a very promising catalyst for hydrogen production from formic acid decomposition reaction.
Conclusions
Catalysts based on zeolites (BETA, ZSM-5 and Y) containing Pd nanoparticles have been successfully prepared and tested for hydrogen production from formic acid dehydrogenation. The results suggested that the porous structure of the zeolite plays an import role in the catalytic performance, being better in the case of samples based on BETA zeolite. The presence of higher external surface in BETA compared to those values obtained for ZSM-5 and Y zeolites helped to obtain well-distributed Pd nanoparticles. This fact, together with the high interaction between the acid sites of BETA zeolite and the formic acid molecules, as deduced from DRIFT spectra analysis, allowed us to obtain a very promising catalyst for hydrogen production from formic acid decomposition.Acknowledgements
The authors would like to acknowledge the Ministerio de Economía y Competitividad, GV and FEDER (CTQ2012/31762 and PROMETEOII/2014/010) for the financial support. Miriam Navlani-García thanks the University of Alicante for the PhD studentship. Martin Martis thanks the Japan Society for the Promotion of Science (JSPS) for the fellowship.Notes and references
- J. A. Ortega Méndez, C. R. López, E. Pulido Melián, O. González Díaz, J. M. Doña Rodríguez, D. Fernández Hevia and M. Macías, Appl. Catal., B, 2014, 147, 439 CrossRef PubMed.
- A. Léon, Hydrogen technology in Mobile and Portable Applications, Springer-Verlag, Berlin, Heidelberg, 2008 Search PubMed.
- M. Jordá-Beneyto, F. Suárez-García, D. Lozano-Castelló, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2007, 45, 293 CrossRef PubMed.
- V. Tozzini and V. Pellegrini, Phys. Chem. Chem. Phys., 2013, 15, 80 RSC.
- B. Panella, M. Hirscher and S. Roth, Carbon, 2005, 43, 2209 CrossRef CAS PubMed.
- P. M. Budd, A. Butler, J. Selbie, K. Mahmood, N. B. McKeown, B. Ghanem, K. Msayib, D. Book and A Walton, Phys. Chem. Chem. Phys., 2007, 9, 1802 RSC.
- J. Germain, J. M. J. Frechet and F. Svec, J. Mater. Chem., 2007, 17, 4989 RSC.
- H. W. Langmi, D. Book, A. Walton, S. R. Johnson, M. M. Al-Mamouri, J. D. Speight, P. P. Edward, I. R. Harris and P. A. Anderson, J. Alloys Compd., 2005, 637, 404 Search PubMed.
- D. Bae, H. Park, J. S. Kim, J.-B. Lee, O. Y. Kwon, K.-Y. Kim, M. K. Song and K. T. No, J. Phys. Chem. Solids, 2008, 69, 1152 CrossRef CAS PubMed.
- J. L. C. Rowsell and O. M. Yagh, Angew. Chem., Int. Ed., 2005, 44, 4670 CrossRef CAS PubMed.
- L. Pan, M. B. Sander, X. Huang, J. Li, M. Smith, E. Bittner, B. Bockrath and J. K. Johnson, J. Am. Chem. Soc., 2004, 126, 1308 CrossRef CAS PubMed.
- H. Lee, J.-W. Lee, D. Y. Kim, J. Park, Y.-T. Seo, H. Zeng, I. L. Moudrakovski, C. I. Ratcliffe and J. A. Ripmeester, Nature, 2005, 434, 743 CrossRef CAS PubMed.
- R. Anderson, A. Chapoy and B. Tohidi, Langmuir, 2007, 23, 3440 CrossRef CAS PubMed.
- F. Cuevas, D. Korablov and M. Latroche, Phys. Chem. Chem. Phys., 2012, 14, 1200 RSC.
- B. Bogdanovic, A. Reiser, K. Schlichte, B. Spliethoff and B. Tesche, J. Alloys Compd., 2002, 345, 77 CrossRef CAS.
- S. K. Singh, Z.-H. Lu and Q. Xu, Eur. J. Inorg. Chem., 2011, 2232 CrossRef CAS.
- M. Nielsen, E. Alberico, W. Baumann, H.-J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85 CrossRef CAS PubMed.
- A. F. Dalebrook, W. Gan, M. Grasemann, S. Moret and G. Laurenczy, Chem. Commun., 2013, 49, 8735 RSC.
- K. Tedsree, T. Li, S. Jones, C. W. A. Chan, K. M. K. Yu, P. A. J. Bagot, E. A. Marquis, G. D. W. Smith and S. C. E. Tsang, Nat. Nanotechnol., 2011, 6, 302 CrossRef CAS PubMed.
- R. Farrauto, S. Hwang, L. Shore, W. Ruettinger, J. Lampert, T. Giroux, Y. Liu and O. Ilinich, Annu. Rev. Mater. Res., 2003, 33, 1 CrossRef CAS.
- K. Mori, M. Dojo and H. Yamashita, ACS Catal., 2013, 3, 1114 CrossRef CAS.
- M. Martis, K. Mori, K. Fujiwara, W.-S. Ahn and H. Yamashita, J. Phys. Chem C, 2013, 117, 22805 CAS.
- J. Park, C. Aliaga, J. R. Renzas, H. Lee and G. Somorjai, Catal. Lett., 2009, 1, 129 Search PubMed.
- M. G. Weir, M. R. Knecht, A. I. Frenkel and R. M. Crooks, Langmuir, 2010, 26, 1137 CrossRef CAS PubMed.
- L. Adak, K. Chattopadhyay and B. C. Ranu, J. Org. Chem., 2009, 74, 3982 Search PubMed.
- K. V. Kovtunov, V. V. Zhivonitko, L. Kiwi-Minsker and I. V. Koptyug, Chem. Commun., 2010, 46, 5764 RSC.
- S. Niembro, A. Shafir, A. Vallribera and R. Alibés, Org. Lett., 2008, 10, 3215 CrossRef CAS PubMed.
- X. Jiang, G. Wei, X. Zhang, W. Zhang, P. Zheng, F. Wen and L. Shi, J. Mol. Catal. A: Chem., 2007, 277, 102 CrossRef CAS PubMed.
- R. Contreras-Cáceres, J. Pacifico, I. Pastoriza-Santos, J. Pérez-Juste, A. Fernández-Barbero and L. M. Liz-Marzán, Adv. Funct. Mater., 2009, 19, 3070 CrossRef.
- P. M. Uberman, L. A. Pérez, G. I. Lacconi and S. E. Martín, J. Mol. Catal. A: Chem., 2012, 245, 363 Search PubMed.
- I. Miguel-García, Á. Berenguer-Murcia and D. Cazorla-Amorós, Appl. Catal., B, 2010, 98, 161 CrossRef PubMed.
- Y. Ping, J.-M. Yan, Z.-L. Wang, H.-L. Wang and Q. Jiang, J. Mater. Chem. A, 2013, 1, 12188 CAS.
- P. Sponholz, D. Mellmann, H. Junge and M. Beller, ChemSusChem, 2013, 6, 1172 CrossRef CAS PubMed.
- J. Čejka, G. Centi, J. Perez-Pariente and W. J. Roth, Catal. Today, 2012, 179, 2 CrossRef PubMed.
- http://www.iza-online.org/ .
- J. M. López, M. V. Navarro, T. García, R. Murillo, A. M. Mastral, F. J. Varela-Gandía, D. Lozano-Castelló, A. Bueno-López and D. Cazorla-Amorós, Microporous Mesoporous Mater., 2010, 130, 239 CrossRef PubMed.
- S. Radhakrishnan, G. Thoelen, J. Franken, J. Degreve, C. E. A. Kirschhock and J. A. Martens, ChemCatChem, 2013, 5, 576 CrossRef CAS.
- J. Jae, E. Mahmoud, R. F. Lobo and D. G. Vlachos, ChemCatChem, 2014, 6, 508 CrossRef CAS.
- S. Domínguez-Domínguez, Á. Berenguer-Murcia, D. Cazorla-Amorós and Á. Linares-Solano, J. Catal., 2006, 243, 74 CrossRef PubMed.
- P. Lu, T. Teranishi, K. Asakura, M. Miyake and N. Toshima, J. Phys. Chem. B, 1999, 103, 9673 CrossRef CAS.
- M. Boudart, Kinetics of Heterogeneous Catalytic Reactions, Princeton University Press, Princeton, NJ, 1984 Search PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- J. M. Newsam, M. M. J. Trecy, W. T. Koetsier and C. B. de Gruyter, Proc. R. Soc. London, Ser. A, 1988, 420, 375 CrossRef CAS.
- A. Corma, M. Moliner, Á. Cantín, M. J. Díaz-Cabañas, J. L. Jordá, D. Zhang, J. Sun, K. Jansson, S. Hovmöller and X. Zou, Chem. Mater., 2008, 20, 3218 CrossRef CAS.
- I. Miguel-García, Á. Berenguer-Murcia, T. García and D. Cazorla-Amorós, Catal. Today, 2012, 187, 2 CrossRef PubMed.
- G. Rubasinghege, S. Ogden, J. Baltrusaitis and V. H. Grassian, J. Phys. Chem. A, 2013, 117, 11316 CrossRef CAS PubMed.
- M. Navlani-García, B. Puértolas, D. Lozano-Castelló, D. Cazorla-Amorós, M. V. Navarro and T. García, Environ. Sci. Technol., 2013, 47, 5851 CrossRef PubMed.
- S. Bordiga, B. Civalleri, G. Spoto, C. Pazé, C. Lamberti, P. Ugliengo, A. Zecchina and S. Bordiga, J. Chem. Soc., Faraday Trans., 1997, 93(21), 3893 RSC.
- A. Mattsson, S. Hu, K. Hermansson and L. Österlund, J. Chem. Phys., 2014, 140, 034705 CrossRef PubMed.
- C. Iuga, C. I. Sainz-Díaz and A. Vivier-Bunge, J. Phys. Chem. C, 2012, 116, 2904 CAS.
- X. Zhou, Y. Huang, W. Xing, C. Liu, J. Liao and T. Lu, Chem. Commun., 2008, 3540 RSC.
| This journal is © The Royal Society of Chemistry 2015 |