
Homogeneous catalytic oxidation of styrene and styrene derivatives with hydrogen peroxide in the presence of transition metal-substituted polyoxotungstates
Tiago A. G.
Duarte
a,
Ana C.
Estrada
b,
Mário M. Q.
Simões
*a,
Isabel C. M. S.
Santos
*a,
Ana M. V.
Cavaleiro
b,
M. Graça P. M. S.
Neves
a and
José A. S.
Cavaleiro
a
aQOPNA, Departamento de Química, Universidade de Aveiro, 3810-193 Aveiro, Portugal. E-mail: ivieira@ua.pt; msimoes@ua.pt; Fax: +351 234 370 084; Tel: +351 234 370 200 Tel: +351 234 370 713
bCICECO, Departamento de Química, Universidade de Aveiro, 3810-193 Aveiro, Portugal
First published on 22nd August 2014
Abstract
The tetrabutylammonium (TBA) salts of the Keggin-type polyoxotungstates with general formula [XW11M(H2O)O39](n−m)−, where X = P, B or Si and M = Mn, Fe or Co, were evaluated as catalysts in the oxidation of styrene, α-methylstyrene, p-methylstyrene, α,p-dimethylstyrene, p-chlorostyrene, p-nitrostyrene, and p-methoxystyrene under mild conditions, using aqueous H2O2 as an eco-sustainable oxidant. In this study, the influence of the catalysts and of the different styrene substituents on the oxidation reaction profile was evaluated in terms of conversion and selectivity. For all the performed catalytic studies, the main product results from the oxidative cleavage of the vinyl double bond, except in the case of the oxidation of p-methoxystyrene catalysed by BW11Mn, for which p-methoxyphenol is the main product. The catalysts BW11Mn and SiW11Co give rise to 100% conversion for almost all of the substrates, excluding p-methoxystyrene and p-nitrostyrene for both catalysts and α,p-dimethylstyrene only in the case of BW11Mn. The selectivity for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C cleavage products resulting from the oxidative cleavage of the vinyl double bond can be as high as 98%, reaching 98% conversion for p-nitrostyrene when SiW11Co was used as a catalyst. Possible pathways are discussed and the oxidation of a few presumed intermediates was carried out. The systematic study of several substituted styrene derivatives suggests a possible reactivity order for these compounds in the catalytic system considered.
C cleavage products resulting from the oxidative cleavage of the vinyl double bond can be as high as 98%, reaching 98% conversion for p-nitrostyrene when SiW11Co was used as a catalyst. Possible pathways are discussed and the oxidation of a few presumed intermediates was carried out. The systematic study of several substituted styrene derivatives suggests a possible reactivity order for these compounds in the catalytic system considered.
1. Introduction
The oxidation of alkenes is an important transformation in the production of fine chemicals. One of the common routes involves a strong oxidant in excess (e.g. peroxyacids, manganese dioxide, chromic acid, potassium dichromate or selenium dioxide), producing large amounts of undesired and/or toxic wastes, most significantly when applied to industrial processes.1,2 The strategies to overcome these drawbacks have led to the use of environmentally benign oxidants in line with the development of appropriate catalysts.2–6It is known that the metal-catalysed oxidation of alkenes can give rise to a whole variety of organic products, namely, epoxides and diols. In addition, depending on the catalyst, alkenes can be converted into aldehydes and/or ketones by oxidative cleavage of the CC double bond, similarly to what happens in ozonolysis.7,8 The particular case of styrene oxidation is of considerable interest for academia research and also for industry due to the important products that can be obtained such as styrene oxide, benzaldehyde or phenylacetaldehyde.9–12
Several homogeneous or heterogeneous systems have been reported for the oxidation of styrene and its derivatives in the presence of different types of polyoxometalates (POMs) using mainly hydrogen peroxide (H2O2),13–33tert-butylhydroperoxide (TBHP),34–37 and molecular oxygen (O2),26,38–40 among other oxidants.34,35,41 The major products usually obtained from styrene in the presence of the reported POMs are styrene oxide and phenylacetaldehyde (apparently resulting from the isomerization of the epoxidation product), along with benzaldehyde and benzoic acid.19,20,22,25,27,30–33,37
Among the several types of POMs, the transition metal monosubstituted Keggin-type polyoxotungstates [XW11M(H2O)O39](n−m)− (X = P, Si, etc.), in which a transition metal cation, Mm+, is coordinated to the binding sites of a lacunary polyoxotungstate anion [XW11O39]n−, are an extraordinarily versatile class of complexes with high catalytic activity in a variety of organic reactions. Moreover, these POMs are able to activate hydrogen peroxide, turning it reactive enough to promote important reactions such as hydroxylation, epoxidation, oxidative dehydrogenation and oxidative cleavage processes, as widely demonstrated in our earlier work.27,32,42–52 The use of aqueous H2O2 in the oxidation of organic substrates is very attractive from the point of view of synthetic organic chemistry, since aqueous H2O2 is an environmentally clean and easy to handle reagent.3,53–55 To our knowledge, studies on the oxidation of styrene and its derivatives using aqueous H2O2 in the presence of transition metal-substituted Keggin-, Dawson-, or sandwich-type polyoxotungstates are still rather limited in number.13,23,25,27,31,33 Following our interest in the use of polyoxotungstates as catalysts for the oxidative transformations of organic compounds,27,32,42–52 we report here the oxidation of styrene and of some derivatives (Scheme 1) with H2O2, in acetonitrile, in the presence of tetrabutylammonium (TBA) salts of [XW11MIII(H2O)O39](n−m)−, X = P, Si, B and M = MnIII, FeIII or CoII (abbreviated as XW11M). The oxidation of some of the reaction products and/or possible intermediates was also studied in order to define the possible reaction routes.
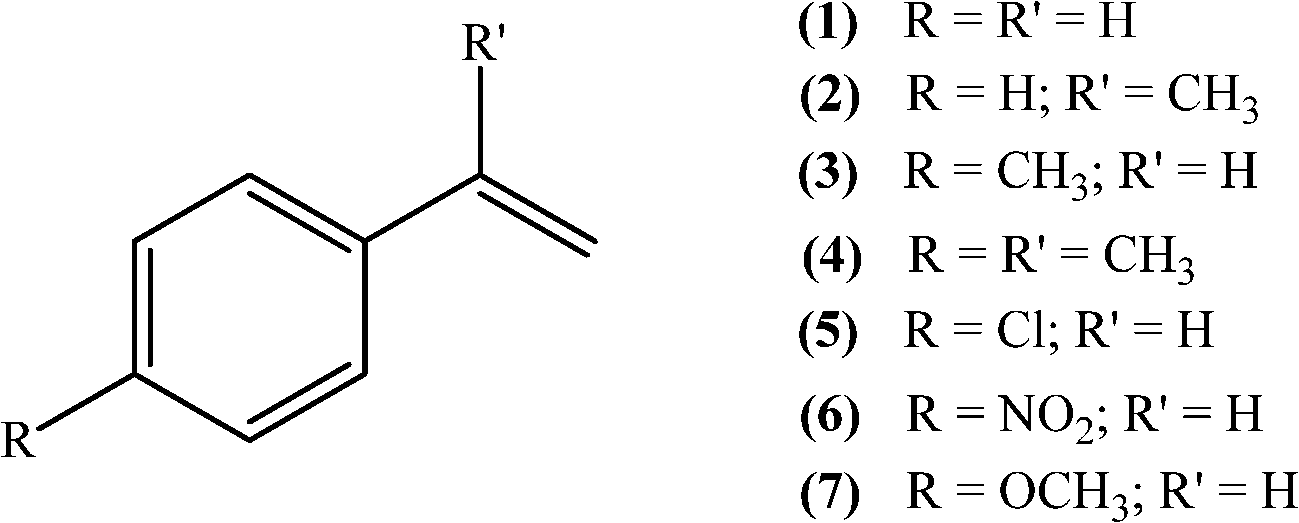 | ||
| Scheme 1 Substrates studied in the oxidation with H2O2 in the presence of transition metal-substituted polyoxotungstates. | ||
2. Experimental section
2.1. Reagents and synthetic procedures
Acetonitrile, 30% (w/w) aqueous hydrogen peroxide, styrene, α-methylstyrene, p-methylstyrene, α,p-dimethylstyrene, p-chlorostyrene, p-nitrostyrene, p-methoxystyrene, styrene oxide and all other reagents and solvents, obtained from commercial sources, were used as received or distilled and dried using standard procedures. 1-Phenylethane-1,2-diol was obtained by acidification (6 M HCl) of styrene oxide followed by liquid–liquid extraction with dichloromethane.The tetrabutylammonium (TBA) salts TBA4Hn−m−4[XW11M(H2O)O39]·xH2O, with X = B, P or Si, M = MnIII, FeIII or CoII, n−m = charge of the anion, and x = 0–4, were prepared and characterized according to previously described procedures.42,56–59 The obtained compounds were characterized by elemental analysis (C, H, N, W, Fe, Mn, P, Si), thermogravimetry and infrared spectroscopy, and the results were in agreement with previously published values.42,58–60
2.2. Typical procedure for the oxidation reactions
Based on the experimental conditions developed in previous studies,27,32 the reactions were typically carried out as follows: the substrate (1.0 mmol), the catalyst (3 μmol), corresponding to a substrate/catalyst (S/C) molar ratio of 333, 30% (w/w) aqueous H2O2 (408 μl, 4 mmol) and 3.0 mL of acetonitrile were stirred in a closed vessel at 80 °C for 6 h. The conversion values presented (SD ≤10) are the result of at least two concordant assays. Blank reactions were performed for all the substrates, confirming that no oxidation or almost no oxidation occurs unless both the catalyst and H2O2 are present.2.3. Instruments and methods
The GC-FID analyses were performed on a Varian Star 3900 chromatograph equipped with a fused silica capillary DB-5 type column (30 m × 0.25 mm i.d.; 0.25 μm film thickness) using helium as the carrier gas (30 cm s−1). The chromatographic conditions for all the substrates and their derivatives were as follows: initial temperature 90 °C; first temperature rate 5 °C min−1 until 140 °C; second temperature rate 50 °C min−1 until 250 °C; injector temperature 250 °C; detector temperature 270 °C. Aliquots were taken from the reaction mixtures, at regular intervals, using a 10 μl syringe. All the reaction products were identified by GC/MS analysis and by co-injection of commercially available standards. GC/MS was performed using a Finnigan Trace GC/MS (ThermoQuest CE Instruments) using helium as the carrier gas (35 cm s−1) and a fused silica capillary DB-5 type column (30 m × 0.25 mm i.d.; 0.25 μm film thickness). The percentages of each compound in the reaction mixtures were estimated from the corresponding chromatographic peak areas, using cyclohexylmethanol as the internal standard (added after the reaction).3. Results and discussion
The oxidation of styrene (1), α-methylstyrene (2), p-methylstyrene (3), α,p-dimethylstyrene (4), p-chlorostyrene (5), p-nitrostyrene (6), or p-methoxystyrene (7) by hydrogen peroxide was carried out in acetonitrile at 80 °C, in the presence of the TBA salts of [XW11M(H2O)O39](n−m)−, X = P, Si or B; M = FeIII, MnIII or CoII. Except when reaction time is referred, the results discussed here were obtained after 6 h of reaction. Substrate oxidation is negligible or not observed in the absence of a catalyst, indicating that hydrogen peroxide alone is unable to oxidize these substrates.3.1. Oxidation of styrene (1)
The results presented in Table 1 show that styrene (1) in the presence of the selected POMs is, in general, efficiently oxidized with hydrogen peroxide. The catalytic activity based on the percentage of conversion of styrene follows the order: BW11Mn (100% after 5 h) ~ SiW11Co (100%) > BW11Fe (80%) > BW11Co (78%) > PW11Co (60%) > SiW11Fe (37%) > PW11Fe (32%) > SiW11Mn (11%) ~ PW11Mn (10%).| Catalyst | Conv.b,c (%) | Selectivityc (%) | CC cleavage (8 + 9 + 12) |
||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
|||
| a Reaction conditions: substrate (1.0 mmol), catalyst (3 μmol) and 30% (w/w) aqueous H2O2 (4.0 mmol) were stirred in 3.0 mL of CH3CN at 80 °C. b Numbers in brackets are the hours when the reaction reaches 100%, if different from 6 h. c Determined by GC-FID. | |||||||||
| BW11Mn | 100(5) | — | 58 | 2 | 5 | 22 | 7 | 6 | 80 |
| PW11Mn | 10 | — | 100 | — | — | — | — | — | 100 |
| SiW11Mn | 11 | — | 82 | 18 | — | — | — | — | 82 |
| BW11Co | 78 | — | 71 | 6 | 6 | 12 | 5 | — | 83 |
| PW11Co | 60 | — | 74 | 6 | 6 | 7 | 7 | — | 81 |
| SiW11Co | 100 | — | 64 | 6 | 5 | 15 | 6 | 4 | 79 |
| BW11Fe | 80 | 3 | 65 | 2 | 3 | 8 | — | 9 | 76 |
| PW11Fe | 32 | — | 78 | 3 | 4 | 5 | — | 10 | 83 |
| SiW11Fe | 37 | — | 87 | 4 | 9 | — | — | — | 87 |
| No catalyst | 6 | — | 100 | — | — | — | — | — | 100 |
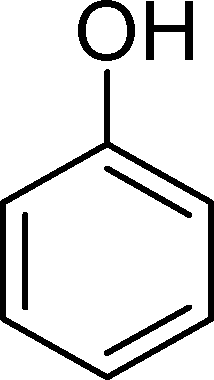
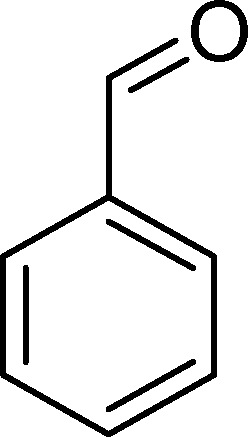
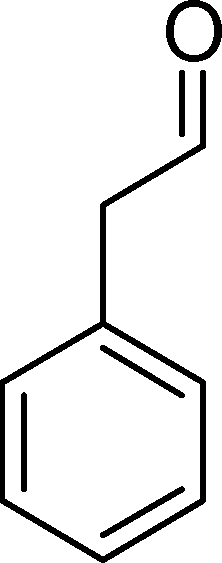
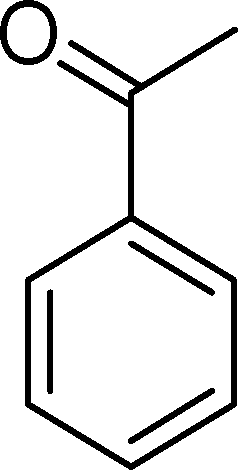
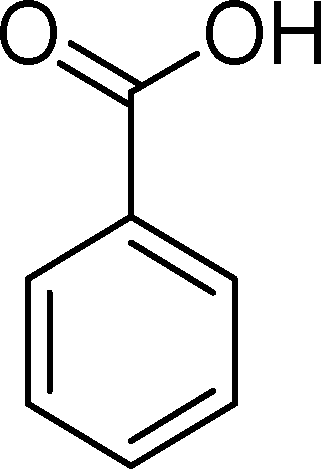
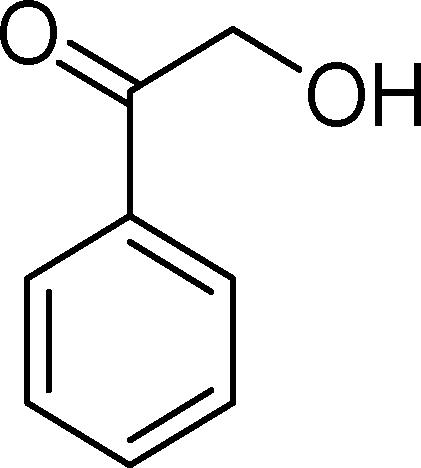
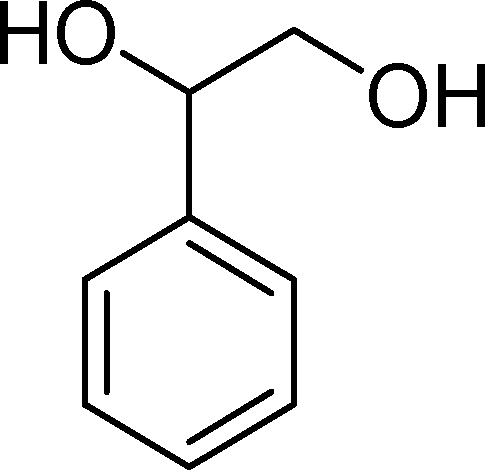
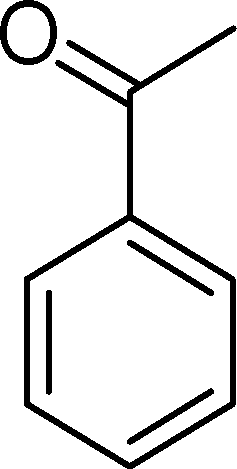
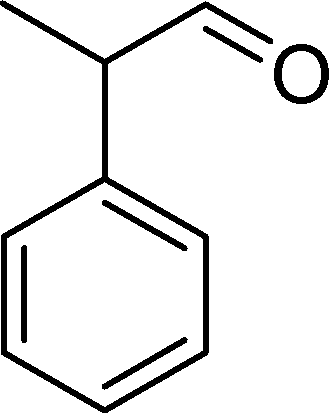
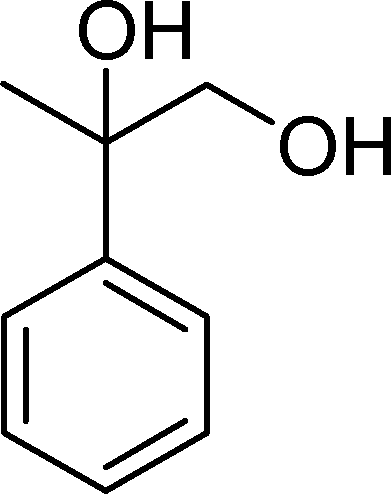
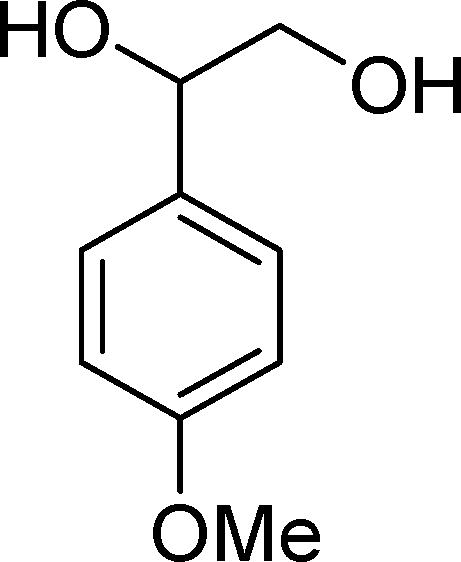
The analysis of the products obtained shows that the oxidation of 1 occurs only at the vinyl double bond and no aromatic functionalization is detected. With all the catalysts, the main product is benzaldehyde (9), reaching selectivities between 58% and 100%. The highest yield for this compound was 64%, obtained with SiW11Co, and all BW11M anions originated yields above 50%. Considering the combined amounts of benzaldehyde (9) and benzoic acid (12) resulting from its oxidation (combined selectivity between 73% and 100%), it is apparent that the principal occurrence is the cleavage of the vinyl carbon–carbon double bond. From the point of view of the mechanism for this oxidation and taking into account also the other products obtained, namely, phenylacetaldehyde (10) (0–18%), acetophenone (11) (0–9%), 2-hydroxy-1-phenylethanone (13) (0–7%) and 1-phenylethane-1,2-diol (14) (0–10%), the corresponding styrene epoxide (15) was considered a putative intermediate, as represented in Scheme 2. In order to clarify this assumption, the oxidation of styrene oxide (15) was carried out in the presence of the most efficient catalysts BW11Mn and SiW11Co under the same conditions used in the oxidation of styrene (1). The results obtained (Table 2) show that the major product of this reaction is the corresponding diol (14) for BW11Mn (47% selectivity and 54% conversion after 6 h) and phenylacetaldehyde (10) for SiW11Co (59% selectivity and 29% conversion after 6 h). Benzaldehyde (9) was obtained with 20% and 8% selectivity in the presence of BW11Mn and SiW11Co, respectively. Considering the amount of diol (14) obtained in these reactions, namely, 47% for BW11Mn and 33% for SiW11Co, its oxidation was carried out under the same conditions in the presence of SiW11Co. After 6 h of reaction (27% conversion, Table 2), the major products detected were hydroxyketone (13) with 51% selectivity and benzaldehyde (9) with 44% selectivity.
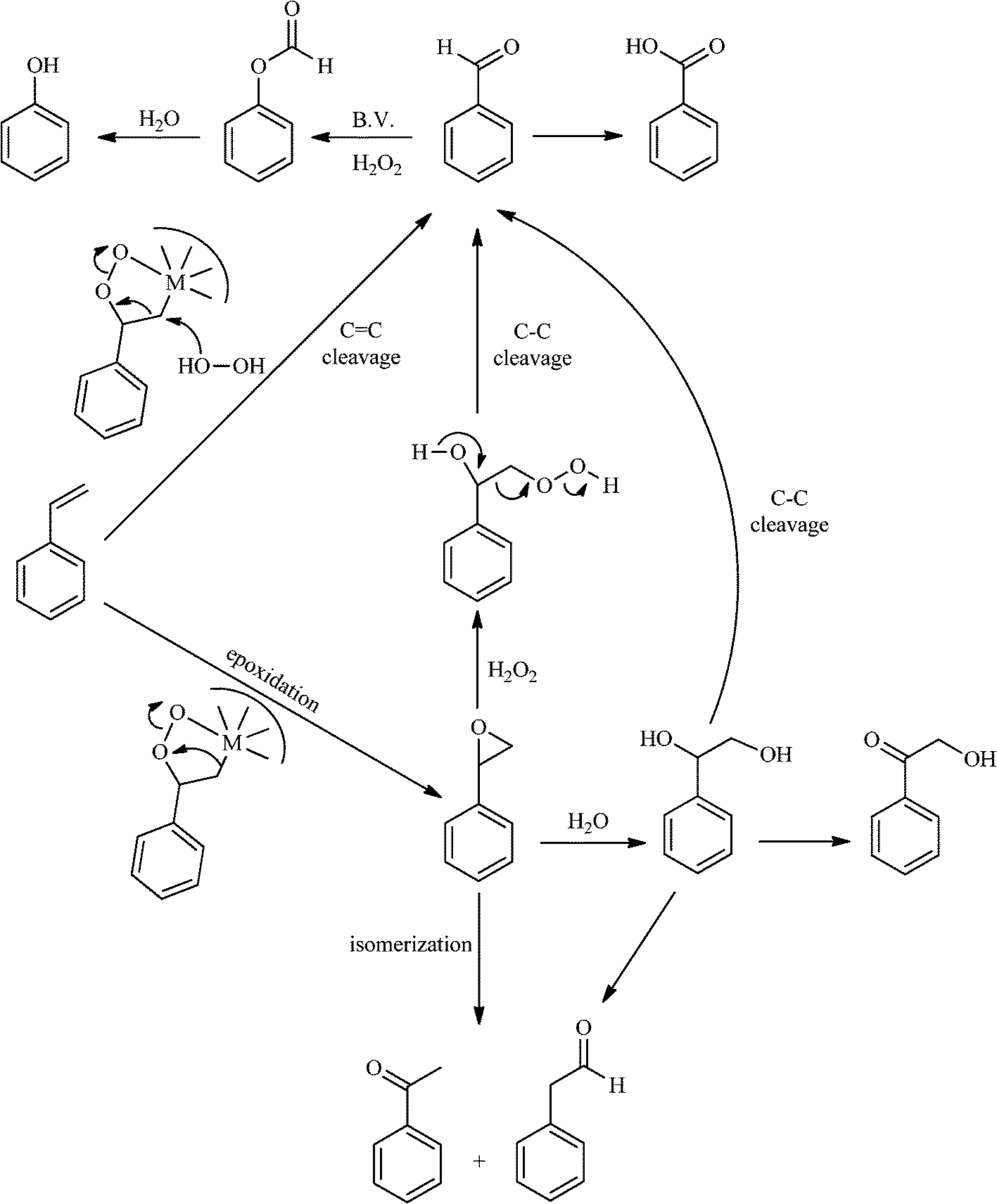 | ||
| Scheme 2 General reaction pathways for the oxidation of styrene (1) with H2O2 catalysed by polyoxometalates (POMs). | ||
| Catalyst | Substrate | Conv.b (%) | Selectivityb (%) | ||||
|---|---|---|---|---|---|---|---|
| a Reaction conditions: substrate (1.0 mmol), catalyst (3 μmol) and 30% (w/w) aqueous H2O2 (4.0 mmol) were stirred in 3.0 mL of CH3CN at 80 °C. b Determined by GC-FID. | |||||||
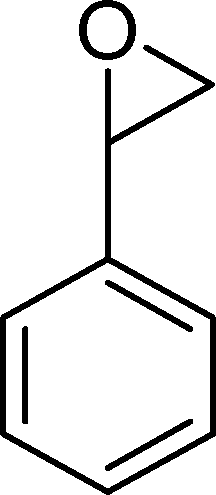 (15) (15) |
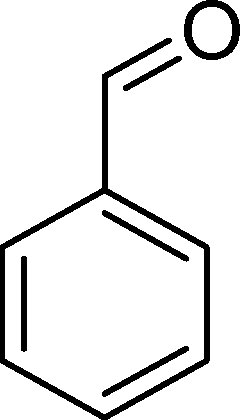 (9) (9) |
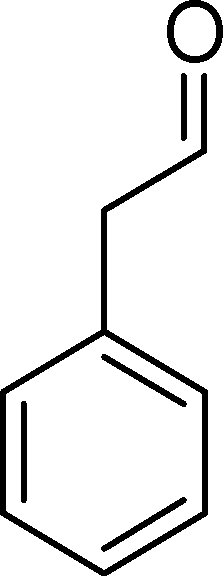 (10) (10) |
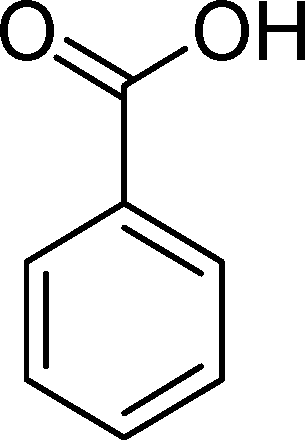 (12) (12) |
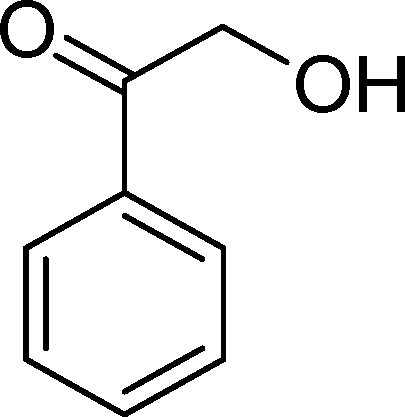 (13) (13) |
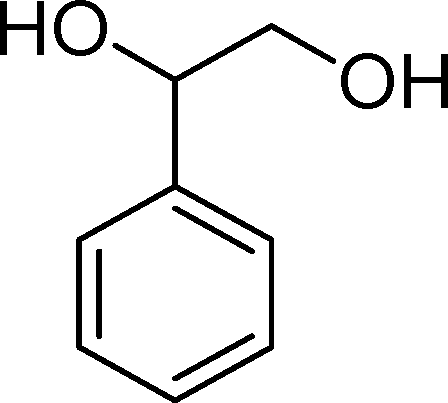 (14) (14) |
||
| BW11Mn | 54 | 20 | 16 | 3 | 14 | 47 | |
| SiW11Co | 29 | 8 | 59 | — | — | 33 | |
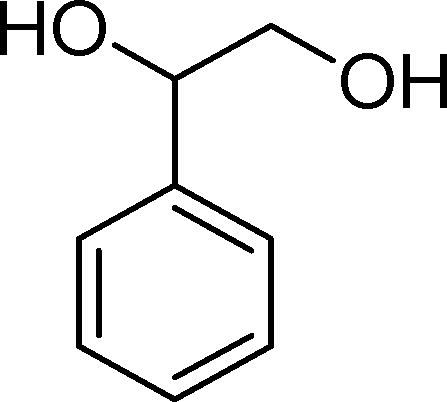 (14) (14) |
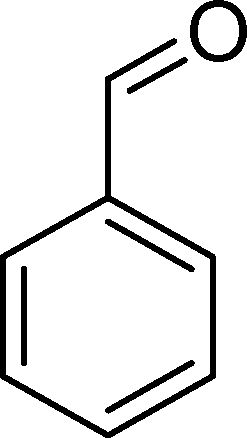 (9) (9) |
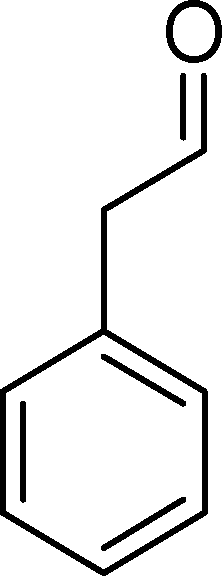 (10) (10) |
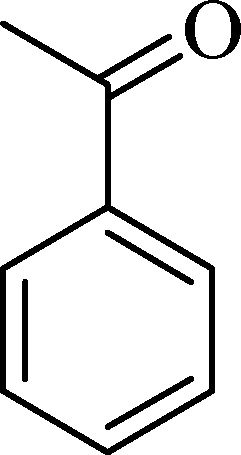 (11) (11) |
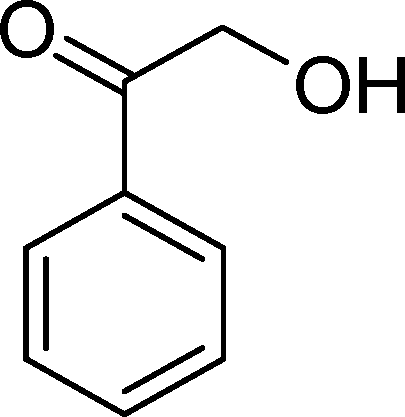 (13) (13) |
|||
| SiW11Co | 27 | 44 | 1 | 4 | 51 | ||
From this study, it can be concluded that styrene oxide (15) (although not detected in the oxidation of styrene reported here) and diol (14) are potential intermediates in the formation of benzaldehyde (Scheme 2). The oxidative cleavage of 1,2-diols is well known from the literature, namely, in the presence of hydrogen peroxide and polyoxometalates.61–63 Benzaldehyde can be formed by C–C cleavage of the diol (after hydrolysis of the epoxide) and by epoxide ring opening by direct action of H2O2, which results in the formation of methanal and benzaldehyde (9). Methanal (formaldehyde) was detected both in the reactor gas phase and in the solution by using GC-FID. In fact, it was possible to detect methanal in solution after cooling down the reaction mixture at −20 °C. Moreover, since the reactions were performed in closed vessels, it was also possible to analyse directly the headspace of the reactor and detect a small amount of methanal by GC. The first step leading to the formation of the epoxide is usually described in the literature23,64,65 as the formation of an intermediate peroxide or hydroperoxide complex with the catalyst, in which H2O2 is coordinated to a metal centre of the POM. The oxygen transfer from the bonded peroxide to styrene may occur by different mechanisms,65,66 namely, through the formation of a 5-membered cyclic intermediate involving a metal centre (M = W, Fe, Mn, Co). We propose that this intermediate could also react with a H2O2 molecule, resulting in the direct formation of benzaldehyde (Scheme 2). This third pathway for the CC cleavage is suggested since, based on the results in Table 2, the epoxide and the diol may not be responsible for all the benzaldehyde formed.
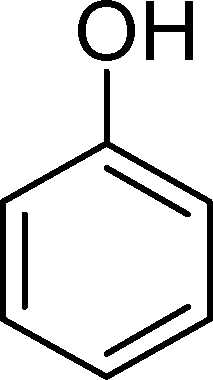
Phenol (8) was also detected in a very small amount and only in the presence of BW11Fe. The formation of phenols is well established in the Baeyer–Villiger (B.V.) oxidation of aromatic aldehydes.67–71 Thus, benzaldehyde can be oxidized to the corresponding formate ester, which may be hydrolyzed to the corresponding phenol (Scheme 2). Considering also the formation of phenol (8) as resulting from benzaldehyde (9) Baeyer–Villiger oxidation, the selectivity for the CC cleavage rises to 76% in the case of BW11Fe.
The results obtained in the oxidation of styrene with H2O2 in the presence of tert-butanol, a hydroxyl (HO˙) radical scavenger, indicate that the reactions do not involve radicals. In fact, the reaction profiles with tert-butanol are exactly the same as without tert-butanol, after 6 h, maintaining all other reaction conditions.
The reports found in the literature on the oxidation of styrene with H2O2 in the presence of some of the POMs used in this work23,27,33 or related metal-substituted anions24,25,31,32 indicate different results, depending on the conditions used. Cobalt was used in some studies23,25,31,33 and the results obtained here with SiW11Co at 6 h compare well with others previously described.33 Other catalysts seem to yield interesting results, namely, the borotungstates, which, to our best knowledge, have not been considered in any previous study.
3.2. Oxidation of α-methylstyrene (2), p-methylstyrene (3), α,p-dimethylstyrene (4), p-chlorostyrene (5), p-nitrostyrene (6), and p-methoxystyrene (7)
The results obtained in the oxidation of the substituted styrenes 2 to 7 with H2O2, in the presence of the monosubstituted polyoxotungstates, under the same conditions described for styrene, are shown in Tables 3–8, respectively. Generally, the oxidation of the styrene derivatives seems to follow a course similar to that of styrene, as depicted in Scheme 2. Considering the data in Tables 3–8, the analysis of the products obtained from each substrate shows some regularity, as described next.| Catalyst | Conv.b,c (%) | Selectivityc (%) | CC cleavage (8 + 11) |
|||
|---|---|---|---|---|---|---|
|
|
|
|
|
|||
| a Reaction conditions: substrate (1.0 mmol), catalyst (3 μmol) and 30% (w/w) aqueous H2O2 (4.0 mmol) were stirred in 3.0 mL of CH3CN at 80 °C. b Numbers in brackets are the hours when the reaction reaches 100%, if different from 6 h. c Determined by GC-FID. d Reaction runs with 2.0 mmol of H2O2. e Reaction runs at 60 °C. | ||||||
| BW11Mn | 100(5) | 2 | 64 | 6 | 27 | 66 |
| BW11Mn | 76d | 1 | 63 | 5 | 30 | 64 |
| BW11Mn | 78e | 5 | 57 | 10 | 26 | 62 |
| PW11Mn | 23 | — | 84 | 16 | — | 84 |
| SiW11Mn | 22 | — | 100 | — | — | 100 |
| BW11Co | 87 | 4 | 81 | 10 | 5 | 85 |
| PW11Co | 80 | — | 87 | 8 | 5 | 87 |
| SiW11Co | 100(3) | 1 | 84 | 9 | 6 | 85 |
| BW11Fe | 65 | 4 | 81 | 7 | 8 | 85 |
| PW11Fe | 30 | — | 88 | 2 | 10 | 88 |
| SiW11Fe | 69 | — | 88 | 12 | — | 88 |
| No catalyst | 8 | — | 100 | — | — | 100 |
| Catalyst | Conv.b,c (%) | Selectivityc (%) | CC cleavage (18 + 21) |
|||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|||
| a Reaction conditions: substrate (1.0 mmol), catalyst (3 μmol) and 30% (w/w) aqueous H2O2 (4.0 mmol) were stirred in 3.0 mL of CH3CN at 80 °C. b Numbers in brackets are the hours when the reaction reaches 100%, if different from 6 h. c Determined by GC-FID. | ||||||||
| BW11Mn | 100(3) | 59 | 7 | 5 | 19 | 10 | — | 78 |
| PW11Mn | 11 | 100 | — | — | — | — | — | 100 |
| SiW11Mn | 4 | 100 | — | — | — | — | — | 100 |
| BW11Co | 76 | 72 | 8 | 2 | 12 | 6 | — | 84 |
| PW11Co | 74 | 66 | 13 | — | 10 | 8 | 3 | 76 |
| SiW11Co | 100(2) | 65 | 4 | 4 | 18 | 9 | — | 83 |
| BW11Fe | 100 | 59 | 10 | 4 | 19 | 8 | — | 78 |
| PW11Fe | 51 | 65 | 15 | 5 | 6 | 9 | — | 71 |
| SiW11Fe | 59 | 73 | 14 | 2 | 6 | 5 | — | 79 |
| No catalyst | 5 | 100 | — | — | — | — | — | 100 |
| Catalyst | Conv.b,c (%) | Selectivityc (%) | CC cleavage (20 + 24) |
|||
|---|---|---|---|---|---|---|
|
|
|
|
|
|||
| a Reaction conditions: substrate (1.0 mmol), catalyst (3 μmol) and 30% (w/w) aqueous H2O2 (4.0 mmol) were stirred in 3.0 mL of CH3CN at 80 °C. b Numbers in brackets are the hours when the reaction reaches 100%, if different from 6 h. c Determined by GC-FID. | ||||||
| BW11Mn | 96 | 17 | 51 | 18 | 5 | 68 |
| PW11Mn | 60 | 3 | 86 | 11 | — | 89 |
| SiW11Mn | 25 | — | 90 | 9 | 1 | 90 |
| BW11Co | 76 | 11 | 63 | 17 | 4 | 74 |
| PW11Co | 88 | — | 89 | 8 | 3 | 89 |
| SiW11Co | 100(2) | 7 | 60 | 16 | 10 | 67 |
| BW11Fe | 65 | 5 | 86 | 9 | — | 91 |
| PW11Fe | 28 | — | 80 | 20 | — | 80 |
| SiW11Fe | 80 | 7 | 76 | 12 | 2 | 83 |
| No catalyst | 4 | — | 100 | — | — | 100 |
| Catalyst | Conv.b,c (%) | Selectivityc (%) | CC cleavage (27 + 30) |
|||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|||
| a Reaction conditions: substrate (1.0 mmol), catalyst (3 μmol) and 30% (w/w) aqueous H2O2 (4.0 mmol) were stirred in 3.0 mL of CH3CN at 80 °C. b Numbers in brackets are the hours when the reaction reaches 100%, if different from 6 h. c Determined by GC-FID. | ||||||||
| BW11Mn | 100(4) | 62 | 5 | 4 | 22 | 3 | 4 | 84 |
| PW11Mn | 6 | 100 | — | — | — | — | — | 100 |
| SiW11Mn | 6 | 100 | — | — | — | — | — | 100 |
| BW11Co | 95 | 66 | 6 | 6 | 22 | — | — | 88 |
| PW11Co | 82 | 73 | 4 | 5 | 13 | — | 5 | 86 |
| SiW11Co | 100(5) | 69 | 5 | 8 | 18 | — | — | 87 |
| BW11Fe | 93 | 70 | 6 | 8 | 15 | — | 1 | 85 |
| PW11Fe | 62 | 72 | 7 | 11 | 10 | — | — | 82 |
| SiW11Fe | 56 | 84 | 5 | 5 | 6 | — | — | 90 |
| No catalyst | 4 | 100 | — | — | — | — | — | 100 |
| Catalyst | Conv.b (%) | Selectivityb (%) | CC cleavage (33 + 36) |
|||
|---|---|---|---|---|---|---|
|
|
|
|
|
|||
| a Reaction conditions: substrate (1.0 mmol), catalyst (3 μmol) and 30% (w/w) aqueous H2O2 (4.0 mmol) were stirred in 3.0 mL of CH3CN at 80 °C. b Determined by GC-FID. | ||||||
| BW11Mn | 93 | 54 | 1 | 7 | 38 | 92 |
| PW11Mn | 0 | — | — | — | — | — |
| SiW11Mn | 4 | 100 | — | — | — | 100 |
| BW11Co | 52 | 68 | — | 17 | 15 | 83 |
| PW11Co | 51 | 65 | — | 7 | 28 | 93 |
| SiW11Co | 98 | 68 | — | 2 | 30 | 98 |
| BW11Fe | 86 | 64 | 2 | 5 | 29 | 93 |
| PW11Fe | 57 | 72 | — | 12 | 16 | 88 |
| SiW11Fe | 62 | 74 | — | 6 | 20 | 94 |
| No catalyst | 0 | — | — | — | — | — |
| Catalyst | Conv.b (%) | Selectivityb (%) | CC cleavage (37 + 38 + 41) |
||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
|||
| a Reaction conditions: substrate (1.0 mmol), catalyst (3 μmol) and 30% (w/w) aqueous H2O2 (4.0 mmol) were stirred in 3.0 mL of CH3CN at 80 °C. b Determined by GC-FID. | |||||||||
| BW11Mn | 75 | 34 | 22 | 17 | — | — | — | 16 | 56 |
| PW11Mn | 20 | — | 61 | 20 | — | — | 8 | 11 | 61 |
| SiW11Mn | 12 | — | 78 | 22 | — | — | — | — | 78 |
| BW11Co | 39 | 11 | 62 | 18 | — | 5 | — | 4 | 78 |
| PW11Co | 86 | 10 | 56 | 10 | 5 | 10 | — | 9 | 76 |
| SiW11Co | 95 | 16 | 42 | 24 | 4 | 9 | 2 | 2 | 67 |
| BW11Fe | 78 | — | 57 | 18 | 2 | 6 | — | 17 | 63 |
| PW11Fe | 38 | 18 | 38 | 21 | — | — | — | 18 | 56 |
| SiW11Fe | 56 | 8 | 53 | 16 | 4 | 6 | 3 | 9 | 67 |
| No catalyst | 4 | — | 100 | — | — | — | — | — | 100 |
The highest conversions are obtained in the presence of BW11Mn and SiW11Co, frequently reaching 100%; other POMs also show excellent efficiency (namely, BW11Fe, BW11Co, and PW11Co), depending on the substrate. The major product is either a substituted benzaldehyde or acetophenone (and substituted acetophenone), resulting from the cleavage of the vinyl double bond. The only exception was observed in the oxidation of p-methoxystyrene (7) in the presence of BW11Mn that yielded p-methoxyphenol (37) as the major product (Table 8). Phenols were obtained mainly in the oxidation of 2, 4 and 7. With the best catalysts (BW11Mn, SiW11Co, BW11Fe, BW11Co, and PW11Co), the selectivity for substituted benzaldehyde or acetophenone (and substituted acetophenone) ranges (with a few exceptions) between 60% and 89%. PW11Mn and SiW11Mn were generally poor catalysts, yielding the lowest conversions, with one exception (PW11Mn, with 60% conversion in the oxidation of 4). Yet, the reactions could be highly selective, like in the oxidation of α-methylstyrene (2) (SiW11Mn, 22% conversion, 100% acetophenone, Table 3) or of p-methylstyrene (3) (PW11Mn, 11% conversion, 100% p-methylbenzaldehyde, Table 4).
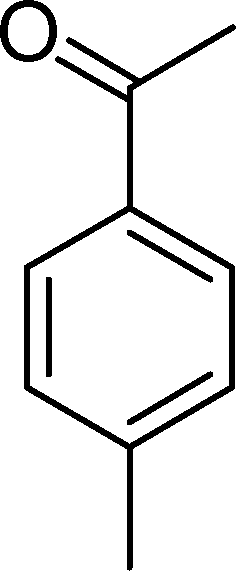
In particular, the oxidation of α-methylstyrene (2) affords acetophenone (11) as the major product with 8 (0–4%), 16 (0–16%) and 17 (0–27%) as minor products (Table 3). SiW11Co and BW11Mn catalyse this oxidation with 100% conversion after 3 and 5 h, respectively. With almost all catalysts, the reaction is very much selective for acetophenone (11) (81–100% selectivity), since few secondary products are formed. This may be due to the existence of the α-methyl group, which may hamper the formation of the hydroxyketone or the isomerisation of the epoxide at the benzylic position. Moreover, unlike benzaldehydes, which may be oxidized to the equivalent benzoic acids, acetophenone is not easily over-oxidized under the present conditions. The best compromise between conversion and selectivity is observed for SiW11Co (100% conversion and 84% selectivity for acetophenone after 3 h of reaction).
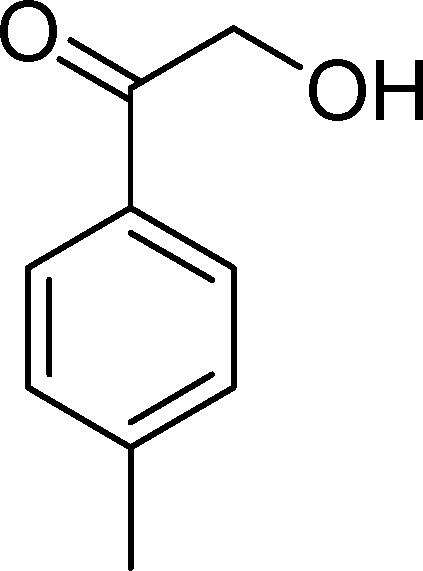
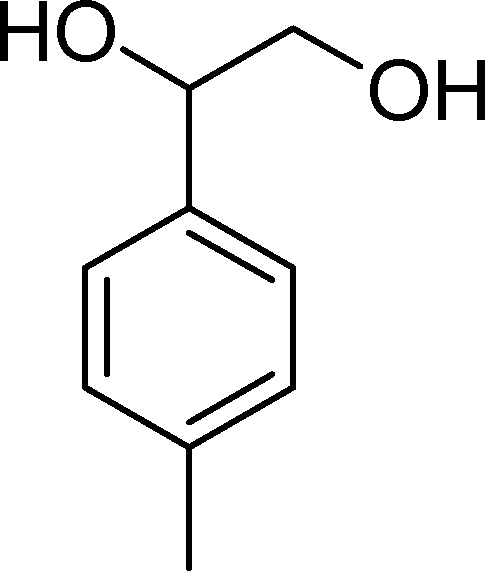
The oxidation of p-methylstyrene (3) yields p-methylbenzaldehyde (18) (the major product, with selectivity above 59%), p-methylbenzoic acid (21) and a few other minor products (Table 4). The best catalysts were SiW11Co (100% conversion and 65% selectivity for 18 after 2 h) and BW11Mn (100% conversion after 3 h). Considering the amount of p-methylbenzaldehyde (18) together with p-methylbenzoic acid (21), the cleavage of the vinyl double bond is responsible for an overall selectivity of 78% (BW11Mn and BW11Fe) and 83% (SiW11Co).
The cleavage of the vinyl double bond in α,p-dimethylstyrene (4) affords p-methylacetophenone (20), with selectivity values between 51% (BW11Mn) and 90% (SiW11Mn), and three other products (Table 5). PW11Co seems to be the most promising catalyst since it gives rise to 88% conversion concomitantly to 89% selectivity for p-methylacetophenone (20).
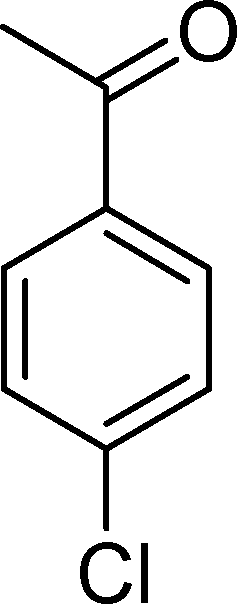
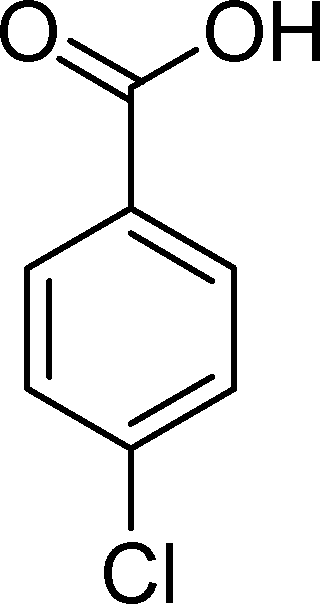
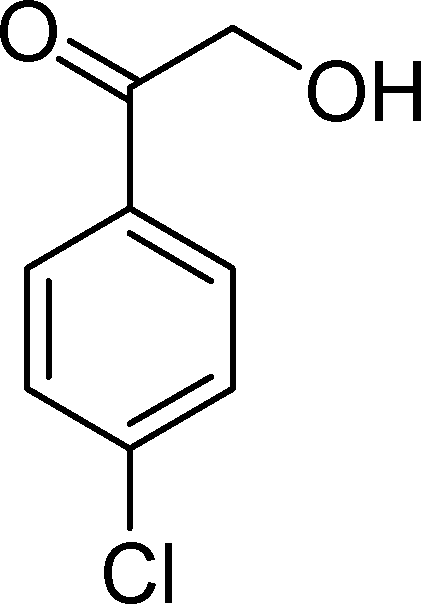
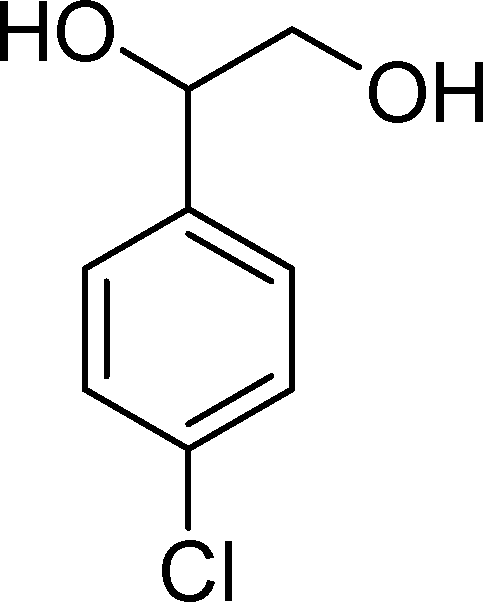
The results obtained in the oxidation of p-chlorostyrene (5) show that the catalysts BW11Mn and SiW11Co are able to totally convert the substrate after 4 and 5 h of reaction, respectively (Table 6). p-Chlorobenzaldehyde (27) was obtained as the major product, together with the corresponding acid 30 (selectivity between 0% and 22%). The amount of secondary products 28–32 is dependent on the catalyst. The products resulting from the CC cleavage represent always more than 80%. In this case, the diol pathway (Scheme 2) is irrelevant, and the isomerization seems to be the major secondary pathway, giving rise to 28 and 29.
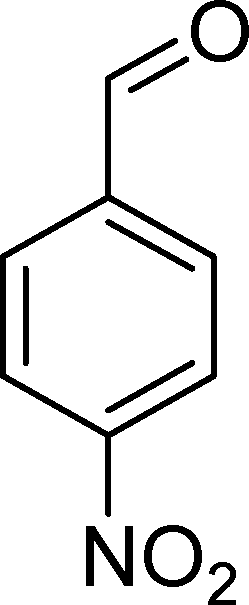
The oxidation of p-nitrostyrene (6) follows a similar profile, affording p-nitrobenzaldehyde (33) as the major product (Table 7). None of the catalysts was able to totally convert this substrate after 6 h of reaction. The selectivity for p-nitrobenzaldehyde (33) varies in the range of 54–74% for the catalysts that show conversions higher than 50%. Generally, the second major product is p-nitrobenzoic acid (36), reaching up to 38% selectivity in the case of BW11Mn and around 30% for PW11Co, SiW11Co, and BW11Fe. In the case of BW11Co, the second major product is p-nitrophenylacetaldehyde (35) with 17% selectivity. The products resulting from CC bond oxidative cleavage (33 plus 36) can reach 98% overall selectivity (SiW11Co), being at least 83% in the case of BW11Co.
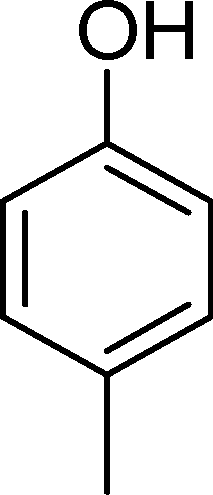
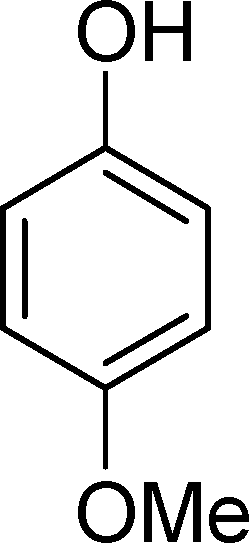
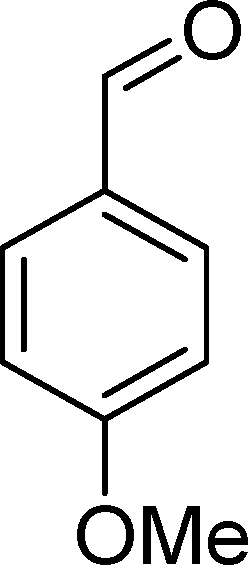
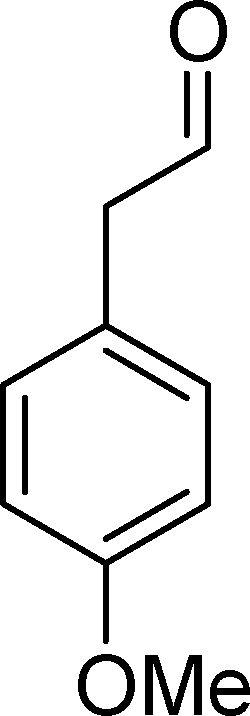
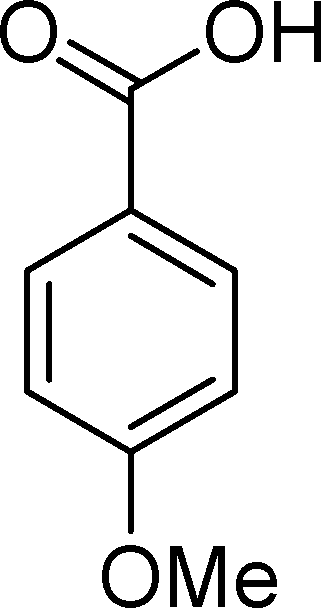
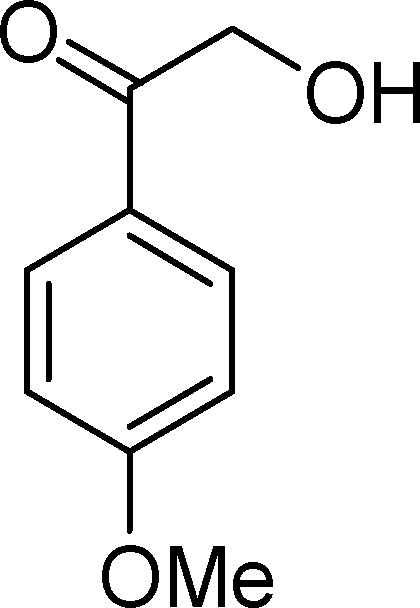
SiW11Co was able to catalyse the oxidization of p-methoxystyrene (7) with a conversion up to 95% after 6 h of reaction and PW11Co, BW11Fe, and BW11Mn give rise to conversions higher than 75% (Table 8). SiW11Co gives only 42% selectivity for the major product, p-methoxybenzaldehyde (38), being better than BW11Mn (22%) and PW11Fe (38%) only. In general, the second major product was p-methoxyphenylacetaldehyde (39), SiW11Co being the most selective for 39 with 24% selectivity. In the case of BW11Mn, p-methoxyphenol (37) was the major product (34% selectivity), unlike for the other catalysts tested. This phenol was also detected in the presence of other POMs but with selectivity varying from 8% to 18%.
The Baeyer–Villiger (B.V.) oxidation of 38 constitutes a very important pathway in the oxidation of 7, since for almost all the catalysts phenol (37) was significant. The formate ester was not detected by GC, but the formation of phenol constitutes evidence of this B.V. pathway from p-methoxybenzaldehyde (38) to p-methoxyphenol (37). The methoxy substituent is probably responsible for this dissimilar behaviour.68
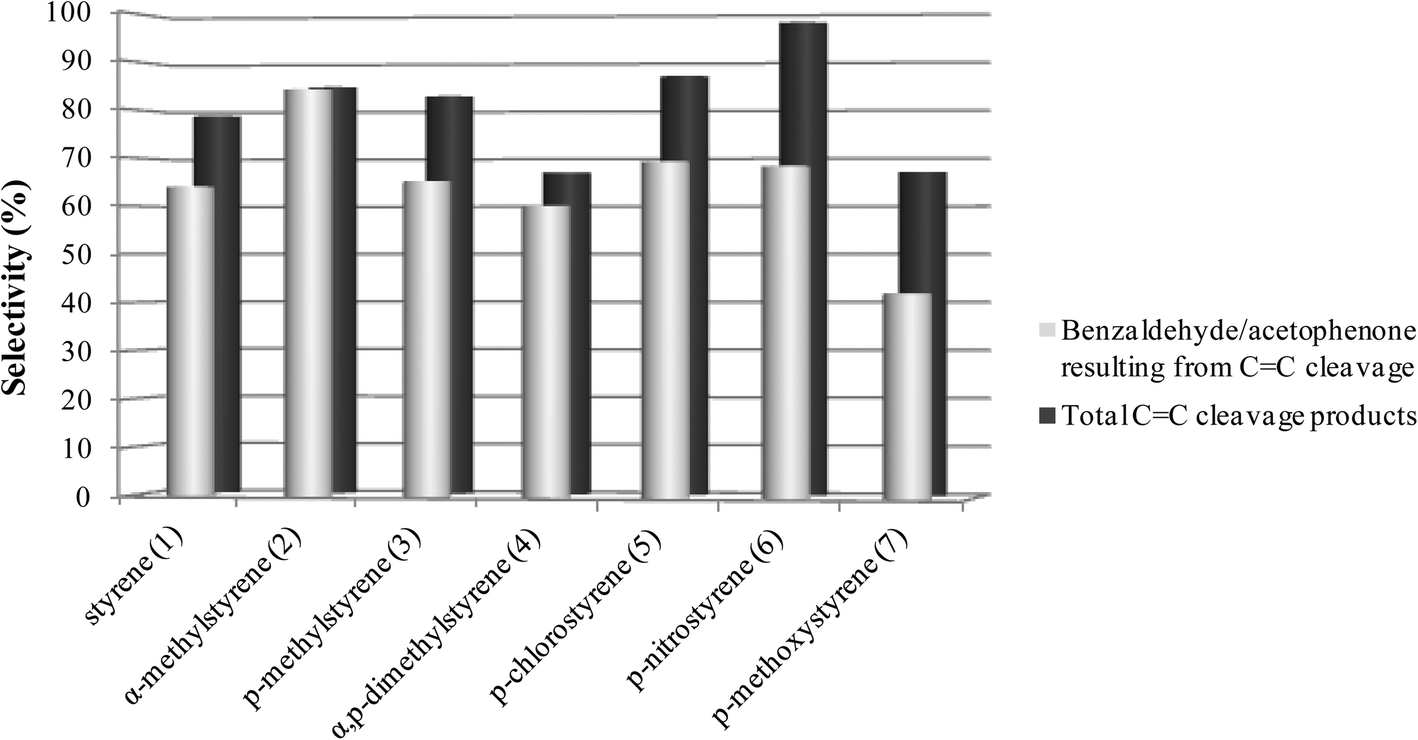
As can be seen in Fig. 1 and 2, for all the substrates studied in the presence of SiW11Co and BW11Mn, the preferential pathway is always the cleavage of the vinyl double bond, with the combined selectivity for the products in the range 56–98%. In the cases where the p-substituent is a chloro or a nitro group, the selectivity for CC cleavage products is higher in comparison with styrene. If the substituent is a methoxy group, the selectivity for the CC cleavage is the lowest. Benzaldehyde, acetophenone and their derivatives account for about 60–99% of the CC cleavage products, depending on the substrates and with only one exception (oxidation of p-methoxystyrene in the presence of BW11Mn). Similar graphs are obtained for other good catalysts (PW11Co, BW11Co, and BW11Fe) and most of these conclusions apply.
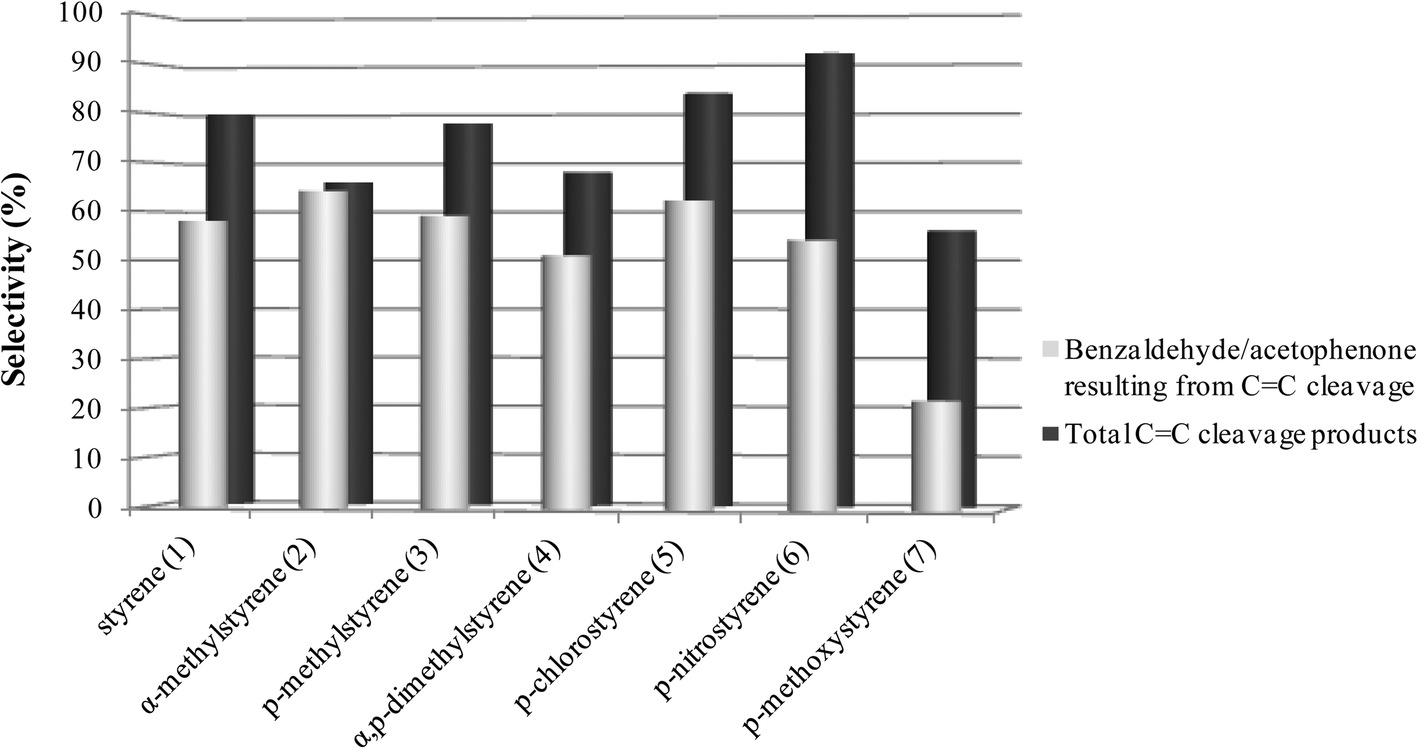 | ||
| Fig. 1 Selectivity for the products resulting from the oxidative cleavage of the vinyl double bond for the reactions catalysed by BW11Mn in the presence of H2O2 at the end of the reactions. | ||
 | ||
| Fig. 2 Selectivity for CC cleavage products resulting from the oxidative cleavage of the vinyl double bond for the reactions catalysed by SiW11Co in the presence of H2O2 at the end of the reactions. | ||
The conversion profile vs. time of reaction for each substrate was evaluated in the presence of BW11Mn and SiW11Co. The comparison of the conversion for different substrates oxidized in the presence of SiW11Co as catalyst is illustrated in Fig. 3. With this catalyst, the reactions are always faster for all the styrenes substituted with one or two methyl groups compared with styrene. This can be explained by the fact that methyl is a weak electron-donating group, which makes the vinyl double bond more reactive. For p-chlorostyrene, the reaction occurs at a similar rate as for styrene. Despite chlorine being an electron-withdrawing group, the reactivities of p-chlorostyrene and styrene are similar. When oxidizing p-methoxy or p-nitro substituted styrenes, the oxidation reactions are slower than those of unsubstituted styrene.
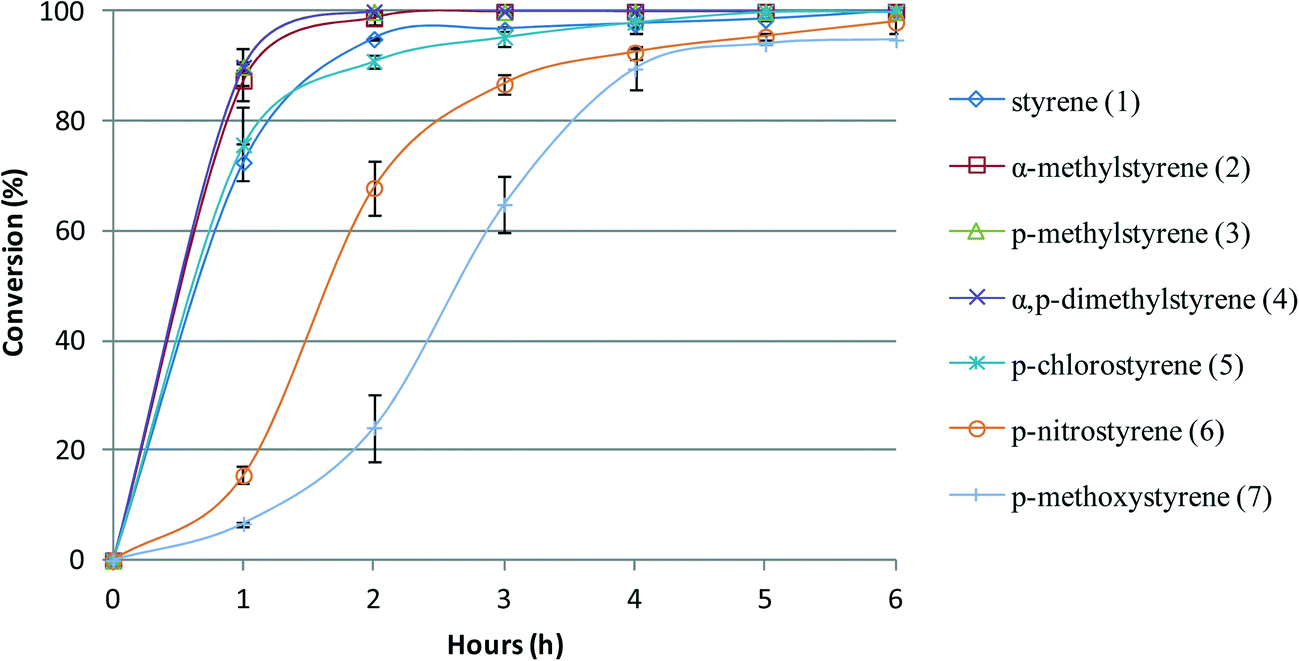 | ||
| Fig. 3 Oxidation reaction conversion profile vs. time of reaction for styrene and styrene derivatives in the presence of SiW11Co, with H2O2 at 80 °C in CH3CN. | ||
The behaviour of p-nitrostyrene is expected in view of the electron-withdrawing character of the nitro group. According to the literature,72 the following order was proposed for the epoxidation reaction of styrenes in the presence of iron(III) porphyrin (FeTPPCl) and PhIO as an oxidant in CH2Cl2 at room temperature: p-OCH3 > p-CH3 > p-H ≅ p-Cl > p-NO2. Despite the differences from the current catalytic conditions, the reactivity pattern in the presence of the selected POMs seems to follow an analogous sequence, excluding p-methoxystyrene. This order is more clearly observed when comparing the values of conversion at 1 or 2 h of reaction, as the values tend to reach 100% at longer reaction times.
The lower reactivity of the vinyl double bond of p-methoxystyrene (7) seems to be atypical. The electron-donor effect of the methoxy group should facilitate the reaction cleavage of the vinyl double bond, contrarily to the observed. This can be explained assuming an interaction of the oxygen of the methoxy group with the POM, either by coordination to a metal or otherwise. In this case, the unshared pairs of electrons from oxygen will not be available for the resonance effect that increases the electronic density at the vinyl group, increasing reactivity. Also, this interaction would possibly block the access to the metal centres needed for catalysis, lowering the reactivity.
The reactions catalysed by BW11Mn are faster in the first hour for all the substrates if compared with styrene itself, except when the substituent is a methoxy group (Fig. 4). The nitro group does not seem to affect the reaction rate during the first hours, but at the end of the reaction, it is clear that the conversion is not complete, as is the case for almost all the other substituents (except for p-methoxystyrene). As expected from the reactions described above, the results show that α-methylstyrene (2) and p-methylstyrene (3) are more reactive than styrene, p-chlorostyrene (5) has no marked difference, and p-nitrostyrene (6) is less reactive. Again, the substrate with the p-methoxy group is the only one with possibly a non-conventional behaviour under the present conditions, being much less reactive than the other substrates. The explanation is possibly the same as for SiW11Co. The results obtained with the two catalysts BW11Mn and SiW11Co may thus be resumed in the following order of substrate reactivity under the conditions of this study: p-CH3, α-CH3, α,p-(CH3)2 > p-H, p-Cl > p-NO2 > p-OCH3.
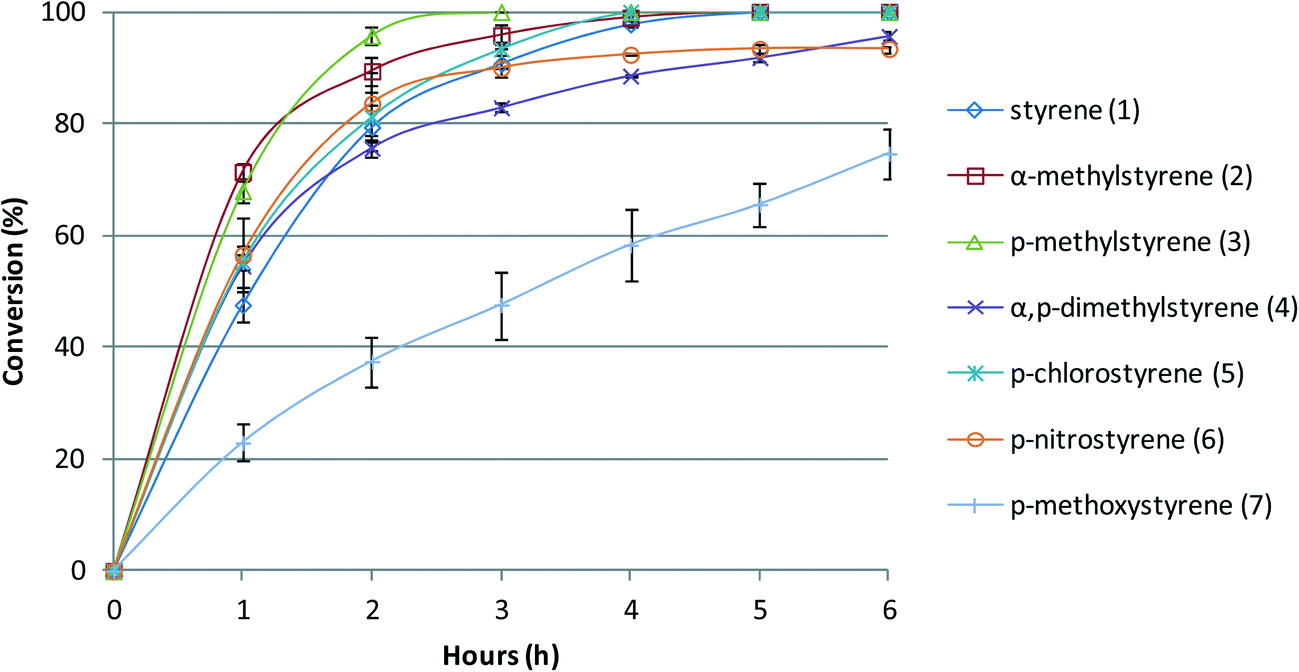 | ||
| Fig. 4 Oxidation reaction conversion profile vs. time of reaction for styrene and styrene derivatives in the presence of BW11Mn, with H2O2 at 80 °C in CH3CN. | ||
As a final remark, we may note that for the more reactive substituted styrenes the time of reaction needed to attain 100% conversion lowers significantly in comparison with styrene. The plots presented in Fig. 3 and 4 illustrate this point clearly.
The conversion of hydrogen peroxide at the end of the styrene oxidation reactions was also evaluated in the presence of all the catalysts and in the absence of a catalyst.73 The comparison is illustrated in Fig. 5. The unproductive dismutation of hydrogen peroxide into water and oxygen seems to be the essential pathway for PW11Mn and SiW11Mn, whereas for BW11Mn and SiW11Co the high conversion of hydrogen peroxide is surpassed by the total conversion of styrene. Opposite to what one might have expected, PW11Fe and SiW11Fe give rise to relatively low percentages of styrene conversion, but the dismutation of hydrogen peroxide seems to be moderate. All the cobalt catalysts associate high to very high conversions of styrene to high conversions of hydrogen peroxide.
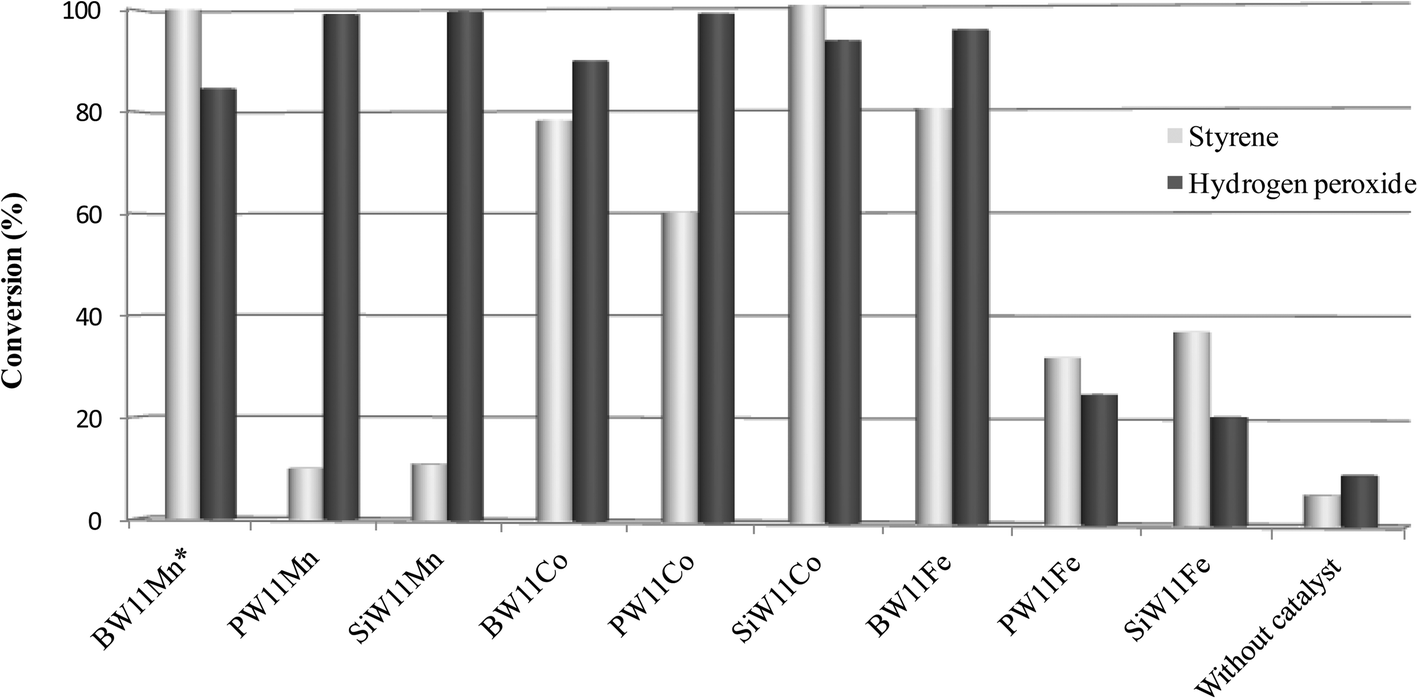 | ||
| Fig. 5 Styrene and hydrogen peroxide conversion after 6 h of reaction (*after 5 h) at 80 °C in CH3CN in the presence of all the catalysts and in the absence of a catalyst. | ||
4. Conclusions
The results presented here reveal a catalytic system based on Mn(III)-, Fe(III)-, or Co(II)-substituted polyoxotungstates for the oxidation of styrene and substituted styrenes in liquid phase, using environmentally benign aqueous H2O2, under very mild conditions. Most of the reactions occurred with high conversion and reasonable selectivity. For all substrates, the major product is almost always the corresponding benzaldehyde or acetophenone, since the oxyfunctionalization involves only the vinyl moiety, and no aromatic ring oxidation occurs. In these oxidation reactions, BW11Mn and SiW11Co are the best catalysts in terms of substrate conversion.The systematic study of several p-substituted styrenes, together with α-methylstyrene, showed that the methyl-substituted styrenes are generally more reactive than styrene. The effect of the styrene substituent influences the reactivity of the vinyl double bond and follows the order p-CH3 > p-H ≅ p-Cl > p-NO2 > p-OCH3. The reactivity found for the p-OCH3 substituted compound was unexpected, since this group is an electron-donating group and an increase in the reactivity of the CC double bond was predictable. A possible explanation is presented.
The products obtained and the interrelations between them are discussed. A schematic mechanism was appointed for styrene, applicable with the necessary adaptations to all other studied substrates. Benzaldehydes are formed by vinyl CC oxidative cleavage and are then oxidized to the corresponding benzoic acids. In a few cases, phenols are formed after Baeyer–Villiger oxidation of the aldehyde. This last B.V. pathway was also registered for the acetophenones resulting from CC oxidative cleavage, since phenols were detected for α-methyl and for α,p-dimethylstyrene. Merging the results of H2O2 conversion (Fig. 5) with the radical scavenger reaction results, the unproductive conversion of H2O2 seems to be mainly dismutation into H2O and O2 and not the H2O2 homolytic cleavage leading to the formation of hydroxyl and hydroperoxyl radicals.
Acknowledgements
We would like to thank Fundação para a Ciência e a Tecnologia (FCT, Portugal), the European Union, QREN, FEDER, and COMPETE for funding the Organic Chemistry Research Unit (QOPNA) (PEst-C/QUI/UI0062/2013) and CICECO (PEst-C/CTM/LA0011/2013).References
- M. Hüdlicky, Oxidations in Organic Chemistry, American Chemical Society, Washington, 1990 Search PubMed.
- Modern Oxidation Methods, ed. J.-E. Bäckvall, Wiley-VCH, Weinheim, 2004 Search PubMed.
- G. Strukul, Catalytic Oxidations with Hydrogen Peroxide as Oxidant, Kluwer, Dordrecht, 1992 Search PubMed.
- Organometallic Oxidation Catalysis, ed. F. Meyer and C. Limberg, Springer, Berlin/Heidelberg/New York, 2007 Search PubMed.
- Mechanisms in Homogeneous and Heterogeneous Epoxidation Catalysis, ed. S. T. Oyama, Elsevier, Amsterdam, 2008 Search PubMed.
- S. Lubis, L. Yuliati, S. L. Lee, I. Sumpono and H. Nur, Chem. Eng. J., 2012, 209, 486 CrossRef CAS PubMed.
- B. Feng, Z. S. Hou, X. R. Wang, Y. Hu, H. Li and Y. X. Qiao, Green Chem., 2009, 11, 1446 RSC.
- V. K. Bansal, P. P. Thankachan and R. Prasad, Appl. Catal., A, 2010, 381, 8 CrossRef CAS PubMed.
- G. Barak and Y. Sasson, J. Chem. Soc., Chem. Commun., 1987, 1266 RSC.
- N. S. Patil, B. S. Uphade, P. Jana, S. K. Bharagava and V. R. Choudhary, J. Catal., 2004, 223, 236 CrossRef CAS PubMed.
- L. Nie, K. K. Xin, W. S. Li and X. P. Zhou, Catal. Commun., 2007, 8, 488 CrossRef CAS PubMed.
- D. M. Gao and Q. M. Gao, Catal. Commun., 2007, 8, 681 CrossRef CAS PubMed.
- R. Ben-Daniel, A. M. Khenkin and R. Neumann, Chem. – Eur. J., 2000, 6, 3722 CrossRef CAS.
- N. K. K. Raj, S. S. Deshpande, R. H. Ingle, T. Raja and P. Manikandan, Catal. Lett., 2004, 98, 217 CrossRef CAS.
- Y. Ding, Q. Gao, G. X. Li, H. P. Zhang, J. M. Wang, L. Yan and J. S. Suo, J. Mol. Catal. A: Chem., 2004, 218, 161 CrossRef CAS PubMed.
- Y. Nakagawa, K. Kamata, M. Kotani, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2005, 44, 5136 CrossRef CAS PubMed.
- K. Kamata, M. Kotani, K. Yamaguchi, S. Hikichi and N. Mizuno, Chem. – Eur. J., 2007, 13, 639 CrossRef CAS PubMed.
- P. Shringarpure and A. Patel, Dalton Trans., 2008, 3953 RSC.
- X. D. Yu, L. L. Xu, X. Yang, Y. N. Guo, K. X. Li, J. L. Hu, W. Li, F. Y. Ma and Y. H. Guo, Appl. Surf. Sci., 2008, 254, 4444 CrossRef CAS PubMed.
- V. Mirkhani, M. Moghadam, S. Tangestaninejad, I. Mohammadpoor-Baltork, E. Shams and N. Rasouli, Appl. Catal., A, 2008, 334, 106 CrossRef CAS PubMed.
- A. M. Al-Ajlouni, O. Saglam, T. Diafla and F. E. Kuhn, J. Mol. Catal. A: Chem., 2008, 287, 159 CrossRef CAS PubMed.
- P. Sharma and A. Patel, J. Mol. Catal. A: Chem., 2009, 299, 37 CrossRef CAS PubMed.
- J. L. Hu, K. X. Li, W. Li, F. Y. Ma and Y. H. Guo, Appl. Catal., A, 2009, 364, 211 CrossRef CAS PubMed.
- Q. Chen, Y. Cui, Q. Sun, W. J. Pan, Q. Chen and Y. Xu, Z. Anorg. Allg. Chem., 2009, 635, 2302 CrossRef CAS PubMed.
- J. Tang, X. L. Yang, X. W. Zhang, M. Wang and C. D. Wu, Dalton Trans., 2010, 39, 3396 RSC.
- P. A. Shringarpure and A. Patel, Dalton Trans., 2010, 39, 2615 RSC.
- A. C. Estrada, M. M. Q. Simoes, I. C. M. S. Santos, M. G. P. M. S. Neves, J. A. S. Cavaleiro and A. M. V. Cavaleiro, Monatsh. Chem., 2010, 141, 1223 CrossRef CAS.
- P. A. Shringarpure and A. Patel, Chem. Eng. J., 2011, 173, 612 CrossRef CAS PubMed.
- S. Pathan and A. Patel, Dalton Trans., 2011, 40, 348 RSC.
- A. Patel and S. Pathan, Ind. Eng. Chem. Res., 2012, 51, 732 CrossRef.
- S. S. Balula, C. M. Granadeiro, A. D. S. Barbosa, I. C. M. S. Santos and L. Cunha-Silva, Catal. Today, 2013, 210, 142 CrossRef CAS PubMed.
- T. A. G. Duarte, I. C. M. S. Santos, M. M. Q. Simoes, M. G. P. M. S. Neves, A. M. V. Cavaleiro and J. A. S. Cavaleiro, Catal. Lett., 2014, 144, 104 CrossRef CAS PubMed.
- S. S. Balula, L. Cunha-Silva, I. C. M. S. Santos, A. C. Estrada, A. C. Fernandes, J. A. S. Cavaleiro, J. Pires, C. Freire and A. M. V. Cavaleiro, New J. Chem., 2013, 37, 2341 RSC.
- R. Neumann and C. Abu-Gnim, J. Chem. Soc., Chem. Commun., 1989, 1324 RSC.
- R. Neumann and C. Abu-Gnim, J. Am. Chem. Soc., 1990, 112, 6025 CrossRef CAS.
- R. Neumann and A. M. Khenkin, Inorg. Chem., 1995, 34, 5753 CrossRef CAS.
- K. Patel, B. K. Tripuramallu and A. Patel, Eur. J. Inorg. Chem., 2011, 1871 CrossRef CAS PubMed.
- N. Mizuno, T. Hirose, M. Tateishi and M. Iwamoto, Chem. Lett., 1993, 1839 CrossRef CAS.
- P. Shringarpure and A. Patel, Inorg. Chim. Acta, 2009, 362, 3796 CrossRef CAS PubMed.
- K. Patel, P. Shringarpure and A. Patel, Supramol. Chem., 2012, 24, 149 CrossRef CAS.
- X. R. Lin, J. Y. Xu, H. Z. Liu, B. Yue, S. L. Jin and G. Y. Xie, J. Mol. Catal. A: Chem., 2000, 161, 163 CrossRef CAS.
- I. C. M. S. Santos, M. S. S. Balula, M. M. Q. Simoes, M. G. P. M. S. Neves, J. A. S. Cavaleiro and A. M. V. Cavaleiro, Synlett, 2003, 1643 CAS.
- M. S. S. Balula, I. C. M. S. Santos, M. M. Q. Simoes, M. G. P. M. S. Neves, J. A. S. Cavaleiro and A. M. V. Cavaleiro, J. Mol. Catal. A: Chem., 2004, 222, 159 CrossRef CAS PubMed.
- M. M. Q. Simoes, I. C. M. S. Santos, M. S. S. Balula, J. A. F. Gamelas, A. M. V. Cavaleiro, M. G. P. M. S. Neves and J. A. S. Cavaleiro, Catal. Today, 2004, 91–2, 211 CrossRef PubMed.
- I. C. M. S. Santos, S. L. H. Rebelo, M. S. S. Balula, R. R. L. Martins, M. M. M. S. Pereira, M. M. Q. Simoes, M. G. P. M. S. Neves, J. A. S. Cavaleiro and A. M. V. Cavaleiro, J. Mol. Catal. A: Chem., 2005, 231, 35 CrossRef CAS PubMed.
- I. C. M. S. Santos, J. A. F. Gamelas, M. S. S. Balula, M. M. Q. Simoes, M. G. P. M. S. Neves, J. A. S. Cavaleiro and A. M. V. Cavaleiro, J. Mol. Catal. A: Chem., 2007, 262, 41 CrossRef CAS PubMed.
- I. C. M. S. Santos, M. M. Q. Simoes, M. S. S. Balula, M. G. P. M. S. Neves, J. A. S. Cavaleiro and A. M. V. Cavaleiro, Synlett, 2008, 1623 CAS.
- A. C. Estrada, M. M. Q. Simoes, I. C. M. S. Santos, M. G. P. M. S. Neves, A. M. S. Silva, J. A. S. Cavaleiro and A. M. V. Cavaleiro, Catal. Lett., 2009, 128, 281 CrossRef CAS.
- A. C. Estrada, M. M. Q. Simoes, I. C. M. S. Santos, M. G. P. M. S. Neves, A. M. S. Silva, J. A. S. Cavaleiro and A. M. V. Cavaleiro, Appl. Catal., A, 2009, 366, 275 CrossRef CAS PubMed.
- A. C. Estrada, M. M. Q. Simoes, I. C. M. S. Santos, M. G. P. M. S. Neves, J. A. S. Cavaleiro and A. M. V. Cavaleiro, ChemCatChem, 2011, 3, 771 CrossRef CAS PubMed.
- A. C. Estrada, I. C. M. S. Santos, M. M. Q. Simoes, M. G. P. M. S. Neves, J. A. S. Cavaleiro and A. M. V. Cavaleiro, Appl. Catal., A, 2011, 392, 28 CrossRef CAS PubMed.
- J. L. C. Sousa, I. C. M. S. Santos, M. M. Q. Simoes, J. A. S. Cavaleiro, H. I. S. Nogueira and A. M. V. Cavaleiro, Catal. Commun., 2011, 12, 459 CrossRef CAS PubMed.
- C. W. Jones, Applications of Hydrogen Peroxide and Derivatives, Royal Society of Chemistry, Cambridge, 1999 Search PubMed.
- W. R. Sanderson, Pure Appl. Chem., 2000, 72, 1289 CrossRef CAS.
- R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977 RSC.
- C. Rocchiccioli-Deltcheff, M. Fournier, R. Franck and R. Thouvenot, Inorg. Chem., 1983, 22, 207 CrossRef CAS.
- M. M. Q. Simoes, C. M. M. Conceicao, J. A. F. Gamelas, P. M. D. N. Domingues, A. M. V. Cavaleiro, J. A. S. Cavaleiro, A. J. V. Ferrer-Correia and R. A. W. Johnstone, J. Mol. Catal. A: Chem., 1999, 144, 461 CrossRef CAS.
- I. C. M. S. Santos, M. M. Q. Simoes, M. M. M. S. Pereira, R. R. L. Martins, M. G. P. M. S. Neves, J. A. S. Cavaleiro and A. M. V. Cavaleiro, J. Mol. Catal. A: Chem., 2003, 195, 253 CrossRef CAS.
- M. S. Balula, J. A. Gamelas, H. M. Carapuca, A. M. V. Cavaleiro and W. Schlindwein, Eur. J. Inorg. Chem., 2004, 619 CrossRef CAS PubMed.
- J. A. Gamelas, F. A. S. Couto, M. C. N. Trovao, A. M. V. Cavaleiro, J. A. S. Cavaleiro and J. D. P. de Jesus, Thermochim. Acta, 1999, 326, 165 CrossRef CAS.
- C. Venturello and M. Ricci, J. Org. Chem., 1986, 51, 1599 CrossRef CAS.
- Y. Ishii, K. Yamawaki, T. Ura, H. Yamada, T. Yoshida and M. Ogawa, J. Org. Chem., 1988, 53, 3587 CrossRef CAS.
- C. D. Brooks, L. C. Huang, M. McCarron and R. A. W. Johnstone, Chem. Commun., 1999, 37 RSC.
- C. L. Hill and C. M. Prosser-McCartha, Coord. Chem. Rev., 1995, 143, 407 CrossRef CAS.
- M. R. Maurya, A. Arya, P. Adao and J. C. Pessoa, Appl. Catal., A, 2008, 351, 239 CrossRef CAS PubMed.
- M. Torrent, M. Sola and G. Frenking, Chem. Rev., 2000, 100, 439 CrossRef CAS PubMed.
- A. Adejare, J. Shen and A. M. Ogunbadeniyi, J. Fluorine Chem., 2000, 105, 107 CrossRef CAS.
- A. Corma, V. Fornes, S. Iborra, M. Mifsud and M. Renz, J. Catal., 2004, 221, 67 CrossRef CAS.
- R. Bernini, A. Coratti, G. Provenzano, G. Fabrizi and D. Tofani, Tetrahedron, 2005, 61, 1821 CrossRef CAS PubMed.
- M. J. H. Moonen, A. H. Westphal, I. M. C. M. Rietjens and W. J. H. van Berkel, Adv. Synth. Catal., 2005, 347, 1027 CrossRef CAS PubMed.
- S. Kirumakki, S. Samarajeewa, R. Harwell, A. Mukherjee, R. H. Herber and A. Clearfield, Chem. Commun., 2008, 5556 RSC.
- J. R. L. Smith and P. R. Sleath, J. Chem. Soc., Perkin Trans. 2, 1982, 1009 RSC.
- A. I. Vogel, A Text-Book of Quantitative Inorganic Analysis Including Elementary Instrumental Analysis, Longmans, London, 3rd edn, 1961, pp. 325–326 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |