
Impact of particle size and metal–support interaction on denitration behavior of well-defined Pt–Cu nanoparticles
Akane
Miyazaki
a,
Kahori
Matsuda
a,
Florica
Papa
b,
Mariana
Scurtu
b,
Catalin
Negrila
c,
Gianina
Dobrescu
b and
Ioan
Balint
*b
aJapan Women's University, Faculty of Science, 2-8-1 Mejirodai, Bunkyo-ku, Tokyo 112-8681, Japan. E-mail: miyazakia@fc.gvu.ac.jp; kaho_rabbit@yahoo.co.jp
bInstitute of Physical Chemistry of the Romanian Academy, Spl. Independentei 202, 060021 Bucharest, Romania. E-mail: frusu@icf.ro; mcarata@icf.ro; gdobrescu@icf.ro; ibalint@icf.ro; Fax: +40 213121147
cNational Institute of Material Physics, P. O. Box MG 7, Magurele, Romania. E-mail: cnegrila@infim.ro
First published on 9th September 2014
Abstract
Well-dispersed Pt–Cu nanoparticles with two average sizes were synthesized: small (≈1.6 nm) and large (≈4.8 nm), respectively. The effects of size and support on the catalytic behavior of the nanoparticles for denitration reaction were analyzed. Both the unsupported and alumina-supported nanoparticles of smaller average size (1.6 nm) were very active, completely converting NO3− and NO2− (an intermediate reaction product) to N2 and NH4+. They were also more selective of N2 compared to the larger particles. The effects of particle size and alumina support on the denitration rate constants were analyzed in detail. The kinetic investigation indicates that the catalytic behavior is strongly related to the size of Pt–Cu nanoparticles as well as to the metal–support interaction. Generally, the larger nanoparticles proved to be less active, but on the other hand, they are less influenced by the support than the smaller ones. The metal–support interaction for bimetallic nanoparticles with smaller average size proved to be a key factor both for nitrate and nitrite reduction. Consideration of the reaction mechanism is made in light of the experimental results.
Introduction
The harmful effect of nitrate ions in drinking water on human health is well documented.1 Technologies proposed for the treatment of drinking water are osmosis, electrodialysis, and ion exchange. Catalytic denitration can be an attractive alternative for water treatment. The potential advantage of catalytic denitration over the other technologies is the simplicity of the required technology and the fact that this alternative does not produce waste with high salt concentrations that can be difficult to dispose of.The pioneering work of Vorlop et al.2 demonstrated that bimetallic systems, composed of a precious metal and a promoter, are able to catalyze the reduction of NO3− to NO2−. Bimetallic catalysts are effective in many reactions due to the synergetic effects taking place between the component metals. Acting individually, the monometallic components of a bimetallic catalyst usually do not display catalytic activity for the reduction of NO3−. One of the most effective systems for catalytic denitration is supported Pd–Cu.3,4 Most of the previously investigated catalysts were prepared by co-impregnating different support materials with the aqueous solutions of the corresponding metal precursors. However, it was observed that the synthesis should be directed so as to obtain intimate contact between the metals by alloy formation.5 Several bimetallic catalytic systems prepared by support impregnation have been investigated for NO3− reduction: Pd–In/Al2O3,6 Pt–In/Al2O3,7 Pt–In/SiO2 or TiO2,8 Pt–Sn/HZSM-5,9 Pt–Cu/Al2O3,10 Rh–Cu/Al2O3,11 Pd–Sn/ZrO2 or TiO2,12etc.
The disadvantages of the impregnation route for catalyst preparation are given as follows: (i) the catalytic active phases are formed with the participation of the support (strong metal–support interaction), (ii) the size and composition of the active phase formed by the interaction (alloying) of the two metals is difficult to control, and (iii) the characterization of the catalyst is demanding because the concentration of the active components compared to the supporting materials is very low (typically a few wt%).
One way to have better control over the composition and size of the catalytic active phase(s) is to first carry out the synthesis of the bimetallic nanoparticles in a controlled manner and then to disperse these nanoparticles over high surface area supports. By this procedure, the size and structure of the bimetallic catalyst can be conveniently directed in the colloidal synthesis stage by adjusting the preparation variables. Secondly, the bimetallic nanoparticles, when separated from the colloidal suspension, can be well characterized by commonly available techniques such as TEM, XRD, XPS, etc. When mild conditions are used, deposition onto the oxide support will usually have little effect on the size of the nanoparticles. There are also other advantages of dispersing metallic nanoparticles onto oxide supports. The metal loading can be increased to relatively high values while maintaining the dispersion (size) and structure of the nanoparticles.13 Thus, a reliable reaction mechanism can be determined by applying kinetic criteria (i.e., Madon-Boudart), which otherwise cannot be applied to conventionally prepared catalytic materials.
The support effect on the catalytic behavior can be better understood if the catalytic runs are carried out in the absence of a supporting material. It is well known that the support has a strong effect on denitration catalytic activity.14
Compared to other bimetallic systems such as Pd–Cu, denitration catalysts based on the bimetallic Pt–Cu couple are less well investigated. The reason is that Pt–Cu-based catalysts are considered less efficient compared to other bimetallic catalysts such as Rh–Cu or Pd–Cu.15 Barrabés et al.16 reported that a Pt–Cu bimetallic catalyst is more selective for N2 formation compared to the Pd–Cu pair. On the other hand, the Pd–Cu catalysts showed higher NO3− reduction activity, but larger amounts of harmful NO2− were produced. The same studies pointed out that Pt–Cu nanospheres are more efficient compared to the corresponding impregnated bimetallic catalysts.
The aims of the present study are (i) to prepare and characterize Pt–Cu bimetallic nanoparticles with a controlled size and to investigate their catalytic behavior in a model structure-sensitive reaction (NO3− reduction with H2), (ii) to take a deeper look at the size effect of the particles, and (iii) to determine the support effects on the kinetic k1 (NO3− reduction) and k2 (NO2−) rate constants.
Experimental section
Materials and methods
The alkaline polyol method was selected to prepare well-defined bimetallic nanoparticles because it is easy and versatile. The synthesis of polyvinylpyrrolidone (PVP)-protected bimetallic nanoparticles was described in detail by Papa et al.17 Briefly, the metal precursors H2PtCl6 (Alfa Aesar) and Cu(CH3COO)2·H2O (Merck), dissolved in an alkaline solution of ethylene glycol (EG), were reduced in two successive steps. The typical concentration of metal ions in EG was 3.8 × 10−2 M. The protective polymer PVP (MW = 8000) was dissolved separately in an alkaline solution of EG (0.25 M NaOH). The concentration of PVP in EG, calculated using monomer base units, was 0.38 M. The synthesis was carried out in a three-necked flask purged with Ar to ensure an inert atmosphere.The Pt4+ (3.8 × 10−2 M)/NaOH (0.25 M)/PVP (0.38 M) solution was heated rapidly to 160 °C under vigorous stirring. The PVP/Pt+ molar ratio in the first reduction step was either 5 or 10. Our previous work pointed out that the ratio between the protective polymer and the platinum is essential for controlling the average size of Pt colloidal nanocrystalline seeds.17 Typically, smaller Pt nanoparticles are obtained with higher PVP/Pt4+ molar ratios. After 1 hour of reaction time at 160 °C, the colloidal suspension of Pt was cooled to room temperature. Cu2+ (0.038 M)/EG solution was then added to the colloidal suspension, and the resulting product yielded a calculated Pt/Cu weight ratio of 2. The reduction of Cu2+ was also carried out at 160 °C for 1 hour. After cooling, the bimetallic nanoparticles were separated by acetone addition followed by refrigeration at −16 °C for 24 h. The separated solid phase at the bottom of the flask was filtered, washed several times with acetone and dried in an oven at 100 °C for 8 h. The recovery yields of the nanoparticles were 95–98 wt%. The bimetallic nanoparticle powders were dispersed by sonication in polar media (i.e., water or alcohols).
Characterization
The obtained nanoparticles were characterized by transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), and X-ray diffraction (XRD).The TEM characterization was performed by using a JEOL 1200 EXS apparatus operated at 200 kV. The analyzed samples were suspended in ethanol and then dispersed on a carbon-coated copper grid.
The XPS analysis was carried out with a VG Esca 3 Mk II spectrometer using an Al Kα radiation source at 1486.7 eV. The 100 mm radius hemispherical electron analyzer was operated at a pass energy of 50 eV. The experimental spectra were fitted with Voight functions (SDP 2.3).
The crystalline structure of the PVP-protected bimetallic powders was analyzed with a D8 Advance (Bruker-AXS) apparatus using CuKα radiation (λ = 1.54 Å). The diffraction patterns were recorded in the 2Θ = 30–135° domain. The experimental data were fitted with the Pawley method to obtain the values of the lattice constants. The average crystallite sizes were calculated from peak broadenings using the Lorentzian function to fit the peak shapes. The catalytic tests were performed with alumina-supported Pt–Cu nanoparticles and with simple, colloidal Pt–Cu nanoparticles. The PVP-protected Pt–Cu nanoparticles, dispersed by sonication in ethyl alcohol, were deposited on high surface area γ-Al2O3 (100 m2 g−1, Aerosil Japan) by physical adsorption. After drying at 100 °C for 4 h, the catalytic materials were calcined at 200 °C for 1 h to remove the protective PVP polymer coating from the nanoparticles. We have performed TG-DTA analysis for Pt–Cu nanoparticles protected with PVP to determine the lowest temperature at which the PVP polymer is removed. According to the experimental data, for a heating rate of 10 °C min−1, around 80% of the PVP is removed at 200 °C. After 1 h at 200 °C, the polymer removal is completed.
The calcination was conducted at low temperature in order to preserve the initial size of the Pt–Cu nanoparticles. The total metal (Pt + Cu) loading in all cases was 1.2 wt% (0.8 wt% Pt and 0.4 wt% Cu). Prior to the catalytic runs, the materials were reduced in H2 at 200 °C for 1 h. Then, 0.3 g of the supported catalyst was dispersed in 150 mL of an aqueous solution containing 2 mM of NO3− (NaNO3). In the case of the catalytic tests conducted with unsupported bimetallic nanoparticles, an equivalent amount (weight) of Pt–Cu nanoparticles (3.6 mg) was dispersed homogeneously into the liquid phase by vigorous agitation. After 20 min of equilibration time, H2 was bubbled (25 cm3 min−1) into the solution containing the suspended catalyst and nitrate ions. The catalytic reactor was topped with a condenser cooled to −10 °C to prevent the ammonia loss by evaporation. The denitration reaction was performed at 25 °C. Aliquots of 2 mL were withdrawn every 30 min, and after filtration, the composition of each was analyzed using an ion chromatography system (Agilent ICS-900). The NO3−, NO2− and NH4+ ions were separated and quantified using the anionic and cationic columns of an ion chromatograph.
Fractal analysis of the bimetallic nanoparticles was carried out on 1026 × 1026 pixel areas of the TEM micrographs. The grey level of each image pixel was converted to height, and the fractal dimension of the equivalent surface was computed.
Results and discussion
Fig. 1 presents TEM micrographs and size distribution histograms of the Pt–Cu nanoparticles obtained for PVP/Pt molar ratios of 5 [Fig. 1A (high magnification) and B (low magnification)] and 10 [Fig. 1C (high magnification) and D (low magnification)]. Looking at the TEM micrographs, it can be observed that well-dispersed Pt–Cu nanoparticles have been obtained at both high and low PVP/Pt ratios. On the other hand, the average size of the Pt–Cu nanoparticles decreased with an increasing PVP/Pt molar ratio. Control of the particle size is a crucial issue in structure-sensitive reactions such as the catalytic reduction of NO3− ions, which involves the formation of a N–N bond.18 According to the size histograms presented in Fig. 1, the mean diameters of the Pt–Cu nanoparticles were 4.8 (Fig. 1E) and 1.6 nm (Fig. 1F) for PVP/Pt molar ratios of 5 and 10, respectively. From here on, for the sake of clarity and simplicity, the larger and smaller diameter Pt–Cu nanoparticles will be referred to as (Pt–Cu)L and (Pt–Cu)S, respectively.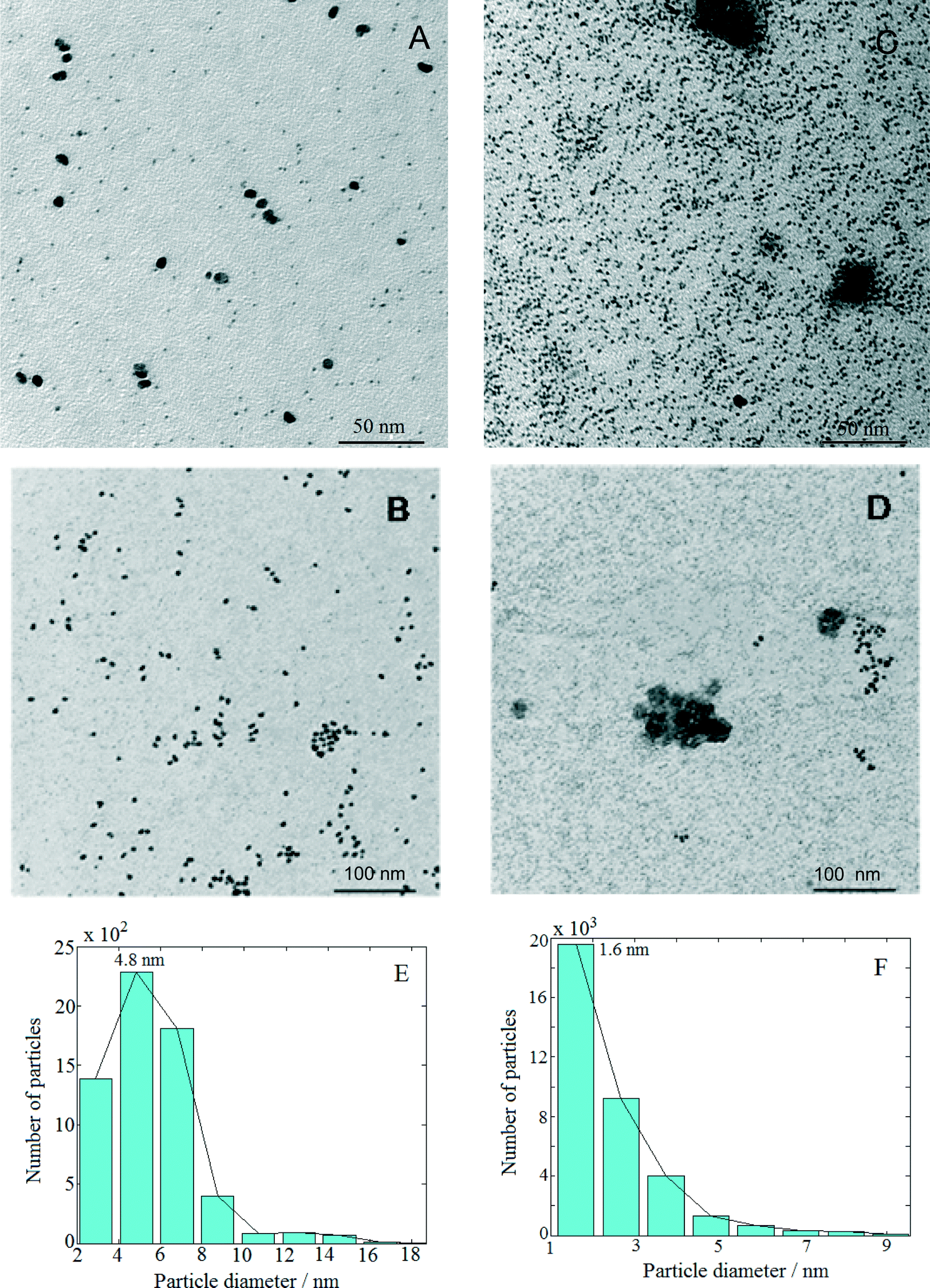 | ||
| Fig. 1 TEM images of large diameter [(Pt–Cu)L] (A and B) and small diameter [(Pt–Cu)S] (C and D) nanoparticles. The PVP/Pt4+ molar ratios were 5 (micrographs A and B with the corresponding size distribution E) and 10 (micrographs C and D with the corresponding size distribution F). The weight ratio between Pt and Cu was 2 in all cases. | ||
Fractal analysis was carried out on 1026 × 1026 pixel square domains selected from the TEM micrographs presented in Fig. 1B and D. The aim was to determine the fractal dimension as well as the self-similarity domains. Two calculation methods were applied: the (i) correlation function and (ii) variable length scale.
The correlation function method19 uses the height correlation function [G(r)] and its scaling law according to eqn (1):
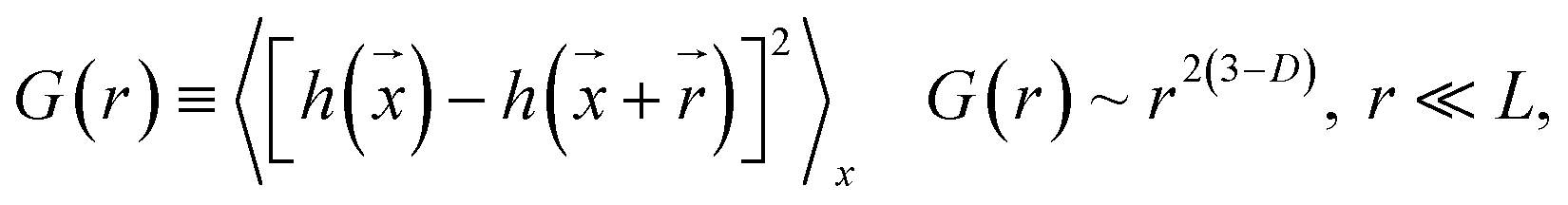 | (1) |
The scaling range where eqn (1) is valid defines the “cut-off” limits and indicates the range of self-affinity (self-similarity); in other words, the range where the surface points are correlated.
The variable length scale method20 consists of computing the root mean square (rms) deviation of the surface for multiple intervals (of length ε or boxes of size ε × ε) and increasing lengths (or box sizes). Rms deviation Rqε, averaged over nε, the number of intervals of length ε, is defined as
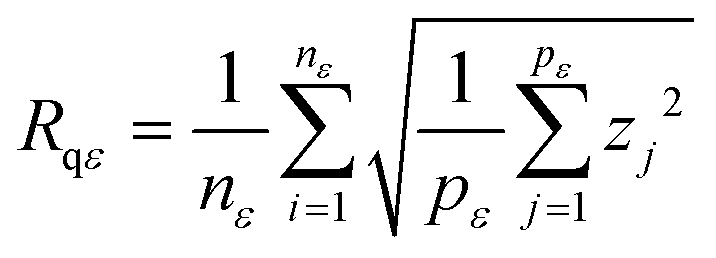 | (2) |
| D = DT − H | (3) |
The results obtained from the fractal analysis are presented in Table 1. Both samples exhibited fractal behavior over a large domain, indicating that the analyzed samples were homogeneous, i.e., the particles were of the same type. The (Pt–Cu)L sample is characterized by two fractal dimensions, 2.5 at the low scale (1.4–2.5 nm) and 2.7 for a self-similarity domain of 2.5–5 nm. After correlating these results with the particle size distribution, one can assign these dimensions to a mixed-fractal structure composed of different sets of particles: small particles (lighter particles in the image), with sizes between 1.4 and 2.5 nm, and large particles (darker particles in the image), with sizes between 2.5 and 5.0 nm. There are correlations between these particles, resulting in a fractal dimension of 2.68, over a large domain (5.0–27.5 nm).
| Sample | Calculation method | Fractal dimension | Correlation coefficient | Self-similarity domain/nm |
|---|---|---|---|---|
| (Pt–Cu)L | Correlation function | 2.50 ± 0.01 | 0.991 | 1.4–2.5 |
| 2.70 ± 0.01 | 0.981 | 2.5–5.0 | ||
| Variable length scale | 2.68 ± 0.01 | 0.984 | 5.0–27.5 | |
| (Pt–Cu)S | Correlation function | 2.40 ± 0.01 | 0.998 | 1.4–2.2 |
| Variable length scale | 2.73 ± 0.01 | 0.996 | 7.5–12.5 | |
| 2.88 ± 0.01 | 0.992 | 12.5–22.5 |
The sample (Pt–Cu)S exhibited different fractal behavior. The small particles, ranging between 1.4 and 2.2 nm, had a homogeneous structure composed of medium-range (fractal dimension = 2.73) and long-range correlated particles (fractal dimension = 2.88).
According to the self-similarity data in Table 1, the size of (Pt–Cu)L and (Pt–Cu)S ranged between 2 and 5 nm and between 1 and 2 nm, respectively.
XRD analysis is an essential characterization tool used to obtain information on the average size, composition and chemical state of crystalline domains of bimetallic nanoparticles. While the nanometric-sized crystallites typically produce characteristic XRD peaks, the subnanometric-sized crystals do not, likely because these smaller particles lack sufficient size for detection by XRD.
The XRD patterns of (Pt–Cu)L and (Pt–Cu)S protected with PVP are presented in Fig. 2A and B, respectively. The sharp and well-defined XRD peaks of (Pt–Cu)L suggest that the nanocrystalline domains of the sample were characterized by a larger average diameter. In contrast, the broad XRD peaks of (Pt–Cu)S indicate a smaller average size of nanocrystalline domains. As can be seen in Fig. 2, the peaks are slightly shifted from the characteristic positions of Pt(fcc). The characteristic reflections of a Cu phase could not be observed in the XRD patterns. The average crystallite size, determined from the fit of the XRD peaks with a Lorentzian function of the Scherrer equation, was 6.7 nm for (Pt–Cu)L and 1.6 nm for (Pt–Cu)S. These values are in good agreement, within experimental error, with the TEM observations: d(TEM) = 4.8 nm for (Pt–Cu)L and 1.6 nm for (Pt–Cu)S (see Table 2).
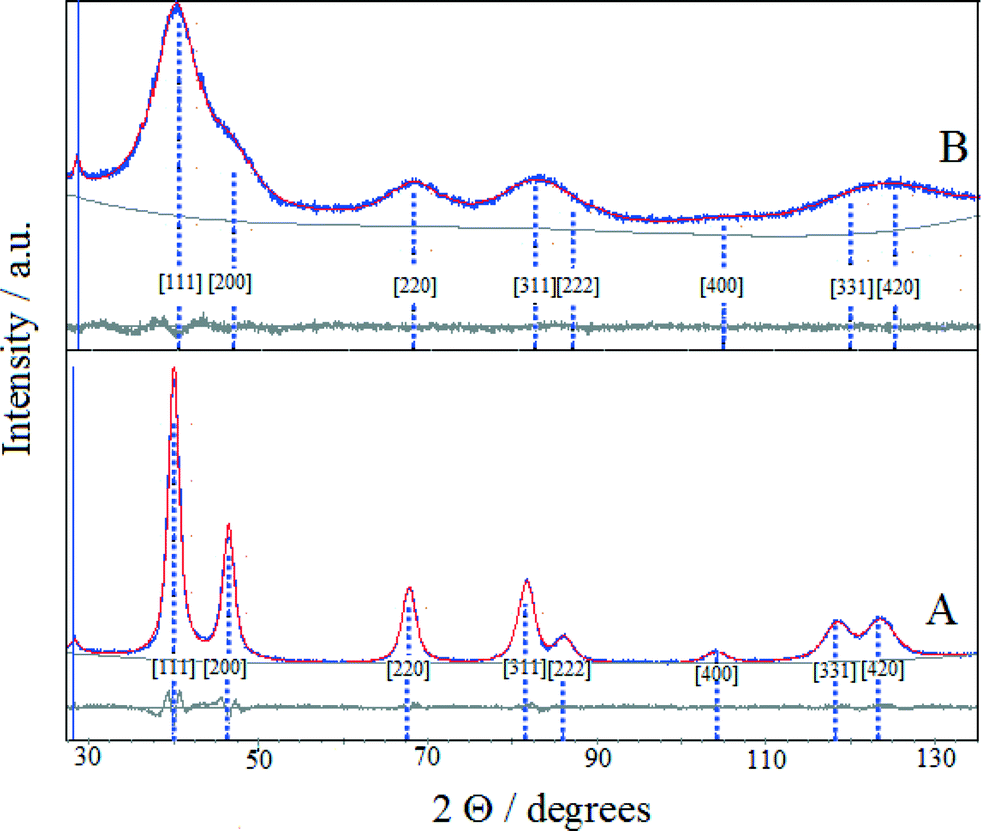 | ||
| Fig. 2 The XRD patterns of (Pt–Cu)L (A) and (Pt–Cu)S (B) protected with PVP. The experimental and Pawley simulated XRD patterns are represented by blue and red traces, respectively. The light gray traces reflect the accuracy of the experimental pattern fit with the Pawley software. The blue peak markers are the characteristic positions of Pt(fcc) diffraction peaks. | ||
| Samples | d XRD | Lattice const. (a)/Å | Lattice strain/G | XRD comp.b/at% | XPS comp.c/at% | Oxidation state | d TEM /nm | ||
|---|---|---|---|---|---|---|---|---|---|
| Pt | Cu | Pt | Cu | ||||||
| a Lorentzian average crystallite size calculated by the Scherrer method. b The average composition of the nanocrystals was calculated from XRD data according to Vegard's law: aalloy = x·aPt + (1 − x)aCu, where a represents the lattice constant and x is the fraction of the element; aPt = 3.920 Å, aCu = 3.610 Å. c The relative composition of the bimetallic nanoparticles calculated from XPS data. d Mean particle size estimated from TEM micrographs. | |||||||||
| (Pt–Cu)L | 6.7 | 3.913 | 1.210 | 97.7 | 2.3 | 37.8 | 62.2 | Pt0, Cu0 | 4.8 |
| (Pt–Cu)S | 1.6 | 3.899 | 4.95 | 93.2 | 6.8 | 41.5 | 58.5 | Pt0, Cu+ (major), Cu0 (minor) | 1.6 |
The lattice constants (a) of the nanocrystalline particles were determined by fitting the experimental patterns with Pawley XRD data processing software. As can be seen in Fig. 2, the patterns obtained from the fit of the experimental data overlap well the experimental patterns. The determined values of the lattice constants (a) were 3.913 and 3.899 Å for (Pt–Cu)L and (Pt–Cu)S, respectively. The shift of platinum peak positions is usually attributed to lattice contraction caused by alloy formation when Pt atoms (atomic radius = 1.373 Å) are substituted by smaller Cu atoms (atomic radius = 1.278 Å) in the fcc structure. The average compositions of the crystalline nanodomains were derived from the experimentally determined lattice constant by applying Vegard's law (see equation in Table 2). The composition of (Pt–Cu)L and (Pt–Cu)S was 97.7 at% Pt–2.3 at% Cu (Pt43Cu) and 93.2 at% Pt–6.8 at% Cu (Pt14Cu), respectively. Taking into account that the theoretical composition of the bimetallic nanoparticles should be 39.4 at% Pt–60.6 at% Cu (corresponding to Pt/Cu weight ratio = 2), it is clear that a significant amount of amorphous copper was present as a segregated phase. It should be emphasized that the presence of copper in stoichiometric amounts in both samples was evidenced by XPS results (see Table 2). Since the copper phase(s) was not detectable by XRD, it most likely formed a thin layer on the platinum-rich seeds. It was reported that by decreasing the average size of particles spontaneous alloying process may occur even for immiscible elements. For example, Kusada21 reported the formation of Ag–Rh alloy when the average size of nanoparticles was decreased to nanometric domain. The Ag–Rh alloy formed between the two immiscible neighboring elements showed remarkable hydrogen storage ability due to a newly resulted electronic structure resembling that of Pd. In line with the abovementioned example, as the average particle size decreased from 4.8 to 1.6 nm, the amount of copper alloyed with Pt in the nanocrystalline domains of nanoparticles increased from 2.3 to 6.8 at%.
XPS analysis was performed to get relevant information on the chemical state and composition of the investigated Pt–Cu nanoparticles. The atomic percentages were calculated using normalized areas of the XPS peaks. The atomic sensitivity factors were derived from the electronic cross section of each element using the transmission and inelastic mean free path corrections. Note that the average size of the analyzed nanoparticles was close to [in the case of (Pt–Cu)L] or smaller [in the case of (Pt–Cu)S] than the penetration depth of the XPS beam (10 to 20 surface atomic layers, representing around 5 nm). Thus, the XPS-derived compositional data in Table 2 give information from not only the surface but also from the entirety (bulk and surface) of the analyzed bimetallic nanoparticles. The slight deviation from the theoretical stoichiometry (39.4 at% Pt–60.6 at% Cu) observed for (Pt–Cu)L (38.8 at% Pt–62.2 at% Cu) and (Pt–Cu)S (41.5 at% Pt–58.5 at% Cu) nanoparticles can be explained by the error (10–15%) associated with the XPS method. The XPS data show that copper was not leached from the system during the preparation stage and that the copper distribution within the sample was uniform.
The energy of the C1s peak at 285 eV was used as a reference for the calculation of the core-level binding energies. The XPS spectra of (Pt–Cu)L in the Cu2p3/2 and Pt4f regions are presented in Fig. 3A and B, respectively. The peak at 933.14 eV in the Cu2p3/2 region indicates Cu+/Cu states for copper.22 The absence of the shake-up satellite peak in the 942–948 eV regions is proof that Cu2+ species were not present.
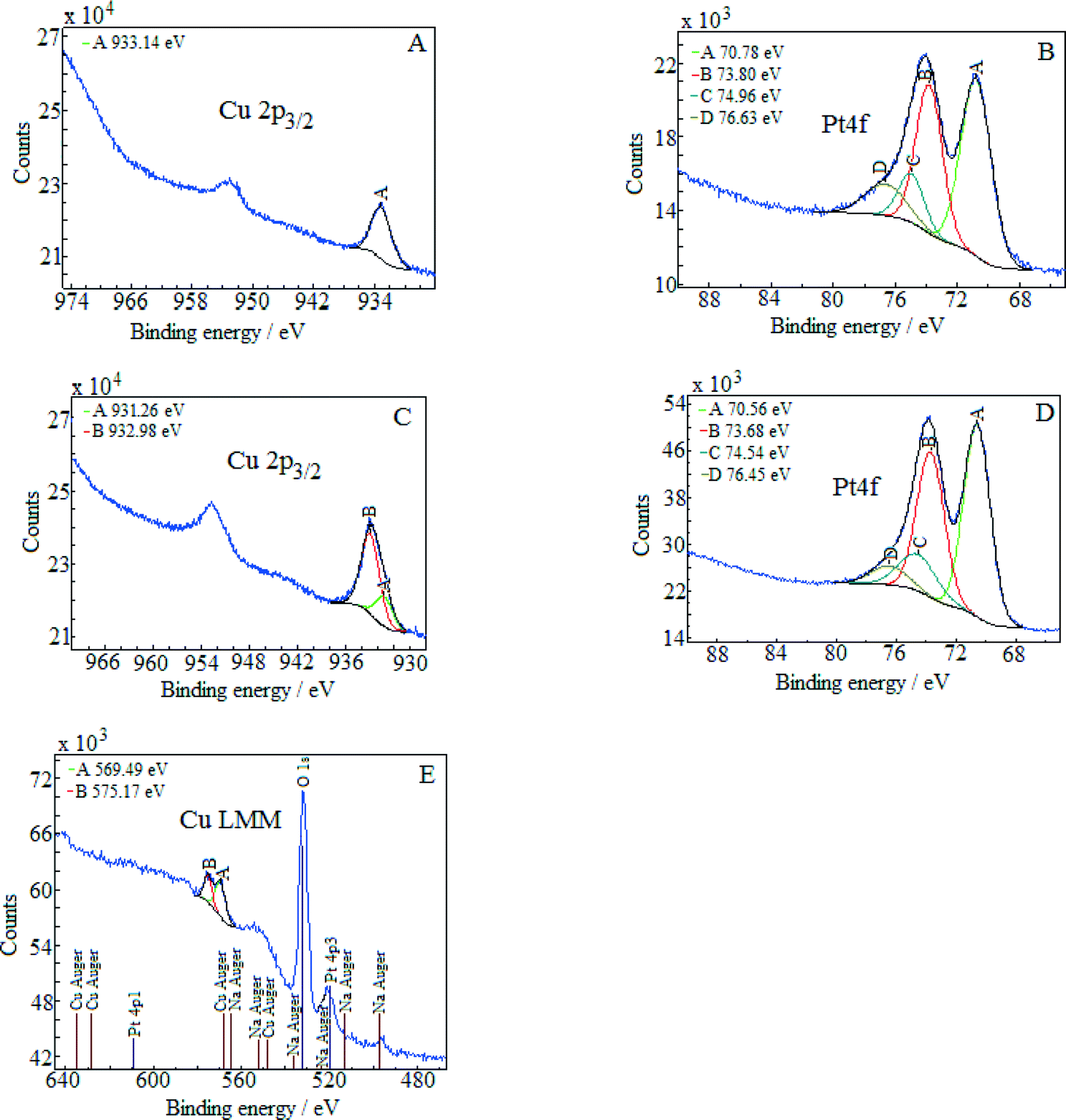 | ||
| Fig. 3 XPS core-level spectra of the Pt4f and Cu2p3/2 regions determined for (Pt–Cu)L (spectra A and B) and for (Pt–Cu)S (spectra C and D). E shows the Cu LMM Auger spectrum of the (Pt–Cu)S nanoparticles. | ||
The platinum doublet A (at 70.78 eV, Pt4f7/2) and B (at 73.8 eV, Pt4f5/2) in the Pt 4f region of Fig. 3B can be attributed to the Pt0 state.23 The pair of peaks, C at 74.96 eV and D at 76.63 eV, of the Cu3p (3p3/2 and 3p1/2) region in the same figure is representative of metallic copper.24
The XPS spectra of (Pt–Cu)S are presented in Fig. 3C (Cu 2p3/2 region) and D (Pt 4f region). In this case, the Cu 2p3/2 region contains two peaks, one located at 931.26 eV (peak A) and the other at 932.98 eV (peak B). According to the literature, Cu+ and Cu0 should have very close binding energies in the Cu2p3/2 region.25 Usually, these two forms cannot be discriminated. However, two peaks in the XPS spectra in Fig. 3C can be clearly fitted. The interpretation is that the small peak (A) at 931.26 represents the metallic Cu alloyed with Pt and the larger one (peak B) at 932.98 eV represents Cu+ species. For our specific particle structure, the discrimination between the two copper species was possible probably because the areas of metallic Cu alloyed with Pt (in the core) and Cu+ (located on the surface of the particles) were charged differently. The charge difference between the nanocrystalline domains (containing Cu0) and amorphous domains (containing Cu+) caused the slight shift in the XPS maxima of the two species, leading to the appearance of two peaks.26,27
The absence of the shake-up satellite peak is consistent with the absence of Cu2+ species. In order to determine the copper species that is responsible for peaks A and B, the Cu LMM Auger spectra were recorded and are presented in Fig. 3E. The Auger spectra are used to discriminate between the Cu+ and Cu0 species since both species exhibit approximately the same binding energy in the Cu 2p3/2 region. The metallic Cu peak should be located at ≈568 eV and the Cu+ peak at 570 eV. The Auger spectra revealed only the presence of Cu+ species (Fig. 3E, peak A at 569.5 eV). The signal of Cu0 should be at around 568 eV but was too weak to be detected, suggesting that the amount of metallic copper fell below the detection limit of Auger investigation. However, from the XRD measurement, it is clear that metallic copper was present and alloyed with Pt in the crystalline domains of (Pt–Cu)S nanoparticles (see Table 2, XRD composition: 93.2 at% Pt–6.8 at% Cu).
In order to justify the financial and scientific efforts undertaken to synthesize novel materials with special properties, applications for these materials with significant practical relevance must be identified. One possible application with environmental significance is the removal of NO3− ions from polluted surface and ground waters. Thus, the catalytic behavior of alumina-supported Pt–Cu nanoparticles for NO3− reduction with H2 was investigated to assess the potential of these nanomaterials with controlled structure and size for practical applications. We compared the activity of alumina-supported nanoparticles with large (4.8 nm) and small (1.6 nm) average sizes to evaluate the size effect on catalytic denitration. Another aim was to take a look at the particle size dependency of the metal–support interaction, which is of crucial importance for developing new efficient catalytic materials. The results of catalytic tests are presented in Fig. 4.
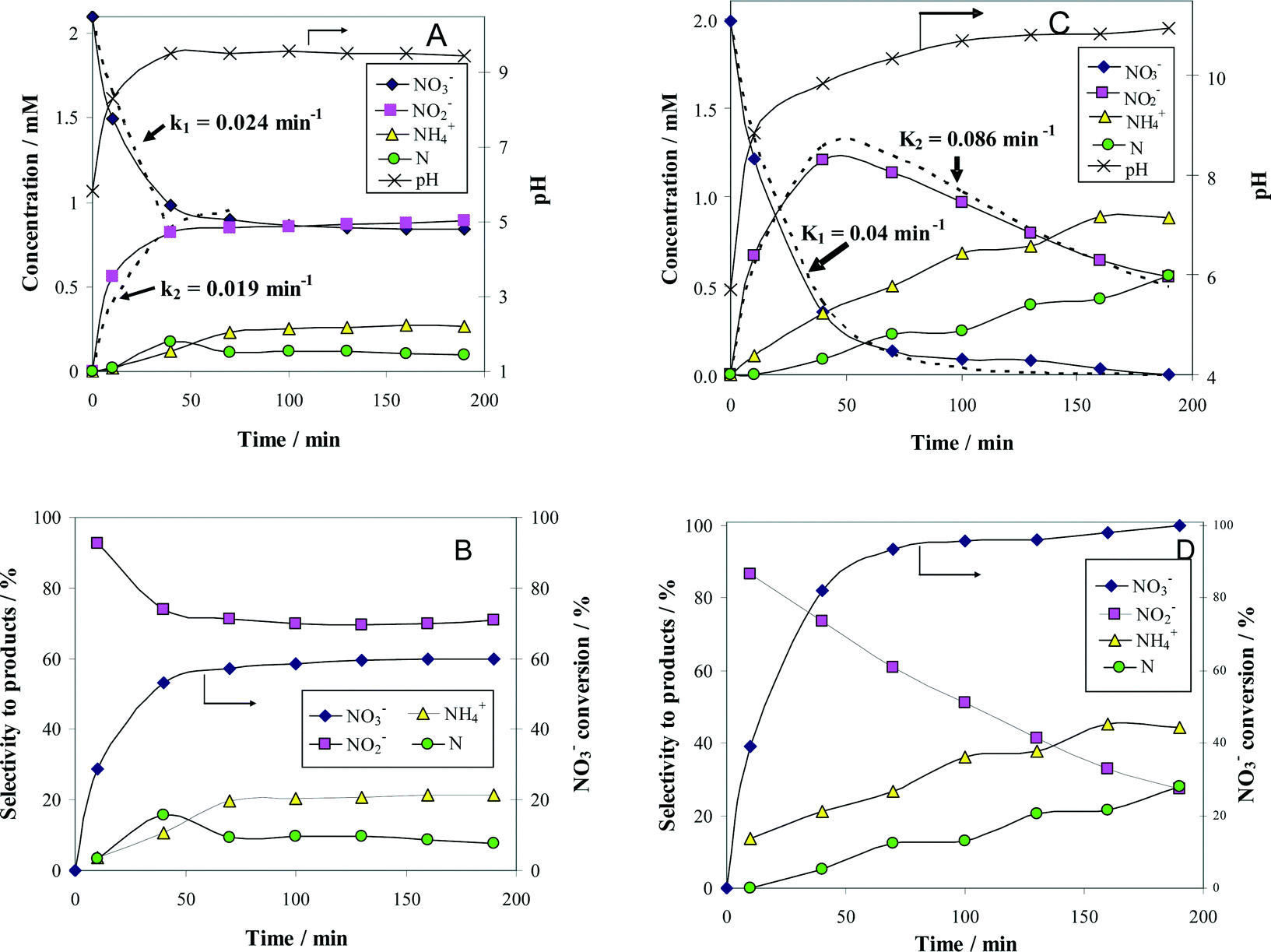 | ||
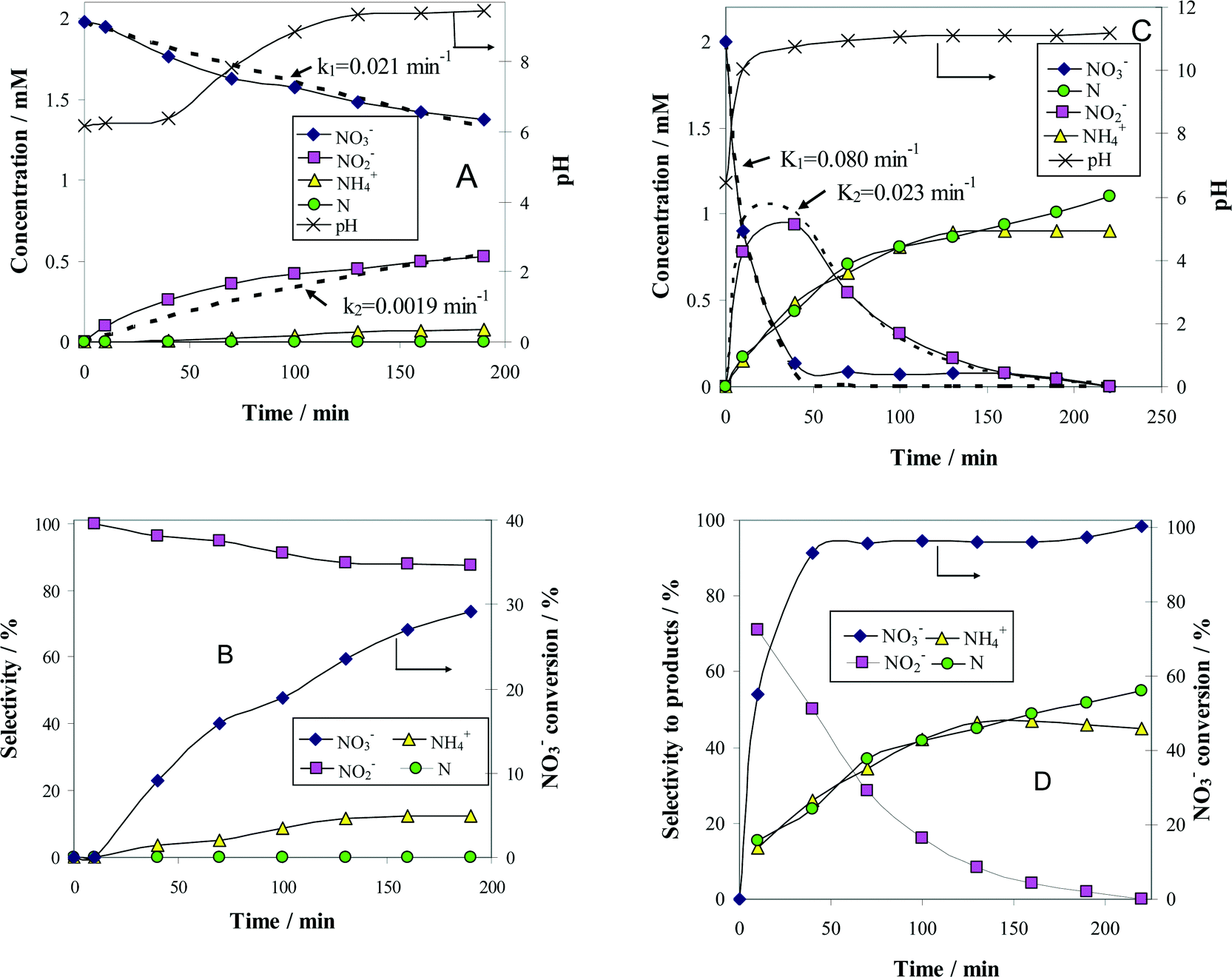
| Fig. 4 Time course of the reactant (NO3−) and product (NO2−, N2 and NH4+) concentrations (A and C), as well as the associated selectivity for products (B and D) during the denitration reaction over 1.2 wt% (Pt–Cu)L/Al2O3 (A and B) and 1.2 wt% (Pt–Cu)S/Al2O3 (C and D). The k1 and k2 values of the apparent reaction rate constants were calculated from the best fit of the experimental data (see dotted lines in A and C). Reaction conditions: Wcat = 0.3 g; Vsol = 150 mL; C0(NO3−) = 2 mM; T = 25 °C. | ||
The change with time of reactant and product concentrations over (Pt–Cu)L/Al2O3 nanoparticles of 4.8 nm is presented in Fig. 4A. It can be observed that the catalyst deactivates with time. After 70 min of reaction time, the decrease in NO3− concentration was negligible. The same trend was observed for the formation of the intermediary reaction product NO2−, as well as for the final products N2 and NH4+.
The Gibbs standard free reaction energies (ΔG0r) at 298 K associated with NO3− conversion to intermediate (NO2−) and final reaction products (N2 and NH4+) are given as follows:28
| NO3− + 2H+ → NO2− + H2O ΔG0r = −163.2 kJ mol−1 |
| NO3− + 6H+ → 1/2N2 + 3H2O ΔG0r = − 599.2 kJ mol−1 |
| NO3− + 10H+ → NH4+ + 3H2O ΔG0r = − 678.5 kJ mol−1 |
The negative ΔG0r values observed for all the reaction steps involved in NO3− transformation to products indicate that these processes, occurring spontaneously, are favored from the thermodynamic point of view. In other words, the reactions associated with NO3− reduction are irreversible at room temperature. In consequence, the absence of NO3− conversion is not due to the attainment of steady-state conditions but to catalyst deactivation. Probably, the formation of an oxide layer at the surface of the bimetallic (Pt–Cu)L was the most likely reason for catalyst deactivation.
It is clear that the oxidation state of copper is a critical issue in the denitration process. We have conducted additional experiments to better understand the effect of oxygen on the activity of catalysts. The catalytic data (not presented here) show that the exposure to air at room temperature had little impact on the activity of (Pt–Cu)L/Al2O3, regardless of the contact time. In contrast, the activity was completely lost after calcination in air at 200 °C for 1 h. Similar deactivation after calcination at high temperature was observed in the case of (Pt–Cu)S/Al2O3.
The concentration of N2 was not measured directly but was indirectly calculated from a nitrogen mass balance. Nitrogen is represented in the graphs as monoatomic N. The evolution of pH with time was also monitored and plotted in the same figure on the secondary Y-axis.
There are relatively few published data on the kinetics of the denitration reaction involving well-defined bimetallic nanoparticles. Witońska et al.11 reported in a previous work that the reduction of NO3− to NO2− over an impregnated Rh–Cu/Al2O3 catalyst follows a first-order decay. The present study was a good opportunity to measure the kinetic constants for a Pt–Cu system and to analyze the way in which the metal–support interaction as well as the particle size effects is reflected by the rate constant k values. It is the first study showing how the rate constants of the individual reaction steps are influenced by nanoparticle average size and by the metal–support interaction. The kinetics of denitration were investigated by considering the following, widely accepted, stepwise processes:15
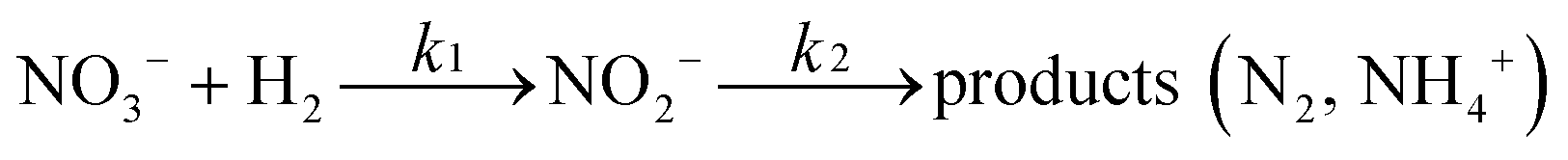 | (4) |
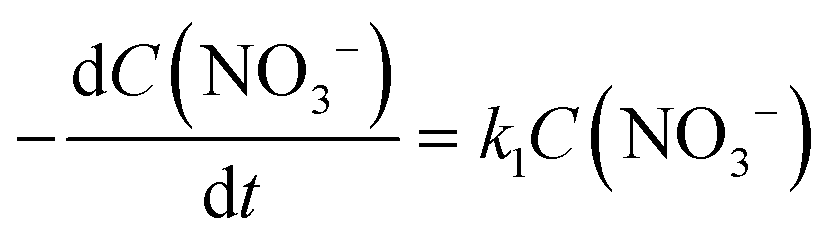 | (5) |
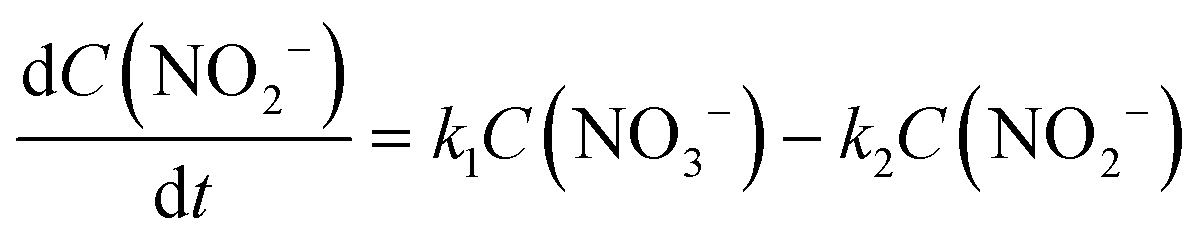 | (6) |
The integral solutions of differential rate eqn (5) and (6) are
| C(NO3−) = C0(NO3−)e−k1t | (7) |
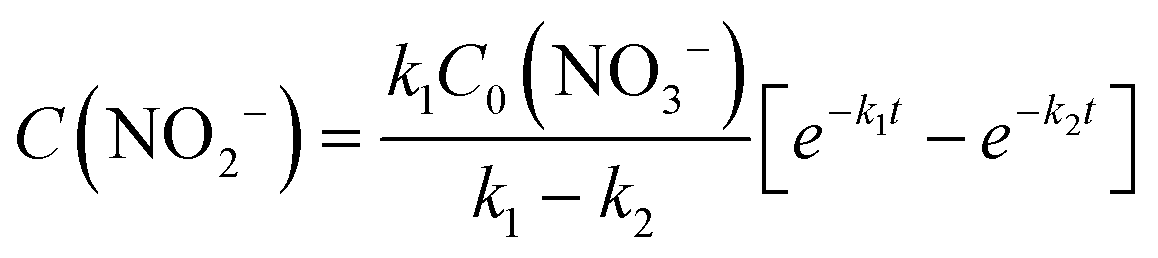 | (8) |
 | ||
| Fig. 5 Time course of reactant (NO3−) and product (NO2−, N2 and NH4+) concentrations (A and C), as well as the associated selectivity for products (B and D) for the denitration reaction involving (Pt–Cu)L (A and B) and (Pt–Cu)S (C and D) nanoparticles suspended in a liquid phase. The k1 and k2 apparent rate constant values were calculated from the best fit of the experimental data. Reaction conditions: Wcat = 3.6 mg; Vsol = 150 mL; C0(NO3−) = 2 mM; T = 25 °C. | ||
The mass transfer limitations were considered. Typically, each kinetic constant was measured for two mixing rates. The unchanged kinetic constant values and final product distributions indicated that the reaction was not affected by mass transfer limitations.
Hydrogen was not included in the rate expression, as the rates were found to be independent of the H2 concentration. The explanation is that the activation energy for H2 chemisorption on Pt is close to zero such that H2 adsorption/desorption rates onto catalytic sites were high and the H2 concentration was constant as the solution was saturated with H2.
The conversion (X) of NO3−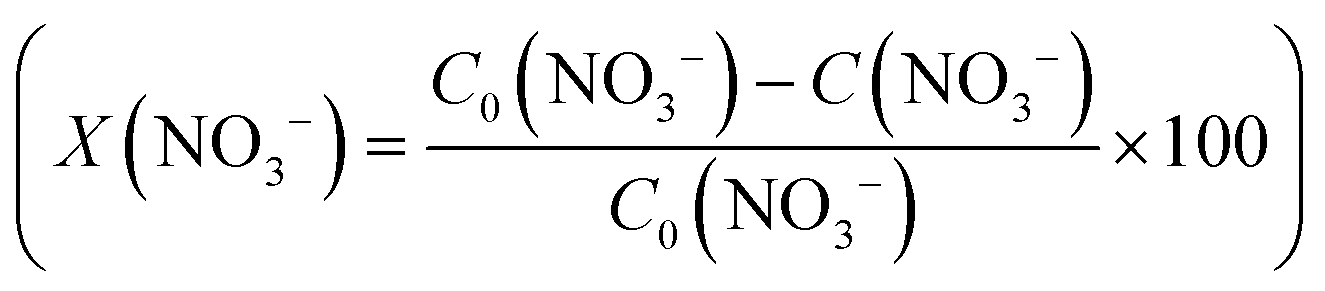 and the selectivity (S) for products (i.e.
and the selectivity (S) for products (i.e.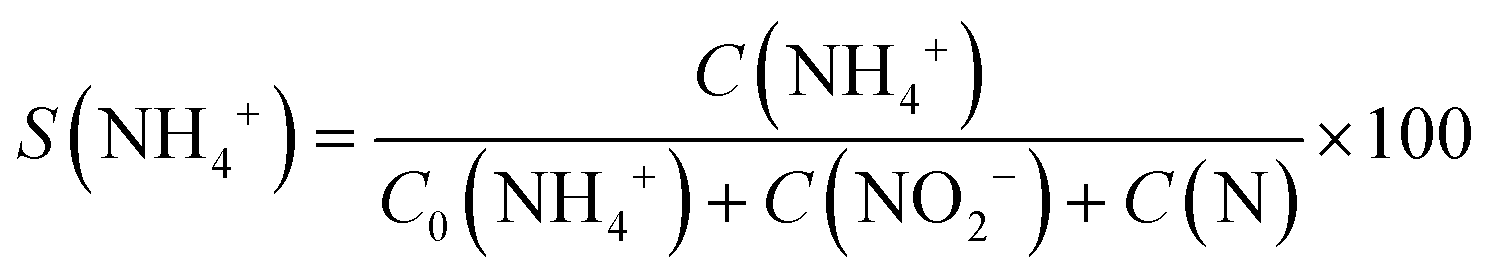 ) are presented in Fig. 4B. The conversion of NO3− over (Pt–Cu)L/Al2O3 was limited to ≈60%. The selectivities of NO2−, NH4+ and N at the end of the reaction time were 70.8%, 21.4% and 7.8%, respectively. Over (Pt–Cu)L/Al2O3, the selectivity of NH4+ was significantly higher (21.4%) compared to that of N (7.8%).
) are presented in Fig. 4B. The conversion of NO3− over (Pt–Cu)L/Al2O3 was limited to ≈60%. The selectivities of NO2−, NH4+ and N at the end of the reaction time were 70.8%, 21.4% and 7.8%, respectively. Over (Pt–Cu)L/Al2O3, the selectivity of NH4+ was significantly higher (21.4%) compared to that of N (7.8%).
The results of the denitration catalytic tests for (Pt–Cu)S/Al2O3 are presented in Fig. 4C and D. The reaction conditions were identical to those used for (Pt–Cu)L/Al2O3. A comparison was performed to determine the effect of different sized nanoparticles on the catalytic potential of Pt–Cu/Al2O3 in the denitration reaction. In Fig. 4C, it can be observed that (Pt–Cu)S/Al2O3 was very active in the conversion of NO3− to NO2−. At the end of the catalytic run, NO3− was completely transformed into reaction products. The greater catalytic activity of (Pt–Cu)S/Al2O3 for NO3− reduction is reflected by the significantly higher k1 value (0.04 min−1) compared to only 0.024 min−1 for (Pt–Cu)L/Al2O3. The small Pt–Cu nanoparticles immobilized on alumina were more active and selective than those previously reported in the literature. Soares et al.15 reported that with an optimum Pt/Cu ratio, of 1% Pt–1% Cu supported on active carbon, the conversion of NO3− was 31% and the selectivity of NO2− was 17%.
Another difference between the two investigated materials is that the concentration of NO2− reached a maximum and then decreased with time according to the kinetic model of successive first-order reactions described by eqn (6) and (8). From the best fit of the experimental data, the calculated k2 value for (Pt–Cu)S/Al2O3 was 0.086 min−1 (see Fig. 4C) compared to only 0.019 min−1 for (Pt–Cu)L/Al2O3 (Fig. 4A). The final concentrations of NO2− and NH4+ were 0.55 and 0.88 mM, respectively.
The NO3− conversion and reaction selectivities of NO2−, NH4+ and N at the end of the reaction time were 27.5%, 44.4% and 28.0%, respectively (see Fig. 4D). The selectivity of NH4+ was about twice that of N.
It is well established that the support plays an essential role in catalysis, influencing the catalytic behavior of the metal particles impregnated on it. The strength of the metal–support interaction is usually difficult to quantify depending on the preparation method, metal precursors, nature of the oxide supports used, etc. It was suggested that the strong metal–support interaction is unavoidable for catalysts prepared by support impregnation.29,30 In some cases, in contrast to the impregnated materials, the metal–support interaction was minimized by depositing the metal nanoparticles on oxide supports. For example, the negative effect of alumina acidity on the catalytic activity of Ru for ammonia synthesis was minimized by using colloidal Ru nanoparticles of ≈5 nm.31
Epron et al.10 reported that the interaction between the platinum and copper of an impregnated Pt–Cu/γ-Al2O3 catalyst was influenced by reducing and oxidizing pretreatment conditions. A reducing treatment of Pt–Cu/Al2O3 induced the diffusion of copper inside the bimetallic particles, enriching the surfaces in platinum.
However, to the best of our knowledge, the support and size effects on the Pt–Cu system was not investigated previously. It is important to design experiments to decouple the effects of support and nanoparticle size on catalytic performance. The effect of support was eliminated by performing an activity test with the simple colloidal Pt–Cu nanoparticles. Prior to the catalytic runs, the Pt–Cu nanoparticles were dispersed into a liquid phase containing NO3− ions. The total amount of nanoparticles was similar (3.6 mg) to the tests performed with supported catalysts (0.3 g of Pt–Cu/Al2O3). The comparative results of the catalytic runs involving (Pt–Cu)S and (Pt–Cu)L are presented in Fig. 5.
The change with time of the NO3−, NO2−, NH4+ and N (N2) concentrations in the presence of (Pt–Cu)L nanoparticles (average size of 4.8 nm) dispersed in a liquid-phase is presented in Fig. 5A. The pH evolution with time is presented in the same figure. In contrast to the (Pt–Cu)L/Al2O3 catalyst, which deactivated after 70 min (Fig. 4A), the conversion of NO3− over the unsupported (Pt–Cu)L is slow but steady (see Fig. 5A). On the other hand, the values of reaction constant k1 were comparable: 0.021 and 0.024 min−1 for the unsupported and supported nanoparticles, respectively (see Fig. 4A and 5A). The disappearance of NO3− was closely followed by the formation of intermediate product NO2− with a rate constant k2 = 0.019 min−1. Nitrogen was absent, and only small amounts of NH4+ were detected at the end of the reaction time (0.07 mM). The total conversion of NO3− was only 29%, and the selectivities of NO2−, NH4+ and N at the end of the reaction time were 87.5%, 12.5% and 0%, respectively (Fig. 5B).
The free (Pt–Cu)S nanoparticles of ≈1.6 nm showed much better catalytic performance than the alumina-supported particles. Most of the NO3− (≈93%) was rapidly converted to products within the first 40 minutes of the reaction time with an apparent reaction rate constant k1 of 0.08 min−1 (Fig. 5C). In contrast, the k1 value when (Pt–Cu)S/Al2O3 was present was only half, 0.04 min−1 (Fig. 4C). Thus, it can be said that the support negatively influenced the activity of the (Pt–Cu)S nanoparticles. The k2 value for NO2− transformation to the final reaction products (N2 and NH4+) in the presence of the unsupported catalyst was smaller (0.023 min−1, Fig. 5C) compared to that of the supported (Pt–Cu)S (k2 = 0.086 min−1, Fig. 4C). However, despite the lower k2 value, the unsupported (Pt–Cu)S was more efficient at converting NO2− to the final products because the NO2− depletion rate depends on the interplay of both the k1 and k2 values according to eqn (8).
A remarkable result is that NO3− and NO2− were completely converted over the unsupported (Pt–Cu)S. At the end of the reaction time, the selectivities of NO2−, NH4+ and N were 0%, 44.9% and 55%, respectively (see Fig. 5D).
An overview of the catalytic test results for supported and unsupported Pt–Cu nanoparticles is presented in Fig. 6. It can be observed that the large Pt–Cu nanoparticles (4.8 nm) showed low activity for the catalytic reduction of NO3− ions, leaving a large amount of the initial nitrate unconverted at the end of the reaction. The alumina support had a positive effect on the catalytic activity of (Pt–Cu)L, leading to an increase in NO3− conversion. A small amount of N2 was observed in the case of supported (Pt–Cu)L, whereas the unsupported bimetallic nanoparticles produced only a small amount of ammonia as a final reaction product.
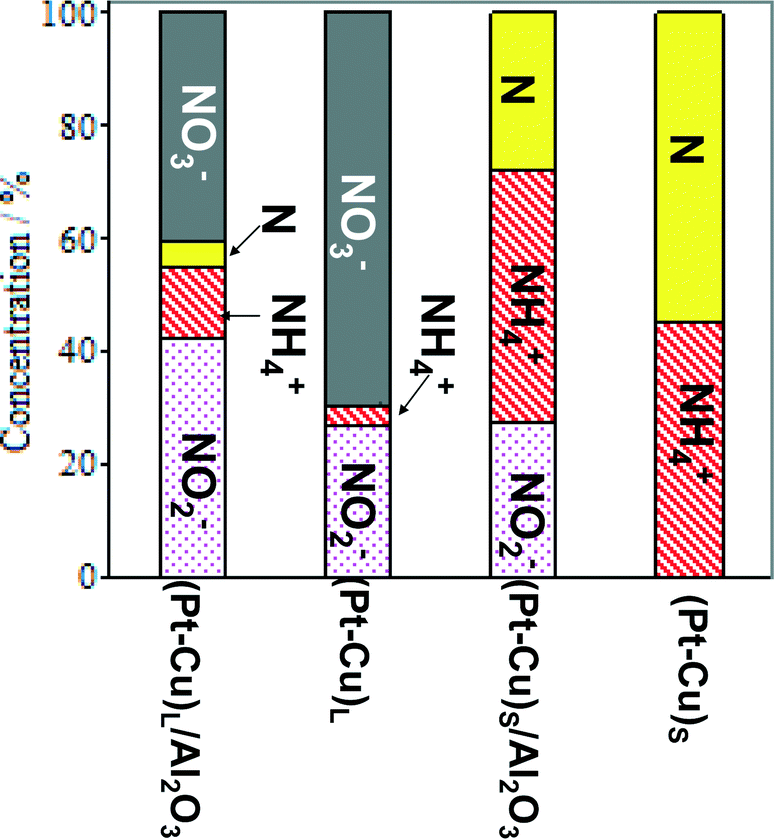 | ||
| Fig. 6 Distribution of reactant and products at the end of time-controlled denitration reactions involving alumina-supported and unsupported (Pt–Cu)L and (Pt–Cu)S nanoparticles. | ||
In contrast to (Pt–Cu)L, the smaller (Pt–Cu)S (1.6 nm average size) proved to be very active, completely transforming the NO3− (Fig. 6). The amounts of NH4+ produced were relatively close to each other for the supported and unsupported bimetallic nanoparticles, but the quantity of N increased in the reactions involving unsupported particles. One significant difference between the compared materials is that in the presence of unsupported (Pt–Cu)S, the NO2− intermediate was completely transformed into the final products (N2 and NH4+). In the case of (Pt–Cu)S/Al2O3, the support inhibited the complete transformation of NO2− into the final products and decreased the selectivity for N2. The catalytic reactivity of nanoparticles as a function of their size is still a debated question. It seems that there are several factors acting simultaneously which result in enhanced catalytic activity observed generally for small nanoparticles. One explanation can be that with decreasing size of nanoparticles, the surface-to-volume ratio increases, leading to a larger number of low-coordinated atoms available for reactants. The size effects might also reflect a metal–support interaction which can be electronic (charge transfer between support and nanoparticles) or structural (strain effects at the interfaces between support and nanoparticles).32
The influence of the alumina support on the k1 and k2 reaction constants was further analyzed. The impact of support on each reduction step was separately determined. The first reduction step consists of the dissociation of H2 on noble metal sites followed by the reduction of NO3− to NO2− on Cu centers.5 Studies dedicated to the investigation of the specific NO3− adsorption sites prior to the first reduction step are scarce. Ebbesen et al.33 investigated by IR spectroscopy (ATR-IR) the adsorption of NO2− and NH4+ from the aqueous phase on Pt/Al2O3 and Pd/Al2O3, in relation to heterogeneous hydrogenation of NO2−. The adsorption of NO2− onto supported metal catalyst was proved by IR spectroscopy. In contrast, the NH4+ ion does not chemisorb on the noble metal but is stabilized by an electrostatic interaction. Taking into account that NO3− and NO2− are resembling molecules, both bearing one negative charge, it can be supposed that NO3− is adsorbed in a similar manner on the noble metal sites (i.e. Pt). Thus, it can be speculated that NO3− adsorbed on Pt sites migrates to Cu0 sites where it is reduced to NO2−. The copper sites are stabilized in a reduced state by intimate contact with a noble metal (i.e. Pd, Pt) via the H spillover mechanism. The mechanism of the second step, which is governed by the k2 rate constant, is different. The participation of H+ in the reduction mechanism of NO2− should be taken into consideration. We have observed that, if the pH value at the end of the NO3− conversion is adjusted to an acidic one by addition of HCl (i.e. pH = 3), the unconverted NO2− is rapidly and selectively reduced to N2 and no extra ammonia is formed. The reduction of NO2− in the presence of HCl is a catalytic reaction because it takes place only in the presence of a catalyst. There are reactions in which the acidity of the support has a strong effect on metal catalytic activity. One typical example is the negative effect played by alumina acidity (Lewis and Brønsted acidity) on the catalytic activity of the supported metals in ammonia synthesis.34
Support effects on nitrate reduction over bimetallic nanoparticles were analyzed in a few previously published studies. For example, Pd–Cu supported on TiO2 exhibited the best performance in terms of NO3− conversion and selectivity of N2.14
The effect of alumina on the catalytic properties of small and large Pt–Cu bimetallic nanoparticles, i.e., on the apparent rate constants k1 and k2, was investigated. The results are compared in Fig. 7.
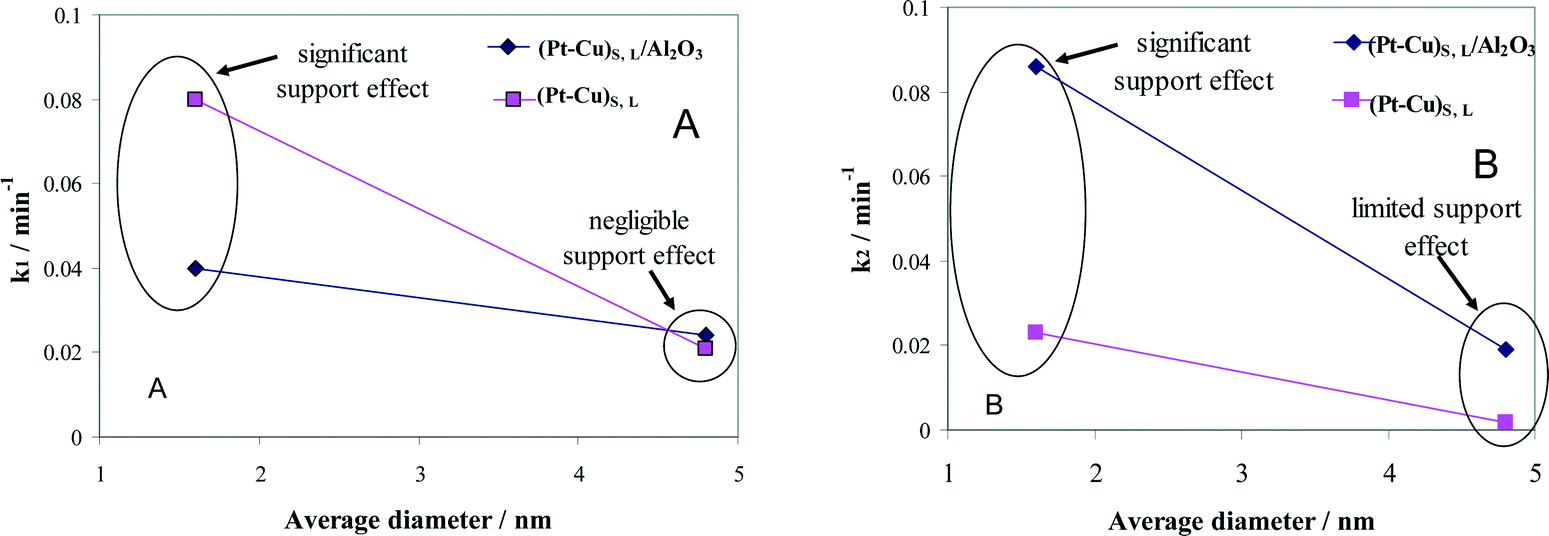 | ||
| Fig. 7 Particle size and support effects on the denitration apparent rate constants k1 (A) and k2 (B). | ||
In Fig. 7A, it can be observed that the alumina support had little effect on the denitration (NO3− → NO2−) activity of the ≈4.8 nm (Pt–Cu)L. The k1 values measured when the large Pt–Cu nanoparticles were used are close to each other regardless of the presence or absence of Al2O3. Conversely, the supported vs. unsupported behavior of the (Pt–Cu)S particles was strongly influenced by the presence of alumina; k1 values decreased from 0.08 for unsupported to 0.04 min−1 for (Pt–Cu)S nanoparticles dispersed on a support.
The metal–support interaction reflected by the k2 constant (NO2− → final products) is presented in Fig. 7B. Both the large and small Pt–Cu nanoparticles were affected by Al2O3. However, the k2 values determined for (Pt–Cu)L were less influenced by alumina compared to (Pt–Cu)S. Interestingly, in this case, the support has a positive effect on the catalytic activity of the bimetallic nanoparticles. Therefore, it can be assumed that the acidity of alumina contributed to the increase in the k2 value, which is a strong argument for the importance of the proton in the reaction mechanism of the second step. Previous studies reported that the NO2− transformation rate is more sensitive to the pH value than that of the reduction of NO3−.35 Proof that the acidic properties of the F− modified CeO2 support are directly involved in nitrate reduction over Pt monometallic particles has been published.36 It was suggested that the addition of CO2 to decrease the pH of the solution might improve the selectivity of N2 in detriment to that of NH4+. Another alternative to increase the selectivity of N2 is to better control the predominant morphology (size and shape) of the metallic nanoparticles. In the presence of cubic Pt nanocrystals of ≈10 nm dispersed on Al2O3, the selectivity of N2 during NO2− reduction was higher compared to supported polycrystalline Pt particles, richer in high index planes and surface defects.37
Conclusions
Well-defined and dispersed Pt–Cu nanoparticles were synthesized by an alkaline polyol method. The synthesis was directed to obtain bimetallic nanoparticles with two predominant average sizes (1.6 nm and 4.8 nm).The present study proved that denitration reaction is a strongly structure-sensitive reaction. Both the size and metal–support interaction, reflected by the apparent rate constants of denitration steps, are crucial in determining the catalytic behavior of Pt–Cu nanoparticles. The supported as well as the unsupported (Pt–Cu)S nanoparticles proved to be very active, completely converting NO3− and NO2− to the final reaction products (N2 and NH4+). They were more selective for N2 compared to their larger counterparts. The simple and supported (Pt–Cu)L nanoparticles deactivated over time, leaving a large amount of NO3− and NO2− unconverted.
The kinetic analysis indicates that the activity of the larger Pt–Cu nanoparticles is less influenced by the support than the smaller particles. The first reduction step over (Pt–Cu)L, which is governed by k1, was not influenced by the Al2O3 support. In contrast, the support showed a strong hindering effect on the activity of the small (Pt–Cu)S nanoparticles during NO3− reduction.
In the second reduction step (NO2− reduction), governed by the k2 reaction constant, the support showed a limited influence on the activity of (Pt–Cu)L and a strong one on that of (Pt–Cu)S. Interestingly, the alumina support had a positive effect on the reaction rate constant k2 for both types of nanoparticles, indicating a different reaction mechanism than that in the first step. It is suggested that the acidity (protons) contributed by the support plays an essential role in converting NO2− to the final products.
Acknowledgements
Financial support through grant PNII-PTPCCA BICLEANBIOS 46/2012 is greatly acknowledged.References
- M. H. Ward, T. M. DeKok, P. Levallois, J. Brender, G. Gulis, B. T. Nolan and J. VanDerslice, Environ. Health Perspect., 2005, 1607–1614 CrossRef CAS
.
- K. J. Vorlop and T. Tacke, Chem. Ing. Tech., 1989, 61, 836–837 CrossRef CAS
- O. S. G. Soares, J. J. Órfão and M. F. R. Pereira, Desalination, 2011, 279, 367–374 CrossRef CAS PubMed
- A. Pintar and J. Batista, Appl. Catal., B, 2006, 63, 150–159 CrossRef CAS PubMed
- J. Sá, S. Gross and H. Vinek, Appl. Catal., A, 2005, 294, 226–234 CrossRef PubMed
- I. Witońska, S. Karski, J. Rogowski and N. Krawczyk, J. Mol. Catal. A: Chem., 2008, 287, 87–94 CrossRef PubMed
- F. Marchesini, S. Irusta, C. Querini and E. Miró, Appl. Catal., A, 2008, 348, 60–70 CrossRef CAS PubMed
- N. Krawczyk, S. Karski and I. Witońska, React. Kinet., Mech. Catal., 2011, 103, 311–323 CrossRef CAS PubMed
- R. Rodríguez, C. Pfaff, L. Melo and P. Betancourt, Catal. Today, 2005, 107, 100–105 CrossRef PubMed
- F. Epron, F. Gauthard and J. Barbier, Appl. Catal., A, 2002, 237, 253–261 CrossRef CAS
- I. Witońska, S. Karski and J. Gołuchowska, Kinet. Catal., 2007, 48, 823–828 CrossRef
- G. Strukul, R. Gavagnin, F. Pinna, E. Modaferri, S. Perathoner, G. Centi, M. Marella and M. Tomaselli, Catal. Today, 2000, 55, 139–149 CrossRef CAS
- I. Balint, A. Miyazaki and K.-I. Aika, Appl. Catal., B, 2005, 59, 72–81 Search PubMed
- K. Wada, T. Hirata, S. Hosokawa, S. Iwamot and M. Inoue, Catal. Today, 2012, 185, 81–87 CrossRef CAS PubMed
- O. S. G. P. Soares, J. J. M. Órfão and M. F. R. Pereira, Appl. Catal., B, 2009, 91, 441–448 CrossRef CAS PubMed
- N. Barrabés, J. Just, A. Dafinov, F. Medina, J. Fierro, J. Sueiras, P. Salagre and Y. Cesteros, Appl. Catal., B, 2006, 62, 77–85 CrossRef PubMed
- F. Papa, C. Negrila, A. Miyazaki and I. Balint, J. Nanopart. Res., 2011, 13, 5057–5064 CrossRef CAS
- B. C. Gates, Chem. Rev., 1995, 95, 511–522 CrossRef CAS
- F. Family and T. Vicsek, J. Phys. A: Math. Gen., 1985, 18, L75–L81 CrossRef
- P. F. Chauvy, C. Madore and D. Landolt, Surf. Coat. Technol., 1998, 110, 48–56 CrossRef CAS
- K. Kusada, M. Yamauchi, H. Kobayashi, H. Kitagawa and Y. Kubota, J. Am. Chem. Soc., 2010, 132, 15896–15898 CrossRef CAS PubMed
- B. R. Strohmeier, D. E. Levden, R. S. Field and D. M. Hercules, J. Catal., 1985, 94, 514–530 CrossRef CAS
- J. Z. Shyu and K. Otto, Appl. Surf. Sci., 1988, 32, 246–252 CrossRef CAS
- G. Ertl, R. Hierl, H. Knözinger, N. Thiele and H. Urbach, Appl. Surf. Sci., 1980, 5, 49–64 CrossRef CAS
- I. Platzman, R. Brener, H. Haick and R. Tannenbaum, J. Phys. Chem. C, 2008, 112, 1101–1108 CAS
- S. Suzer, H. Sezen, G. Ertas and A. Dâna, J. Electron Spectrosc. Relat. Phenom., 2010, 176, 52–57 CrossRef CAS PubMed
- J. Cazaux, J. Electron Spectrosc. Relat. Phenom., 2010, 178, 357–372 CrossRef PubMed
- W. Lindsay, M. Sadiq and L. Porter, Soil Sci. Soc. Am. J., 1981, 45, 61–66 CrossRef CAS
- I. Balint, A. Miyazaki and K.-I. Aika, Chem. Mater., 2001, 13, 932–938 CrossRef CAS
- I. Balint, A. Miyazaki and K.-I. Aika, Chem. Mater., 1999, 11, 378–383 CrossRef CAS
- A. Miyazaki, I. Balint, K.-I. Aika and Y. Nakano, J. Catal., 2001, 204, 364–371 CrossRef CAS
- B. R. Cuenya, Thin Solid Films, 2010, 518, 3127–3150 CrossRef CAS PubMed
- S. D. Ebbesen, B. L. Mojet and L. Lefferts, Langmuir, 2008, 24, 869–879 CrossRef CAS PubMed
- I. Balint and A. Miyazaki, Trans. Mater. Res. Soc. Jpn., 2007, 32, 387 CAS
- Y. H. Liou, C. J. Lin, I. C. Hung, S. Y. Chen and S. L. Lo, Chem. Eng. J., 2012, 181, 236–242 CrossRef PubMed
- N. Barrabés, A. Dafinov, F. Medina and J. Sueiras, Catal. Today, 2010, 149, 341–347 CrossRef PubMed
- A. Miyazaki, T. Asakawa, Y. Nakano and I. Balint, Chem. Commun., 2005, 3730–3732 RSC
| This journal is © The Royal Society of Chemistry 2015 |